Abstract
Regulatory T-cell (Treg) modulation is developing as an important therapeutic opportunity for the treatment of a number of important diseases, including cancer, autoimmunity, infection, and organ transplant rejection. However, as demonstrated with IL-2 and TGN-1412, our understanding of the complex immunological interactions that occur with Treg modulation in both non-clinical models and in patients remains limited and appears highly contextual. This lack of understanding will challenge our ability to identify the patient population who might derive the highest benefit from Treg modulation and creates special challenges as we transition these therapeutics from non-clinical models into humans. Thus, in vivo testing in the most representative animal model systems, with careful progress in the clinic, will remain critical in developing therapeutics targeting Treg and understanding their clinical utility. Moreover, toxicology models can inform some of the potential liabilities associated with Treg modulation, but not all, suggesting a continued need to explore and validate predictive models.
Introduction
Over the last several decades, therapeutic immunomodulation through administration of recombinant cytokines, growth factors, adjuvants, vaccines, and monoclonal antibodies has become part of established clinical practice in the treatment of infection, autoimmunity, allergy, and cancer. Although experience with immunomodulatory therapies has revealed both their potential for remarkable therapeutic benefit, they also induce unique toxicities (Gribble et al., Citation2007). Modulation of regulatory T-cells (Treg) offers a unique therapeutic opportunity due to the pivotal role these cells play in regulating immunity by acting as a primary mediator between immunity and tolerance. This presentation evaluates the promise and challenge of regulatory T-cell modulation through a review of Treg biology, toxicology considerations, and translational challenges.
Unexpected results: IL-2 expands the CD4+ T-cell compartment, but does not impact HIV disease severity
In October 2009, a report was published in the New England Journal of Medicine describing the results of two clinical trials evaluating the utility of recombinant interleukin (IL)-2 (rIL-2) therapy in human immunodeficiency virus (HIV)+ patients treated with anti-retroviral therapy (Abrams et al., Citation2009). Whereas the SILCAAT trial enrolled HIV+ patients with low CD4+ T-cell counts (50–299 cells/mm3, 1695 patients), the ESPRIT trial enrolled patients with higher CD4+ T-cell counts (>300 cells/mm3, 4111 patients). Each trial had evenly balanced arms assigned to treatment with or without rIL-2 to increase CD4+ counts, wherein rIL-2 was administered cyclically as two daily doses for five consecutive days at 8-week intervals. In the SILCAAT trial, rIL-2 was administered as six cycles of 4.5 million IU/dose, and in the ESPRIT trial, rIL-2 was administered as three cycles of 7.5 million IU/dose. Cyclical rIL-2 therapy was subsequently administered to patients so as to maintain CD4+ T-cell counts above predetermined target levels. The primary endpoint was opportunistic disease or death from any cause in both trials, and the median follow-up period was 7–8 years. The cost for these trials was estimated as ∼$100 million.
Results from these trials demonstrated rIL-2-stimulated CD4+ T-cell counts in each patient population as anticipated. At one year, the median increase in CD4+ T-cells was 99 cells/mm3 (SILCAAT) and 185 cells/mm3 (ESPRIT) among patients treated with rIL-2 and anti-retroviral therapy over patients treated with anti-retroviral therapy alone. During the entire follow-up period, CD4+ T-cell counts increased by an average of 53 cells/mm3 (SILCAAT) and 159 cells/mm3 (ESPRIT) among patients treated with rIL-2 and anti-retroviral therapy over patients treated with anti-retroviral therapy alone. Despite this sustained increase in CD4+ T-cell counts, no clinical benefit of rIL-2 was noted as measured by risk of opportunistic infection or death among patients in either trial. Specifically, hazard ratios of 0.91 (95% confidence interval: 0.70–1.18; p = 0.47) and 0.94 (95% confidence interval: 0.75–1.16; p = 0.55) were estimated in the SILCAAT and ESPRIT trials, respectively, where a hazard ratio <1 demonstrates decreased risk of death or disease associated with treatment. These increases in circulating CD4+ T-cells with rIL-2 are consistent with diminished responses in patients with greater baseline deficits in CD4+ T-cell concentrations and with previously reported responses in HIV patients considering the extended treatment-free observation period in both patient populations (used for the denominator) (reviewed in Piscitelli et al., Citation2000).
The results from this study appear to call into question the utility of CD4+ counts as a sufficient surrogate marker of disease severity in patients with HIV, since improved CD4+ T-cell counts did not correlate with reduced risk of disease or death. But, why didn’t the IL-2-mediated increase in CD4+ T-cell counts decrease the risk of infection or death among HIV+ patients in the SILCAAT and ESPRIT studies? One possibility, noted by the authors, is that CD4+ T-cells induced by rIL-2 in these patients have limited or no role in host defense. Another possibility proposed by the authors is that the CD4+ T-cells induced by rIL-2 have some modest clinical benefit that is counteracted by negative effects of rIL-2 (Abrams et al., Citation2009). To gain insight into this issue requires a review of what we know about the immunological activity of IL-2.
IL-2 is a potent T-cell mitogen
Experiments in the mid-1970s revealed the presence of potent mediators in the cell culture supernatant of activated T-cells that stimulated the growth and maintenance of cultured primary T-cells (Morgan et al., Citation1976; Gillis and Smith, Citation1977; Ruscetti et al., Citation1977); the cytokine responsible for this activity was subsequently established as IL-2 (Taniguchi et al., Citation1983). The identification and availability of this cytokine as an experimental tool enabled in vitro experimentation on cloned T-cells with single antigenic specificity to probe the nature of the receptors, cytokines, and signaling pathways involved in T-cell activation and cell fate determination (Oppenheim, Citation2007). Subsequent work also revealed pleiotropic activity of IL-2 in activating NK cells (Henney et al., Citation1981) and B-cells (Zubler et al., Citation1984) in addition to T-cells. This work, which was largely conducted in vitro, resulted in a prevailing model wherein activation of the T-cell, with co-stimulation via CD28 and other molecules, results in IL-2 production and increased expression of IL-2 receptor (IL-2R), which subsequently drive extensive clonal expansion and development of effector function (Malek, Citation2008).
Today rIL-2, marketed as Proleukin® (aldesleukin), is used therapeutically in the treatment of metastatic melanoma and renal cell carcinoma. Clinical experience across multiple studies suggest overall response rates (partial and compete response) of 13–20% in renal cell carcinoma, including a 7% complete response rate (Jeal and Goa, Citation1997; Novartis, Citation2011). Similarly, a 16% overall response rate was observed in patients with metastatic melanoma treated with high dose rIL-2, including complete response in 6% of patients (Atkins et al., Citation1999; Novartis, Citation2011). Although these overall response rates are low, the observation of durable complete responses in a fraction of patients with late stage cancer upon treatment with IL-2 speaks to the dramatic potential for this immunotherapy in cancer treatment. Unfortunately, IL-2 therapy is associated with a number of serious toxicities that require special management, including constitutional symptoms of an acute-phase response (e.g., fatigue, malaise, fever, myalgia, and arthalgia); toxicities to a wide range of organs and systems including the gastrointestinal tract, nervous system, liver, skin, cardiovascular system, kidneys, skin, and the hematological system; and, is associated with potentially severe capillary leak syndrome (Novartis, Citation2011).
Conversely, IL-2 blockade is used to prevent organ transplant rejection. Two therapeutic anti-IL-2R antibodies, Simulect® (basiliximab) and Zenapax® (daclizumab), are currently approved for the prevention of organ transplant rejection. In addition, IL-2 suppression occurs following treatment with glucocorticoids (Leung and Bloom, Citation2003), Prograf® (tacrolimus), and Rapamune® (sirolimus) contributing to their immunosuppressive activity and utility in suppressing organ transplant rejection and other conditions.
IL-2 deficiency results in autoimmunity
Given the potent proinflammatory role of IL-2 identified through the in vitro studies and clinical experience described above, we might anticipate overt immunosuppression in IL-2-deficient animals. However, this is not the case. Mice deficient in IL-2 develop apparently normal B- and T-cell compartments (using CD4 and CD8 to enumerate T-cells), but display impaired ex vivo polyclonal T-cell activation and ability to induce IgM secretion in stimulated B-cells (Schorle et al., Citation1991). Moreover, by 9 week-of-age, IL-2-deficient mice develop inflammation, severe anemia, and an ulcerative colitis-like autoimmunity, and die after 10 week-of-age (Sadlack et al., Citation1993; Kramer et al., 1995; Suzuki et al., Citation1995). Histopathology evaluation of IL-2-deficient mice reveals lymphadenopathy and splenomegaly associated with an increase in B- and T-cell populations, including activated cells. Evaluation of serum demonstrates markedly increased IgG1 and IgE levels and the presence of autoantibodies leading to the observed (hemolytic) anemia. The observed colitis/inflammation in the gut were driven by antigenic stimulation, as mice reared in germ-free environments did not develop the colitis (Sadlack et al., Citation1993). Moreover, the disease could be reduced or eliminated with introduction of IL-2 wild-type bone marrow cells to repopulate thymic T-cells (Kramer et al., 1995). Functionally, IL-2-deficient mice have reduced immunity to various viruses attributable to modest reductions in helper and cytotoxic T-cell responses and marked reductions in NK cell responses (Horak et al., Citation1995), but are capable of rejecting islet allografts (Steiger et al., Citation1995).
Identification of Treg
Taken together, the aggregate data derived from IL-2- or IL-2R-deficient mice demonstrates that IL-2 is not obligatory for the normal early development and ontogeny of the immune system and appears redundant for initiation of immunity. However IL-2 is non-redundant to support development of a unique T-cell population that normalize inflammation and protect the host from autoimmunity by establishing peripheral T-cell homeostasis (Almeida et al., Citation2002; Hori et al., Citation2003; Malek and Bayer, Citation2004). The cells responsible for maintaining peripheral tolerance were subsequently identified as a small fraction of CD4+ or CD8+ T-cells expressing CD25, the α chain of the IL-2 receptor (Sakaguchi et al., Citation1995; Asano et al., Citation1996). These cells, natural Treg, are characterized as a population of thymically-derived T-cells with high expression of CD25 that are responsible for tolerance to self-antigen. A second population of Treg, adaptive Treg, has been described in the literature as a peripherally-derived population arising in response to either tissue-specific or foreign antigens, but with variable CD25 expression (Bluestone and Abbas, Citation2003). Other phenotypic characteristics of Treg include increased expression of CTLA-4, glucocorticoid-inducible tumor necrosis factor receptor (GITR), and OX40 (Fontenot and Rudensky, Citation2005), however, these phenotypic markers are not considered definitive of Treg. It was not until the identification of a mutation in the forkhead/winged transcription factor gene FoxP3 as the underlying defect in the scurfy mouse that a more definitive characterization of Treg was possible, discussed below.
Scurfy (sf) mice present with a lymphoproliferation of CD4+CD8− T-cells, anemia and thrombocytopenia, gastrointestinal bleeding, exfoliative dermatitis, splenomegaly, and lymphadenopathy that results in a wasting disease; these mice characteristically die by 3 weeks-of-age (Lyon et al., Citation1990; Blair et al., Citation1994). The scurfy gene mutation was identified in a new member of the forkhead/winged-helix family of transcription regulators, FoxP3 (Brunkow et al., Citation2001). Subsequent work has confirmed the important role of the transcription factor FoxP3 (aka Scurfin) in the development of Treg (Fontenot et al., Citation2003; Hori et al., Citation2003; Khattri et al., Citation2003; Kim et al., Citation2007, Citation2009; Lahl et al., Citation2007). It is worth noting that the scurfy phenotype shares similar characteristics to those observed in mice deficient in CTLA-4 (Read et al., Citation2000) and transforming growth factor (TGF)-β (Powrie et al., Citation1996; Gorelik and Flavell, Citation2000) that also play important roles in Treg activity.
In humans, two rare genetic disorders have been described, IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome) and XLAAD (X-linked autoimmunity-allergic-dysregulation syndrome) as genetic deficiencies in FoxP3. Patients with these genetic deficiencies present with multisystem autoimmunity and severe atopy, including eczema, food allergy, and eosinophilic inflammation (Chatila et al., Citation2000; Bennett et al., Citation2001; Wildin et al., Citation2002; Li et al., Citation2007). The corresponding autoimmunity arising from defects in FoxP3 observed in both rodents and humans suggests a central and conserved evolutionary role for this pathway in mediating peripheral tolerance.
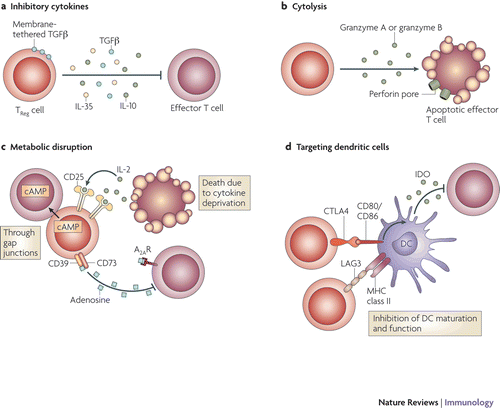
Although FoxP3 is an important mediator of Treg cell development and function, it does not appear to be sufficient for regulatory function, and other factors appear to be involved. Treg appear to exert their immunomodulatory role through multiple mechanisms, including expression of inhibitory cytokines, cytolysis, metabolic disruption, and inhibition of dendritic cell maturation or function (Vignali et al., Citation2008) ().
Figure 1. Basic mechanisms used by Treg cells to modulate immunity. Depiction of the various regulatory T (Treg)-cell mechanisms centered around four basic modes of action. (A) Inhibitory cytokines include interleukin-10 (IL-10), IL-35, and transforming growth factor (TGF)-β. (B) Cytolysis dependent on the granzymes A and B, and perforin. (C) Metabolic disruption includes high-affinity CD25 also know and IL-2 receptor α-dependent cytokine deprivation-mediated apoptosis, cyclic AMP (cAMP)-mediated inhibition, and adenosine receptor 2A (A2AR)-mediated immunosuppression. (D) Targeting dendritic cells (DC) includes mechanisms that modulate DC maturation/function, such as lymphocyte-activation gene 3 (LAG3; also know as CD223)-MHC Class II-mediated suppression of DC maturation, and cytotoxic T-lymphocyte antigen 4 (CTLA4)-CD80/CD86-mediated induction of indoleamine 2,3-dioxygenase (IDO) which is an immunosuppressive molecule made by DC. (Reprinted with permission from Vignali et al., Citation2008).
Thus, the critical role of IL-2 in the development and activity of Treg, coupled with the identified biology of Treg in attenuating immune response, provide a rationale for why the IL-2-mediated expansion of CD4+ T-cells did not improve survival or reduce development of opportunistic disease in patients with HIV in the SILCAAT or ESPRIT trials. These data also suggest that the utility of CD4+ T-cell counts as a surrogate marker of disease activity can be confounded by unusual changes in various sub-populations expressing CD4, including Treg sub-populations and natural killer T (NKT) cells, the latter of which respond to lipid antigens.
Therapeutic utility of modulating Treg
Given the pivotal role Treg play in shaping peripheral tolerance and modulating immunity, one can imagine therapeutic utility in their induction or suppression. Induction of Treg could provide utility in reducing inflammation associated with autoimmune conditions and organ transplant rejection. Potential adverse effects associated with such therapy could include an increase in opportunistic infection or cancer associated with impaired host surveillance and immunity. Alternatively, suppression of Treg could reduce tolerance in the treatment of cancer or chronic infection. In this case, potential side effects could manifest as inflammation or autoimmunity. Moreover, a role for Treg has been identified in maternal–fetal tolerance and maintenance of pregnancy (Mellor and Munn, Citation2000; Aluvihare et al., Citation2004); thus, suppression of Treg could lead to an increased rate of fetal loss; conversely whether induction of Treg could reduce fetal loss in mothers with a history of miscarriage remains an open question.
A model of cancer immunosurveillance
A role for immunity in host protection from cancer can be traced to pioneering experiments by William Coley in the late 1800s, using infection with live or attenuated pathogens in the treatment of inoperable cancers, predominantly sarcomas and lymphomas (Wiemann and Starnes, Citation1994; Hoption Cann et al., Citation2003). Using pathogen vaccination, Coley’s treatment was associated with >10 years disease-free prior to loss from follow-up in 34% and 45% of patients treated for soft tissue sarcomas (n = 84) and lymphomas (n = 33), respectively (Wiemann and Starnes, Citation1994). Such a vaccine would likely result in production of a broad range of cytokines, including tumor necrosis factor (TNF)-α, IL-2, and interferon (IFN)-α, and result in activation of cytotoxic T-cells, macrophages and other innate and adaptive immune cells (Wiemann and Starnes, Citation1994). Modern era vaccines, such as those developed by Dendreon (Higano et al., Citation2009), BioVex (Kaufman and Bines, Citation2010), and Bavarian Nordic (Kantoff et al., Citation2010), shape the immune response towards specific tumor-specific antigens rather than provoke a systemic inflammation in the hopes of avoiding the toxicity while maintaining the anti-tumor immunity.
Review of the epidemiologic literature likewise supports a role for an intact immunity to protect the host from cancer. Among patients treated with various immunosuppressive regimens following organ transplantation, long-term cancer rates are generally observed to increase, particularly for skin, lip, and kidney cancers, as well as cancers associated with oncogenic viruses, including lymphomas (Epstein Barr virus), Kaposi’s sarcoma (human herpes virus 8), liver cancer (hepatitis B and C viruses), and anogenital cancers (human papilloma viruses) (Hanto et al., Citation1985; Penn, Citation1994, Citation2000; Euvrard et al., Citation1997). Patients with human immunodeficiency virus (HIV) present with an increased risk of virally-associated cancers, including lymphomas, Kaposi’s sarcoma, liver cancer, anogenital cancers, as well as lung cancer (Engels et al., Citation2008). Finally, various animal models support a role for innate and adaptive immunity in host protection from chemically- and virally-induced cancers, and rejection of tumors. These studies highlight a particular role for αβT-cells and γδT-cells, NK cells, and macrophages in host defense against induced tumors (Dunn et al., Citation2002).
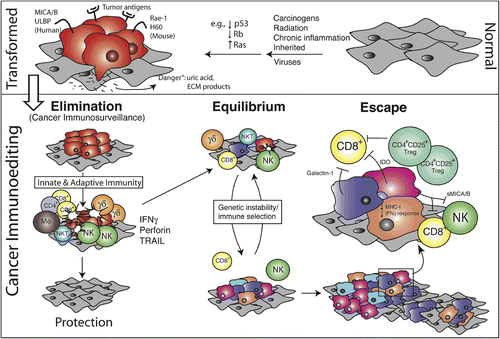
Through experiments to understand the nature of immunity against induced and spontaneous tumors in animal models, Dunn, Schreiber and colleagues proposed a comprehensive model of cancer immunosurveillance (Dunn et al., Citation2002, Citation2004) that builds upon the immunological surveillance model of CitationBurnet (1970). The Dunn–Schreiber model postulates that induction of cancerous cells leads to the expression of unique tumor antigens that initiate host immunity. This immunity can eliminate the tumor, reach a state of equilibrium with the tumor, or allow the tumor to escape (resulting in clinical disease). During the equilibrium phase, immunity applies a selective pressure against the tumor such that susceptible cells are killed and resistant cells, derived from genetic or epigenetic alterations, achieve a selective advantage. These new tumor variants may be recognized by an adaptive immune response, or they may not, thus leading to tumor escape ().
Figure 2. Dunn–Schreiber model of tumor immunosurveillance in involves elimination, tumor editing, and tumor escape. Normal cells (gray) subject to common oncogenic stimuli ultimately undergo transformation and become tumor cells (red) (Top). Even at early stages of tumorigenesis, these cells may express distinct tumor-specific markers and generate proinflammatory “danger” signals that initiate the cancer immunoediting process (Bottom). In the first phase of elimination, cells and molecules of innate and adaptive immunity, which comprise the cancer immunosurveillance network, may eradicate the developing tumor and protect the host from tumor formation. However, if this process is not successful, the tumor cells may enter the equilibrium phase where they may be either maintained chronically or immunologically sculpted by immune “editors” to produce new populations of tumor variants. These variants may eventually evade the immune system by a variety of mechanisms and become clinically detectable in the escape phase. (Reprinted with permission from Dunn et al., Citation2004).
How can the Dunn–Schreiber model of immunosurveillance be reconciled with the curious case of IL-2? IL-2 is used in the treatment of metastatic melanoma and renal cell carcinoma, resulting in durable responses in a fraction of patients with both conditions. These data are consistent with a role for IL-2 as an important cytokine that drives effector T-cell activity. However, IL-2 is an important cytokine for the development and expansion of Treg. So, can the IL-2-mediated expansion of Treg be reconciled against the anti-tumor activity of IL-2?
Could Treg expansion drive anti-tumor immunity?
There is a growing literature suggesting that Treg participate in tumor escape. Treg are increased in a variety of human tumors, including non-small cell lung cancer (Woo et al., Citation2001), breast and pancreatic cancers (Liyanage et al., Citation2002), and gastric and esophageal cancers (Ichihara et al., Citation2003). Furthermore, tumors can produce factors that induce Treg and suppress anti-tumor immunity, including indoleamine 2,3-dioxygenase (IDO), IL-10, and TGFβ (Huang et al., Citation1995; Jones et al., Citation2002; Zou, Citation2006; Munn and Mellor, Citation2007; Godin-Ethier et al., Citation2009). Finally, elimination of Treg improves tumor clearance in various animal models (Onizuka et al., Citation1999; Shimizu et al., Citation1999; Sutmuller et al., Citation2001; Ko et al., Citation2005).
Hirschhorn-Cymerman et al. (Citation2009) recently reported on studies of OX40 ligation by OX-86 in combination with cyclophosphamide, demonstrating improved anti-tumor immunity, as measured by tumor area and survival, in a mouse xenograft model with B16 melanoma cells. Paradoxically, these animals also presented with increased peripheral Treg, whether treated with OX-86 alone or in combination with cyclophosphamide. Closer evaluation of these animals revealed hyper-activation of Treg in the tumor with subsequent apoptosis, suggesting that these cells could be specifically deleted in the tumor, resulting in improved anti-tumor activity. Whereas such activity may be postulated to arise following high dose IL-2 therapy in patients, such activity has not been reported.
Alternatively, a broad literature exists on the role of a proinflammatory environment on the promotion of tumor growth and survival. Specifically, a proinflammatory environment can directly promote tumor growth and survival, promote angiogenesis and metastasis, assist the tumor in subverting adaptive immunity, and alter the tumor response towards hormones and chemotherapeutic agents (Mantovani et al., Citation2008). Under such circumstances, the IL-2-mediated expansion of Treg could alter the local environment of the tumor by suppressing those factors that promote tumor growth and survival.
These data highlight the challenges in interpreting the literature regarding the nature of tumor microenvironment and the role of immunity in supporting or suppressing tumor growth, and suggest the context of inflammation plays a strong role in individual tumor response over time. Indeed, Treg have been shown to play an important role in tumor escape in early, but not late, stages of tumor growth (Elpek et al., Citation2007). Thus, IL-2 likely induces effects on both effector T-cells and Treg in a given individual, and whether this drives anti-tumor immunity will be shaped by other host-specific factors. Such a model is supported by a study reported by Imai et al. (Citation2007), wherein depletion of Treg prior to IL-2 therapy enhanced anti-tumor immunity in a mouse xenograft model.
Implications of Treg therapy in the treatment of immunological disorders and cancer
It is clear that the context surrounding antigen detection and presentation, including co-stimulation and cytokines, is centrally important for establishing immunity or tolerance. As shown for cancer, interventional treatments select for tumor variants that escape tumor surveillance, and effective therapy at one phase of disease may not be effective at another. Because a specific ‘cancer’ is comprised of a population of genetic and phenotypic variants, it is challenging to make broad generalizations regarding treatment design. Ultimately, the beneficial effects of immunotherapy, including modulation of Treg, will depend on an ability to identify ‘responder’ populations, or those most likely to derive benefit at a given stage of their disease. Because our non-clinical models do not generally reflect the clinical history of patients, it is unlikely that they will fully inform the potential efficacy or risks of our experimental therapeutics.
The data presented above also demonstrate that the central importance of the microenvironment at the immune synapse provides challenges for us when interpreting experimental results. For example, the proinflammatory nature of IL-2 that is identified through in vitro study is not borne out in vivo, where IL-2 deficiency leads to a proinflammatory condition. Thus, we must remain congnizant of the limitations of our experimental systems, which likely contributes to the conflicting results that are reported in the literature. Also, as identified by Hirschhorn-Cymerman et al. (Citation2009), monitoring effects in the periphery, wherein an increase in peripheral Treg in response to OX40 ligation and cyclophosphamide was observed, did not reflect therapeutic activity in the tumor, where Treg were reduced.
Our understanding of Treg biology is relatively immature, yet these cells are developing as a critical therapeutic target in establishing (or tipping) the balance between immunity and tolerance. The intent of this talk has been to highlight the promises and complexities in considering Treg therapy in treating immunological disorders and cancer. Using IL-2 therapy as a common thread, this talk has highlighted the challenges and complexities in understanding the myriad interactions of this cytokine in shaping the host immune response, which is mediated in part via modulation of Treg. Similar complexities are expected in developing therapies targeting Treg, including developing an understanding of the role of Treg in a given disease in the context of the broader immunological states, and how to appropriately tailor therapy to avoid toxicity. One need look no further than the well-publicized toxicity of TGN-1412 after a single dose in healthy volunteers to be reminded of the risks associated with attempting T-cell modulation (Suntharalingam et al., Citation2006).
The toxicological implications associated with Treg modulation have been recently reviewed (Fort and Narayanan, Citation2010). Theoretically, one could consider Treg induction in the treatment of autoimmunity (Kim et al., Citation2007), to establish tolerance to a transplanted organ (Boros and Bromberg, Citation2009), or in the treatment of spontaneous abortions (Leber et al., Citation2010). In this case, potential side effects could include increased risk of infection or cancer associated with impaired immunosurveillance. From a pre-clinical perspective, it is likely that non-clinical species could inform these risks, although standard 2-year carcinogenicity studies have <50% positive predictive value for identifying cancer associated with immunosuppression (Bugelski et al., Citation2010). Alternatively, one could consider Treg suppression in the treatment of cancer or chronic infection, as described above (Eastwood et al., Citation2010). Potential toxicities associated with such therapies would include systemic inflammation or autoimmunity, particularly in response to challenge, and rejection of pregnancy via loss of maternal–fetal tolerance. As demonstrated with TGN-1412, non-clinical species may be poor predictors for systemic inflammation and autoimmunity in humans (Gribble et al., Citation2007). The basis for this interspecies difference remains unresolved, but may be attributed to an evolutionary divergence in T-cell regulation (Nguyen et al., Citation2006; Soto et al., Citation2010; Eastwood et al.).
In summary, Treg modulation is developing as a critical area for immunotherapy research, with applications in a wide variety of diseases. However, because our understanding of the underlying biology around Treg and their myriad interactions is relatively immature, and because the immune response in any individual is highly contextual, we must continuously challenge ourselves to understand those factors that contribute to both defining the patient population that might derive the highest benefit as well as those that may be at highest risk from such therapies. Moreover, there is emerging data that non-clinical models are poor predictors for human risk for some immune-mediated toxicities. As a result, Treg modulation can be expected to require special attention and caution when transitioning from pre-clinical to clinical evaluation, which is recognized in recent guidance from the European Medicines Agency (CHMP, Citation2007). Thus, it is in our interest to continue efforts to develop and validate models or systems that can inform human risk from such therapies.
Declaration of interest:.The Author is an employee of Amgen, Inc. The Author reports no conflicts of interest. The Author alone is responsible for the content and writing of the paper.
References
- Abrams, D., Lévy, Y., Losso, M. H., Babiker, A., Collins, G., Cooper, D. A., Darbyshire, J., Emery, S., Fox, L., Gordin, F., Lane, H. C., Lundgren, J. D., Mitsuyasu, R., Neaton, J. D., Phillips, A., Routy, J. P., Tambussi, G. and Wentworth, D.; INSIGHT-ESPRIT Study Group; SILCAAT Scientific Committee. 2009. Interleukin-2 therapy in patients with HIV infection. N. Engl. J. Med. 361:1548–1559.
- Almeida, A. R., Legrand, N., Papiernik, M. and Freitas, A. A. 2002. Homeostasis of peripheral CD4+ T-cells: IL-2R α and IL-2 shape a population of regulatory cells that controls CD4+ T-cell numbers. J. Immunol. 169:4850–4860.
- Aluvihare, V. R., Kallikourdis, M. and Betz, A. G. 2004. Regulatory T-cells mediate maternal tolerance to the fetus. Nat. Immunol. 5:266–271.
- Asano, M., Toda, M., Sakaguchi, N. and Sakaguchi, S. 1996. Autoimmune disease as a consequence of developmental abnormality of a T-cell subpopulation. J. Exp. Med. 184:387–396.
- Atkins, M. B., Lotze, M. T., Dutcher, J. P., Fisher, R. I., Weiss, G., Margolin, K., Abrams, J., Sznol, M., Parkinson, D., Hawkins, M., Paradise, C., Kunkel, L. and Rosenberg, S. A. 1999. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 17:2105–2116.
- Bennett, C. L., Christie, J., Ramsdell, F., Brunkow, M. E., Ferguson, P. J., Whitesell, L., Kelly, T. E., Saulsbury, F. T., Chance, P. F. and Ochs, H. D. 2001. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27:20–21.
- Blair, P. J., Bultman, S. J., Haas, J. C., Rouse, B. T., Wilkinson, J. E. and Godfrey, V. L. 1994. CD4+CD8– T-cells are the effector cells in disease pathogenesis in the scurfy (sf) mouse. J. Immunol. 153:3764–3774.
- Bluestone, J. A. and Abbas, A. K. 2003. Natural versus adaptive regulatory T-cells. Nat. Rev. Immunol. 3:253–257.
- Boros, P. and Bromberg, J. S. 2009. Human FOXP3+ regulatory T-cells in transplantation. Am. J. Transplant. 9:1719–1724.
- Brunkow, M. E., Jeffery, E. W., Hjerrild, K. A., Paeper, B., Clark, L. B., Yasayko, S. A., Wilkinson, J. E., Galas, D., Ziegler, S. F. and Ramsdell, F. 2001. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 27:68–73.
- Bugelski, P. J., Volk, A., Walker, M. R., Krayer, J. H., Martin, P., andDescotes, J 2010. Critical review of preclinical approaches to evaluate the potential of immunosuppressive drugs to influence human neoplasia. Int. J. Toxicol 29(5):435–466.
- Burnet, F. M. 1970. The concept of immunological surveillance. Prog. Exp. Tumor. Res. 13:1–27.
- Chatila, T. A., Blaeser, F., Ho, N., Lederman, H. M., Voulgaropoulos, C., Helms, C. and Bowcock, A. M. 2000. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J. Clin. Invest. 106:R75–R81.
- CHMP, E. 2007. Guideline on strategies to identify and mitigate risks for first-in-human clinical trials with investigational medicinal products (Doc. Ref.EMEA/CHMP/SWP/28367/07). pp.1–12.
- Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J. and Schreiber, R. D. 2002. Cancer immunoeditting: From immunosurveillance to tumor escape. Nat. Immunol. 3:991–998.
- Dunn, G. P., Old, L. J. and Schreiber, R. D. 2004. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 21:137–148.
- Eastwood, D., FIndlay, L., Poole, S., Bird, C., Wadhwa, M., Moore, M., Burns, C., Thorpe, R., andStebbings, R 2010. Monoclonal antibody TGN1412 trial failure explained by species differences in CD28 expression on CD4+ effector memory T-cells. Br. J. Pharmacol. 161:512–526.
- Elpek, K. G., Lacelle, C., Singh, N. P., Yolcu, E. S. and Shirwan, H. 2007. CD4+CD25+ T-regulatory cells dominate multiple immune evasion mechanisms in early but not late phases of tumor development in a B-cell lymphoma model. J. Immunol. 178:6840–6848.
- Engels, E. A., Biggar, R. J., Hall, H. I., Cross, H., Crutchfield, A., Finch, J. L., Grigg, R., Hylton, T., Pawlish, K. S., McNeel, T. S. and Goedert, J. J. 2008. Cancer risk in people infected with human immunodeficiency virus in the United States. Int. J. Cancer. 123:187–194.
- Euvrard, S., Kanitakis, J., Pouteil-Noble, C., Claudy, A. and Touraine, J. L. 1997. Skin cancers in organ transplant recipients. Ann. Transplant. 2:28–32.
- Fontenot, J. D. and Rudensky, A. Y. 2005. A well adapted regulatory contrivance: regulatory T-cell development and the forkhead family transcription factor Foxp3. Nat. Immunol. 6:331–337.
- Fontenot, J. D., Gavin, M. A. and Rudensky, A. Y. 2003. Foxp3 programs the development and function of CD4+CD25+ regulatory T-cells. Nat. Immunol. 4:330–336.
- Fort, M. M. and Narayanan, P. K. 2010. Manipulation of regulatory T-cell function by immunomodulators: A boon or a curse? Toxicol. Sci. 117:253–262.
- Gillis, S. and Smith, K. A. 1977. Long term culture of tumour-specific cytotoxic T-cells. Nature. 268:154–156.
- Godin-Ethier, J., Pelletier, S., Hanafi, L. A., Gannon, P. O., Forget, M. A., Routy, J. P., Boulassel, M. R., Krzemien, U., Tanguay, S., Lattouf, J. B., Arbour, N. and Lapointe, R. 2009. Human activated T-lymphocytes modulate IDO expression in tumors through Th1/Th2 balance. J. Immunol. 183:7752–7760.
- Gorelik, L. and Flavell, R. A. 2000. Abrogation of TGFβ signaling in T-cells leads to spontaneous T-cell differentiation and autoimmune disease. Immunity. 12:171–181.
- Gribble, E. J., Sivakumar, P. V., Ponce, R. A. and Hughes, S. D. 2007. Toxicity as a result of immunostimulation by biologics. Expert. Opin. Drug. Metab. Toxicol. 3:209–234.
- Hanto, D. W., Frizzera, G., Gajl-Peczalska, K. J. and Simmons, R. L. 1985. Epstein-Barr virus, immunodeficiency, and B-cell lymphoproliferation. Transplantation. 39:461–472.
- Henney, C. S., Kuribayashi, K., Kern, D. E. and Gillis, S. 1981. Interleukin-2 augments natural killer cell activity. Nature. 291:335–338.
- Higano, C. S., Schellhammer, P. F., Small, E. J., Burch, P. A., Nemunaitis, J., Yuh, L., Provost, N. and Frohlich, M. W. 2009. Integrated data from 2 randomized, double-blind, placebo-controlled, Phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 115:3670–3679.
- Hirschhorn-Cymerman, D., Rizzuto, G. A., Merghoub, T., Cohen, A. D., Avogadri, F., Lesokhin, A. M., Weinberg, A. D., Wolchok, J. D. and Houghton, A. N. 2009. OX40 engagement and chemotherapy combination provides potent antitumor immunity with concomitant regulatory T-cell apoptosis. J. Exp. Med. 206:1103–1116.
- Hoption Cann, S. A., van Netten, J. P. and van Netten, C. 2003. Dr William Coley and tumour regression: a place in history or in the future. Postgrad. Med. J. 79:672–680.
- Horak, I., Löhler, J., Ma, A. and Smith, K. A. 1995. Interleukin-2 deficient mice: A new model to study autoimmunity and self-tolerance. Immunol. Rev. 148:35–44.
- Hori, S., Nomura, T. and Sakaguchi, S. 2003. Control of regulatory T-cell development by the transcription factor Foxp3. Science. 299:1057–1061.
- Huang, M., Wang, J., Lee, P., Sharma, S., Mao, J. T., Meissner, H., Uyemura, K., Modlin, R., Wollman, J. and Dubinett, S. M. 1995. Human non-small cell lung cancer cells express a type 2 cytokine pattern. Cancer Res. 55:3847–3853.
- Ichihara, F., Kono, K., Takahashi, A., Kawaida, H., Sugai, H. and Fujii, H. 2003. Increased populations of regulatory T-cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin. Cancer. Res. 9:4404–4408.
- Imai, H., Saio, M., Nonaka, K., Suwa, T., Umemura, N., Ouyang, G. F., Nakagawa, J., Tomita, H., Osada, S., Sugiyama, Y., Adachi, Y. and Takami, T. 2007. Depletion of CD4+CD25+ regulatory T-cells enhances interleukin-2-induced anti-tumor immunity in a mouse model of colon adenocarcinoma. Cancer. Sci. 98:416–423.
- Jeal, W. and Goa, K. L. 1997. Aldesleukin (recombinant -2): A review of its pharmacological properties, clinical efficacy and tolerability in patients with renal cell carcinoma. Bio. Drugs. 7:285–317.
- Jones, E. A., Pringle, J. H., Angel, C. A. and Rees, R. C. 2002. Th1/Th2 cytokine expression and its relationship with tumor growth in B-cell non-Hodgkin’s lymphoma (NHL). Leuk. Lymphoma. 43:1313–1321.
- Kantoff, P. W., Schuetz, T. J., Blumenstein, B. A., Glode, L. M., Bilhartz, D. L., Wyand, M., Manson, K., Panicali, D. L., Laus, R., Schlom, J., Dahut, W. L., Arlen, P. M., Gulley, J. L. and Godfrey, W. R. 2010. Overall survival analysis of a Phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 28:1099–1105.
- Kaufman, H. L. and Bines, S. D. 2010. OPTIM trial: A Phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future. Oncol. 6:941–949.
- Khattri, R., Cox, T., Yasayko, S. A. and Ramsdell, F. 2003. An essential role for Scurfin in CD4+CD25+ T-regulatory cells. Nat. Immunol. 4:337–342.
- Kim, J. M., Rasmussen, J. P. and Rudensky, A. Y. 2007. Regulatory T-cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 8:191–197.
- Kim, J., Lahl, K., Hori, S., Loddenkemper, C., Chaudhry, A., deRoos, P., Rudensky, A. and Sparwasser, T. 2009. Cutting edge: depletion of Foxp3+ cells leads to induction of autoimmunity by specific ablation of regulatory T-cells in genetically targeted mice. J. Immunol. 183:7631–7634.
- Ko, K., Yamazaki, S., Nakamura, K., Nishioka, T., Hirota, K., Yamaguchi, T., Shimizu, J., Nomura, T., Chiba, T. and Sakaguchi, S. 2005. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T-cells. J. Exp. Med. 202:885–891.
- Krämer, S., Schimpl, A. and Hünig, T. 1995. Immunopathology of interleukin (IL) 2-deficient mice: Thymus dependence and suppression by thymus-dependent cells with an intact IL-2 gene. J. Exp. Med. 182:1769–1776.
- Lahl, K., Loddenkemper, C., Drouin, C., Freyer, J., Arnason, J., Eberl, G., Hamann, A., Wagner, H., Huehn, J. and Sparwasser, T. 2007. Selective depletion of Foxp3+ regulatory T-cells induces a scurfy-like disease. J. Exp. Med. 204:57–63.
- Leber, A., Teles, A. and Zenclussen, A. C. 2010. Regulatory T-cells and their role in pregnancy. Am J Reprod Immunol 63:445–459.
- Leung, D. Y. and Bloom, J. W. 2003. Update on glucocorticoid action and resistance. J. Allergy. Clin. Immunol. 111:3–22 quiz 23.
- Li, B., Samanta, A., Song, X., Iacono, K. T., Brennan, P., Chatila, T. A., Roncador, G., Banham, A. H., Riley, J. L., Wang, Q., Shen, Y., Saouaf, S. J. and Greene, M. I. 2007. FOXP3 is a homo-oligomer and a component of a supramolecular regulatory complex disabled in the human XLAAD/IPEX autoimmune disease. Int. Immunol. 19:825–835.
- Liyanage, U. K., Moore, T. T., Joo, H. G., Tanaka, Y., Herrmann, V., Doherty, G., Drebin, J. A., Strasberg, S. M., Eberlein, T. J., Goedegebuure, P. S. and Linehan, D. C. 2002. Prevalence of regulatory T-cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 169:2756–2761.
- Lyon, M. F., Peters, J., Glenister, P. H., Ball, S. and Wright, E. 1990. The scurfy mouse mutant has previously unrecognized hematological abnormalities and resembles Wiskott-Aldrich syndrome. Proc. Natl. Acad. Sci. USA. 87:2433–2437.
- Malek, T. R. 2008. The biology of interleukin-2. Annu. Rev. Immunol. 26:453–479.
- Malek, T. R. and Bayer, A. L. 2004. Tolerance, not immunity, crucially depends on IL-2. Nat. Rev. Immunol. 4:665–674.
- Mantovani, A., Allavena, P., Sica, A. and Balkwill, F. 2008. Cancer-related inflammation. Nature. 454:436–444.
- Mellor, A. L. and Munn, D. H. 2000. Immunology at the maternal-fetal interface: lessons for T-cell tolerance and suppression. Annu. Rev. Immunol. 18:367–391.
- Morgan, D. A., Ruscetti, F. W. and Gallo, R. 1976. Selective in vitro growth of T-lymphocytes from normal human bone marrows. Science. 193:1007–1008.
- Munn, D. H. and Mellor, A. L. 2007. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Invest. 117:1147–1154.
- Nguyen, D. H., Hurtado-Ziola, N., Gagneux, P. and Varki, A. 2006. Loss of Siglec expression on T-lymphocytes during human evolution. Proc. Natl. Acad. Sci. USA. 103:7765–7770.
- Novartis. 2011. Proleukin® (aldesleukin) package insert.
- Onizuka, S., Tawara, I., Shimizu, J., Sakaguchi, S., Fujita, T. and Nakayama, E. 1999. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor α) monoclonal antibody. Cancer. Res. 59:3128–3133.
- Oppenheim, J. J. 2007. IL-2: More than a T cell growth factor. J. Immunol. 179:1413–1414.
- Penn, I. 1994. The problem of cancer in organ transplant recipients: An overview. Transplant. Sci. 4:23–32.
- Penn, I. 2000. Post-transplant malignancy: The role of immunosuppression. Drug. Saf. 23:101–113.
- Piscitelli, S. C., Bhat, N. and Pau, A. 2000. A risk-benefit assessment of interleukin-2 as an adjunct to antiviral therapy in HIV infection. Drug. Saf. 22:19–31.
- Powrie, F., Carlino, J., Leach, M. W., Mauze, S. and Coffman, R. L. 1996. A critical role for transforming growth factor-β but not interleukin-4 in the suppression of T-helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J. Exp. Med. 183:2669–2674.
- Read, S., Malmström, V. and Powrie, F. 2000. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J. Exp. Med. 192:295–302.
- Ruscetti, F. W., Morgan, D. A. and Gallo, R. C. 1977. Functional and morphologic characterization of human T cells continuously grown in vitro. J. Immunol. 119:131–138.
- Sadlack, B., Merz, H., Schorle, H., Schimpl, A., Feller, A. C. and Horak, I. 1993. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 75:253–261.
- Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M. and Toda, M. 1995. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155:1151–1164.
- Schorle, H., Holtschke, T., Hünig, T., Schimpl, A. and Horak, I. 1991. Development and function of T-cells in mice rendered interleukin-2 deficient by gene targeting. Nature. 352:621–624.
- Shimizu, J., Yamazaki, S. and Sakaguchi, S. 1999. Induction of tumor immunity by removing CD25+CD4+ T-cells: A common basis between tumor immunity and autoimmunity. J. Immunol. 163:5211–5218.
- Soto, P. C., Stein, L. L., Hurtado-Ziola, N., Hedrick, S. M. and Varki, A. 2010. Relative over-reactivity of human versus chimpanzee lymphocytes: Implications for the human diseases associated with immune activation. J. Immunol. 184:4185–4195.
- Steiger, J., Nickerson, P. W., Steurer, W., Moscovitch-Lopatin, M. and Strom, T. B. 1995. IL-2 knockout recipient mice reject islet cell allografts. J. Immunol. 155:489–498.
- Suntharalingam, G., Perry, M. R., Ward, S., Brett, S. J., Castello-Cortes, A., Brunner, M. D. and Panoskaltsis, N. 2006. Cytokine storm in a Phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 355:1018–1028.
- Sutmuller, R. P., van Duivenvoorde, L. M., van Elsas, A., Schumacher, T. N., Wildenberg, M. E., Allison, J. P., Toes, R. E., Offringa, R. and Melief, C. J. 2001. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25+ regulatory T-cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J. Exp. Med. 194:823–832.
- Suzuki, H., Kündig, T. M., Furlonger, C., Wakeham, A., Timms, E., Matsuyama, T., Schmits, R., Simard, J. J., Ohashi, P. S. and Griesser, H. 1995. Deregulated T-cell activation and autoimmunity in mice lacking interleukin-2 receptor β. Science. 268:1472–1476.
- Taniguchi, T., Matsui, H., Fujita, T., Takaoka, C., Kashima, N., Yoshimoto, R. and Hamuro, J. 1983. Structure and expression of a cloned cDNA for human interleukin-2. Nature. 302:305–310.
- Vignali, D. A., Collison, L. W. and Workman, C. J. 2008. How regulatory T cells work. Nat. Rev. Immunol. 8:523–532.
- Wiemann, B. and Starnes, C. O. 1994. Coley’s toxins, tumor necrosis factor and cancer research: A historical perspective. Pharmacol. Ther. 64:529–564.
- Wildin, R. S., Smyk-Pearson, S. and Filipovich, A. H. 2002. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J. Med. Genet. 39:537–545.
- Woo, E. Y., Chu, C. S., Goletz, T. J., Schlienger, K., Yeh, H., Coukos, G., Rubin, S. C., Kaiser, L. R. and June, C. H. 2001. Regulatory CD4(+)CD25(+) T-cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer. Res. 61:4766–4772.
- Zou, W. 2006. Regulatory T-cells, tumour immunity, and immunotherapy. Nat. Rev. Immunol. 6:295–307.
- Zubler, R. H., Lowenthal, J. W., Erard, F., Hashimoto, N., Devos, R. and MacDonald, H. R. 1984. Activated B-cells express receptors for, and proliferate in response to, pure interleukin 2. J. Exp. Med. 160:1170–1183.