Abstract
Estriol (E3), an endogenous estrogen predominantly produced during human pregnancy, has been suggested to play an important role in modulating the immune system function during pregnancy. The present study sought to investigate the ability of E3 to alter splenocyte functions in non-immunized naïve BALB/c female mice and also in mice injected with complete Freund’s adjuvant (CFA), and the effect of E3 was compared with that of 17β-estradiol (E2). When mice were injected with CFA, their spleen weight index (i.e., wet organ wet/whole body weight) was increased by ~ 300%, but co-administration of E3 almost completely suppressed splenomegaly. E3 also reduced cytokine production and reduced ERK and p38 activation in both splenocytes and peritoneal exudate cells from CFA-treated animals. In comparison, while E2 had a similar but slightly weaker effect than E3 in reducing splenomegaly, it had a rather different effect from E3 on cytokine production and ERK activation in splenocytes and peritoneal exudate cells from CFA-treated mice. Under naïve immunological conditions, E3 and E2 had very similar effects on splenocyte functions. Both of them transiently increased the percentages of splenic CD4+ and CD8+ cells. They also increased the proliferation of splenocytes ex vivo, and stimulated production of interferon-γ and interleukin-2. Altogether, these data show that E3 and E2 have different effects on splenocyte functions when the animals are under experimentally induced inflammatory conditions.
| Abbreviations: | ||
| CFA, | = | complete Freund’s adjuvant; |
| ConA, | = | concanavalin A; |
| IFNγ, | = | interferon-γ; |
| IL-2, | = | interleukin-2; |
| APC, | = | antigen-presenting cells; |
| ERK, | = | extracellular signal-regulated kinase; |
| MAPK, | = | mitogen-activated protein kinase; |
| BrdU, | = | 5-bromo-2-deoxyuridine. |
Introduction
The ability of endogenous estrogens to modulate lymphocyte development and function in the thymus has been recognized for many years (reviewed in Lang, Citation2004; Pernis, Citation2007). In ovariectomized female mice or rats where serum levels of endogenous estrogens are very low, there is a significant enlargement of the thymus compared to intact female animals (Utsuyama and Hirokawa, Citation1989). In contrast, during pregnancy where elevated serum and tissue levels of estrogens are present, there is a marked shrinkage of the thymus in these animals (Clarke and Kendall, Citation1989; Citation1994). Experimentally, treatment of ovariectomized or intact female mice or rats with an estrogen can also induce a strong thymic atrophy similar to what is commonly observed during pregnancy (Novotny et al., Citation1983). It has been suggested that alterations in thymocyte development and function may contribute to the development and maintenance of maternal tolerance toward the immunologically foreign fetus (Aluvihare et al., Citation2004). In addition, estrogen-induced thymic changes may modulate the severity of certain forms of T-cell-mediated autoimmune diseases (such as multiple sclerosis, rheumatoid arthritis, and thyroiditis) (Jansson et al., Citation1994).
In contrast, very little is known at present about the modulating effect of estrogens on splenocyte development and function. The spleen is a major lymph organ; its white pulp is composed primarily of B-cells, T-cells, macrophages, and dendritic cells, each of which has diverse immunological and/or phagocytic functions in the body. Many earlier studies have noted that chronic treatment of naïve (non-immunized) mice or rats with an estrogen had little effect on their spleen weight change, which was very different from the drastic atrophy commonly seen in the thymus. This is, perhaps, one of the reasons why very few previous studies examined the effect of estrogens on the immunological functions of the spleen. In the present study, we sought to further investigate the potential modulating effect of estriol (E3), which is an endogenous estrogen preferentially produced during human pregnancy (Fotsis, Citation1987), on the functions of splenocytes in female mice under different immunological and/or inflammatory conditions, and to also compare its effects with that of 17β-estradiol (E2).
Notably, most of the earlier studies focused on exploring the immune-modulating effects of E2 in mice under different immunological conditions. E3 is usually produced only in a very small quantity in a non-pregnant woman, but it becomes a quantitatively-major estrogen during pregnancy (Fotsis, Citation1987). Interestingly, while E2 has very high binding affinity for both human ERα and ERβ, the relative binding affinities of E3 for human ERα and ERβ are 11% and 35%, respectively, of that of E2 (Zhu et al., Citation2006), thus giving E3 a 3.5-fold relative binding preference for ERβ over ERα. It has been speculated that E3 may play a unique role in modulating the immune system function during pregnancy. In partial support of this hypothesis, a recent study showed that E3 is an important hormonal component that can induce the development of immunotolerance toward the major histocompatibility complex (MHC)-compatible heart allografts as seen during pregnancy in a rat experimental model (Funeshima-Fuji et al., Citation2009). Moreover, E3 was also reported to have a very different effect from E2 in modulating the production of antigen-induced specific antibodies (Ding and Zhu, Citation2008). Here, we report additional observations showing that E3 has a distinctly different effect from E2 in modulating some of the splenocyte functions in mice injected with complete Freund’s adjuvant (CFA).
Materials and methods
Chemicals and reagents
17β-Estradiol (E2; catalog # E8875), estriol (E3; catalog # E1253), cholesterol, sulfanilamide, N-[naphthyl]-ethylenediamine dihydrochloride, lipopolysaccharides (LPS; Escherichia coli type 055:B5), and Complete Freund’s Adjuvant (CFA) containing Mycobacterium tuberculosis (strain H37Rv), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were obtained from Sigma (St. Louis, MO). CFA was emulsified with an equal volume of phosphate-buffered saline (PBS, pH 7.4) before it was injected into the animals. Fetal bovine serum (FBS) was obtained from Invitrogen (Carlsbad, CA), and was pre-treated with dextran-coated charcoal to remove any steroid present in it.
For all in vitro or ex vivo experiments, the estrogen (E3 or E2) was dissolved in 200-proof ethanol at a stock concentration of 5 mM. They were diluted in cell culture medium immediately before use. For all in vivo experiments, estrogen was administered by implanting an estrogen-containing pellet. The pellets containing 10 mg E3 or 1 mg E2 were hand-pressed using the Pellet Presser (Parr Instrument Company, Moline, IL). Each pellet (weighing 25 mg in total) contained 5 mg of sodium chloride (NaCl) and a combined 20 mg of estrogen plus cholesterol. The vehicle pellets contained 5 mg of NaCl and 20 mg of cholesterol. Dry crystals of NaCl, cholesterol, and estrogen were mixed thoroughly by grinding the mixtures to fine powders. Pellets were prepared in our laboratory manually by the same person by applying consistent pressure.
Animal experiments and preparation of splenocytes and peritoneal exudate cells (PEC)
Six- to eight-week-old female BALB/c mice obtained from Harlan (Indianapolis, IN) were housed under specific pathogen-free conditions. The animals were housed in plastic cages in a facility maintained at 22°C with a 50% relative humidity and a 12-h light/12-h dark cycle. All animals were provided access to Harlan Teklad standard chow (8604) and distilled water ad libitum. The animal experiments were carried out according to the National Institutes of Health Guidelines for Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of South Carolina, where most of the authors once worked as employees.
For estrogen treatment, the mice were anesthetized with isoflurane inhalation and then implanted under the dorsal skin with a 25-mg pellet containing 10 mg E3, 1 mg E2, or vehicle alone. One week after pellet implantation, the animals were injected subcutaneously (SC) with 0.2 mL of CFA emulsion, which is an antigen solution emulsified in mineral oil commonly used as a non-specific booster of immunity and inflammation in experimental animal models (Johnston et al., Citation1991; Qin et al., Citation1993). The animals were euthanized 1 week later by CO2 overdose followed by decapitation, their spleens were removed, and splenocytes isolated and prepared for use in ex vivo experiments. Notably, the selection of the different doses of E3 and E2 used in the present study was based on measurements of plasma E3 and E2 levels (some of the data are shown in the Results section). The doses of E3 and E2 used in this study would yield plasma concentrations of free (unbound) estrogens comparable to those seen during late stages of human pregnancy (Zhang et al., Citation2005). Further, the dose of E3 and E2 used would give a near maximum estrogenic hormonal activity based on the 2-day in vivo uterotropic activity assay conducted in ovariectomized female rats (data not shown).
For preparation of the splenocytes, spleens were removed from the animals under aseptic conditions. Single cell suspensions were prepared and cell debris and clumps were removed by filtration. Erythrocytes were lysed with a Tris-HCl-buffered ammonium chloride (0.73% w/v) solution. The remaining mononuclear cells (mostly lymphocytes) were then washed using centrifugation (1200 rpm at 4°C for 5 min) followed by re-suspension in phenol red-free RPMI-1640 medium supplemented with 10% FBS, 100 U penicillin/ml, and 100 μg streptomycin/ml (Sigma).
For preparation of peritoneal exudate cells (PEC), additional sets of mice were first injected IP with 0.2 ml CFA emulsion. The animals were euthanized 4 days later and PEC obtained by peritoneal lavage with ice-cold PBS. After determination of yields and viability (using the Trypan blue exclusion assay), the PEC (at 1 × 105/well) were placed into 96-well plates and pre-treated in vitro with E3 or E2 for 4 h before being stimulated with 100 ng/ml LPS for an additional 24 h (for NO determination) or 30 min (for Western blotting). Cell culture supernatants were subsequently isolated for determination of NO production, while the cells were collected for Western blot analysis of protein expression.
Splenocyte viability and proliferation assays
The viability of splenocytes obtained from mice that were treated in vivo with E3 or E2 was determined in the ex vivo bioassay. Briefly, splenocytes (5 × 106/ml) were cultured in triplicate for 48 h in 96-well plates at 37°C. A 20 µl aliquot of MTT (5 mg/ml) was added to each well for the last 4 h; the cells were then lysed with a buffer containing 10% sodium dodecyl sulfate and 50% N,N-dimethyl formamide. Absorbance in each well was then measured at 570 nm in a microplate reader (BioRad, Model 550, Tokyo, Japan). Similarly, to determine the rate of proliferation of these splenocytes, a [3H]-thymidine incorporation assay was employed. Briefly, splenocytes (5 × 106/ml) were cultured in triplicate and stimulated with 5 μg concanavalin A (ConA)/ml in 96-well plates for 48 h. A 25 µl aliquot of [3H]-thymidine (0.25 μCi/well) was then added to the cells for the last 8 h of culture. The cells were then harvested onto glass fiber filters and the incorporated radioactivity counted using a β-scintillation counter (MicroBeta Trilux, PerkinElmer Life Sciences, Boston, MA).
To determine the direct effect of E3 or E2 on splenocyte viability and proliferation, splenocytes were obtained from untreated mice and then cultured in vitro in the presence of different concentrations of estrogens. The same MTT and [3H]-thymidine incorporation assays as described above were used to determine cell viability and rate of cell proliferation, respectively.
Flow cytometry analysis
Flow cytometry was used to determine the population of various splenocytes. Briefly, the cells were washed twice with ice-cold PBS and a 100 µl aliquot of cell suspension (containing 250,000–500,000 cells) was placed into a 12 × 75 mm polystyrene tube to which 5 µl of properly diluted phycoerythrin (PE)-conjugated anti-mouse CD4, PE-anti-mouse B220, fluorescein isothiocyanate (FITC)-conjugated anti-mouse CD8, or FITC-anti-mouse CD11b monoclonal antibody (mAb; BD PharMingen, San Diego, CA) was then added. Each antibody-containing cell suspension was then incubated for a minimum of 30 min on ice in the dark. Thereafter, the cells were washed twice with ice-cold PBS (1 ml) and suspended in 500 µl ice-cold 1% paraformaldehyde in PBS. Appropriate FITC- and PE-conjugated isotype control antibodies were used as negative controls. Each mAb was specific for the stated surface protein.
Samples were ultimately analyzed using a FACS-Calibur flow cytometer from Becton Dickinson Immunocytometry Systems, Inc (BDIS, San Jose, CA). Instrument performance was standardized weekly using Calibrite beads (BDIS) and the same instrument settings were used for all acquisitions. The assays were sufficiently uniform to use the same forward scatter (FSC), side scatter (SSC), and fluorescence (FL) settings. The sensitivity of the instrument was constant. The acquisition and analysis software for flow cytometry data was CELLQuest Pro from BDIS running on an Apple computer. A minimum of 10,000 events/sample was analyzed.
Measurement of cytokine production
For measuring the production of interleukin 2 (IL-2) and interferon γ (IFNγ) by cultured splenocytes, single cell suspensions (at 5 × 106/ml) from untreated or treated mouse spleens were prepared and cultured (in triplicates) in 96-well plates and stimulated with 5 μg ConA/ml in the presence or absence of E3 or E2 for 24 h (for measuring IL-2) or 48 h (for measuring IFNγ). Supernatants from each culture were then collected and the concentrations of murine IL-2 and IFNγ were determined by ELISA kits (BD PharMingen) following manufacturer’s instructions. The detection sensitivity for IL-2 and IFNγ in each kit was 10 pg/ml.
Measurement of nitric oxide (NO) production
The change in NO production by peritoneal exudate cells (PEC) was reflected through measures of the levels of nitrite (stable reaction product of NO) in the culture medium (Zhou et al., Citation2006). Briefly, PEC (at 1 × 105/well) from untreated or treated mice were cultured (in triplicate) in 96-well plates and stimulated with 1 μg LPS/ml for 24 h. Thereafter, 100 μl of culture medium was removed and mixed with an equal volume of Griess reagent (1% sulfanilamide and 0.1% N-[naphthyl]-ethylenediamine dihydrochloride in 2.5% H3PO4) at room temperature for 10 min. The absorbance of the reaction mixture was then measured at 540 nm in a microplate reader. The nitrite concentration in each sample was then extrapolated from a standard curve prepared in parallel using known concentrations of sodium nitrite (NaNO2).
Western blotting
To study the modulation of splenocyte or PEC functions by estrogens, in this study, the ConA-induced the activation (phosphorylation) of two representative mitogen-activated protein kinases (MAPKs), i.e., the extracellular signal-regulated kinases (ERK1/2) and p38, was determined in cultured splenocytes or PEC obtained from mice with different treatments. Based on earlier studies (Tang et al., Citation2000; Wang et al, Citation2007; Cargnello and Roux, Citation2011), ERK1/2 and p38 were rapidly activated following ConA treatment. In the present study, a 10-min in vitro exposure to ConA was used.
To measure the protein levels of ERK1/2 and p38, splenocytes or PEC were suspended in a lysis buffer containing 20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, supplemented with a mixture of protease inhibitors, pH 7.4 for 15 min at 4°C. The lysates were then centrifuged at 14,000 rpm (4°C) for 5 min, and the resultant supernatants were isolated and then boiled for 5 min in sodium dodecyl sulfate (SDS) buffer containing 50 mM Tris-HCl (pH 6.8), 2% (w/v) SDS, 10% glycerol, 1.2% 2-mercaptoethanol, and 0.02% bromphenol blue. Equal amounts of proteins (~ 20 μg/lane) were then loaded and electrophoresed over a 10% polyacrylamide gel before being electro- transferred to PVDF membranes (BioRad, Hercules, CA). The membranes were treated with 10% non-fat milk for 1 h to block non-specific binding, rinsed, and then incubated with specific primary antibodies (each at 1:2000 dilution in 10% non-fat milk) for 1 h at room temperature with gentle rocking. The membranes were then washed with PBS to remove unbound antibody and then incubated in a solution containing horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (1:2000 dilution) antibodies for 1 h at room temperature with rocking. After gentle washing to remove unbound secondary antibody, the immune complexes that were present were detected via chemiluminescence.
Determination of plasma levels of free E3 and E2
To determine the plasma levels of unbound (free) E3 and E2 in naïve mice implanted with an E3 or E2 pellet, a separate experiment was conducted. The female mice (5–6 per group) were anesthetized using isoflurane and underwent subcutaneous (SC) implantation of a pellet containing the same amount of E3 or E2 as described earlier (i.e., 10 mg E3 or 1 mg E2). The control animals received a vehicle pellet only. Five days after pellet implantation, the animals were euthanized and blood samples were collected. The free plasma E3 and E2 levels in each sample were determined using ELISA kits (Cayman Chemical Company, Ann Arbor, MI) according to the manufacturer’s procedures. The detection sensitivity for E3 and E2 in these ELISA kits ranged from 14–19 pg/ml.
Reproducibility and statistical analysis
Some of the experiments reported in this study were repeated twice and similar results were obtained. In such cases, one set of the representative data is presented. For those experiments with an n ≥ 3, data are expressed as mean (± SD) as indicated. A two-way ANOVA (analysis of variance) test was used to determine statistical difference. A p-value of ≤ 0.05 was used to represent statistical significance.
Results
Effect of E3 and E2 on splenocyte functions in CFA-treated mice
Female BALB/c mice were implanted with a pellet containing 10 mg E3 or 1 mg E2 for 1 week and then received a single SC injection of CFA emulsion. One week later, a marked splenomegaly was observed in CFA-treated mice; the weight index (wet organ weight/whole body weight) in these mice was increased by~ 3-fold compared to that of untreated animals. Mice implanted with E3 or E2 each exhibited a strong suppression of CFA-induced splenomegaly (). E3 had a somewhat stronger effect than E2 in suppressing splenomegaly development.
Figure 1. Effect of E3 and E2 on splenocyte functions in CFA-treated mice. (a) Effect of E3 and E2 on CFA-induced spleen weight change. Spleen weight index here refers to ratio of spleen wet weight to whole animal body weight. Five animals were used in each treatment group. Similar in vivo observations were made in two separate experiments; only one representative data set is shown. (b) Rate of splenocyte proliferation in an ex vivo assay. (c and d) Production of IFNγ and IL-2 by cultured splenocytes. The female BALB/c mice were implanted with a pellet containing E3 or E2, and the animals were injected SC with CFA emulsion 1 week later. A week after CFA injection, the mice were euthanized and spleens of 3–5 animals were pooled for preparation of splenocytes. The splenocytes (5 × 106 cells/ml) were then cultured in the absence or presence of ConA for 24 h (for measuring IL-2) or 48 h (for measuring IFNγ). Data shown are mean (± SD) of triplicates from one representative experiment. Group labels: V = mice treated with vehicle + CFA; E2 = mice treated with E2 + CFA; and E3 = mice treated with E3 + CFA. * p < 0.05.

Ex vivo bioassays showed that splenocytes from mice treated with CFA plus E3 or E2 also had a reduced rate of induced cell proliferation either in the absence or presence of ConA. The effect of E3 was markedly stronger than that of E2 (p < 0.05; ). Interestingly, while the in vivo E3 administration to CFA-treated mice strongly suppressed ConA-induced IFNγ production by their splenocytes, E2 had an opposite effect (). In vivo treatment with E3 or E2 increased ConA-stimulated IL-2 production by splenocytes ex vivo, and the effect of E2 was much stronger than that of E3 (p < 0.05; ).
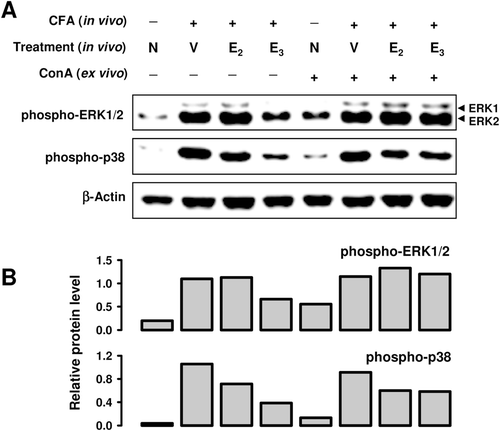
To further study the modulation of splenocyte activation by estrogens, the activation (phosphorylation) of two representative mitogen-activated protein kinases (MAPKs), i.e., the extracellular signal-regulated kinase (ERK) and p38, was determined in cultured splenocytes obtained from CFA-treated mice. E2 treatment in vivo slightly increased the level of phospho-ERK, but slightly decreased the level of phospho-p38 (). In comparison, E3 treatment in vivo markedly reduced the phospho-ERK level, but this effect was diminished when splenocytes were further stimulated with ConA in vitro. E3 treatment in vivo significantly blocked the phosphorylation of p38 as assayed ex vivo in the absence or presence of ConA as a stimulator ().
Figure 2. Effect of E3 and E2 on MAPK activation in splenocytes from CFA-treated mice. Three spleens from each treatment group were pooled for preparation of splenocytes. The splenocytes (at 5 × 106 cells/ml) were then cultured in vitro in the presence or absence of ConA for 10 min. Cells from multiple in vitro treatment wells were pooled together for preparation of whole cell lysates, which were used for measurement of protein levels by Western blot analysis (data shown in a). The blots were re-probed with anti-β-actin antibody for confirmation of equal loading of proteins in each lane. Quantitative values shown in (b) were calculated according to the laser densitometry scanning data (mean of two separate experiments). Since two bands were detected for ERK1/2, the quantitative data were based on their combined changes. The unit for the relative protein level is expressed as the densitometry ratio of the protein of interest to that of β-actin. Group labels: N = mice receiving no treatment; V = mice treated in vivo with vehicle + CFA; E2 = mice treated with E2 + CFA; and E3 = mice treated with E3 + CFA.
Effect of E3 and E2 on CFA-elicited peritoneal exudate cells (PEC)
Earlier studies showed that IP injection of CFA recruits macrophages into the abdominal cavity. For comparison, we examined the modulating effect of E3 and E2 on the functions of CFA-elicited PEC in these mice. PEC were harvested and seeded into 96-well culture plates. Upon stimulation with LPS, release of nitrite from PEC of vehicle-treated mice was increased by ~ 5-fold above control mouse values. Cultured PEC from E2-treated mice released more NO than vehicle-treated animals (p < 0.05; , right part). However, NO release from PEC of E3-treated mice was much lower than that from PEC of E2-treated animals. In fact, the in vivo E3 treatment did not appear to have any appreciable effect on LPS-induced NO release. In the absence of LPS stimulation, the changes were very small (, left part).
Figure 3. Effect of E3 and E2 on nitrite (NO) production (a) and MAPK activation (b and c) in peritoneal exudate cells (PEC). CFA-elicited PEC were collected from three mice treated with CFA + estrogen, and the pooled cells were then cultured in vitro in the presence of 1 μg LPS/ml for 24 h (a) or 30 min (b). Data in (a) are mean (± SD) of triplicate cultures. * p < 0.05. For data in (c), PEC were prepared from mice treated with CFA only (in absence of estrogen). These cells were then cultured in vitro in the presence of 10 nM of E3 or E2 for 4 h, followed by 100 ng LPS/ml stimulation for another 30 min. Cells from multiple treatment wells were pooled together for preparation of whole cell lysates, which were then used for measurement of protein levels by Western blotting. The blots were re-probed with anti-β-actin antibody for confirmation of equal loading of proteins in each lane. The quantitative values shown in lower parts of (b) and (c) were calculated according to laser densitometry scanning data (mean of two separate experiments). Because two bands were detected for ERK1/2, quantitative data were based on their combined changes. The unit for the relative protein level is expressed as the densitometry ratio of the protein of interest to that of β-actin. Group labels for Panel B: V = mice treated with vehicle only; E2 = mice treated in vivo with E2 only; and E3 = mice treated with E3 only.
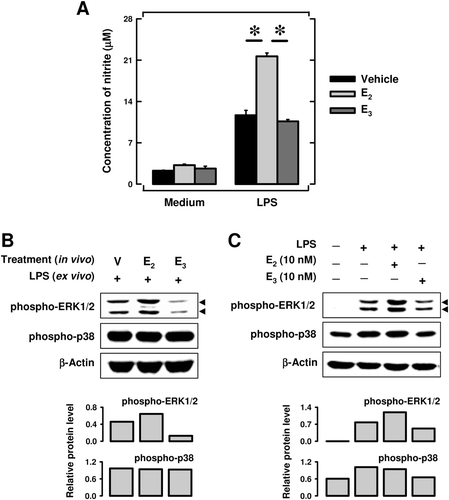
It is known that stimulation of macrophages with LPS results in MAPK activation that, in turn, enhances NO production (Amura et al., Citation1998). Here, we also evaluated the modulating effect of E3 and E2 on MAPK activation in the PEC. As shown in , the protein level of phospho-ERK was up-regulated in PEC obtained from E2-treated mice compared with that in the cells from vehicle-treated control animals, but the level was reduced by E3 treatment. In contrast, the extent of p38 phosphorylation was not significantly different between these treatment groups.
For comparison, we also studied the in vitro direct effect of E3 and E2 on PEC harvested from mice that were not treated with estrogen but were only injected IP with CFA emulsion 4 days earlier. As shown in , exposure to 10 nM E2 in vitro increased the phospho-ERK protein level, whereas exposure to 10 nM E3 slightly decreased its level. E3 also slightly reduced the level of phospho-p38. These data are largely in agreement with the effects of E3 and E2 observed in vivo.
In summary, E3 has a distinctly different effect from E2 in modulating the functions of CFA-elicited PEC. The effect of in vivo treatment of mice with E3 or E2 on the functions of CFA-elicited PEC as seen in the ex vivo bioassays appears to agree with the direct effect of these two estrogens in CFA-elicited PEC in culture.
Effect of E3 and E2 on splenocyte functions in naïve mice
Earlier studies have noted that chronic treatment of naïve (non-immunized) mice or rats with estrogen has little effect on their spleen weight, which is very different from the observations made in the present study with CFA-treated mice. This difference suggests that estrogens may have very different effects under different inflammatory and/or immunological conditions. For comparison, we have also determined in this study the effect of E3 and E2 on splenocyte functions in immunologically-naïve female BALB/c mice. These mice were implanted with a pellet containing 10 mg E3 or 1 mg E2 for 1 or 2 weeks (without CFA treatment). As shown in , treatment of mice with E3 for 1 or 2weeks only slightly decreased the spleen weight index by 8% (p < 0.05) and 20% (p < 0.05), respectively. The spleen weight index in mice treated with 1 mg E2 for 1 or 2 weeks was not appreciably changed ().
Figure 4. Effect of E3 and E2 on spleen weight and lymphocyte populations in naïve mice. (a) Spleen wet weight change after 1 or 2 weeks of E3 or E2 treatment (five mice per group). The spleen weight index refers to ratio of spleen wet weight to whole animal body weight. (b) Spleen cell populations after 1-week estrogen treatment. (c) Spleen cell populations after 2-week estrogen treatment. Female BALB/c mice were implanted with a small pellet containing 10 mg E3 or 1 mg E2 for 2 weeks. Three spleens from each treatment group were then pooled for preparation of splenocytes, that were, in turn, stained with fluorescence-labeled antibodies and subjected to flow cytometry. The experiment was repeated twice and similar results were obtained; a representative data set is shown. Group labels: the open bar (V) = mice treated with vehicle only, the light shaded bar (E2) = mice treated with E2, and the black bar (E3) = mice treated with E3. * p < 0.05.
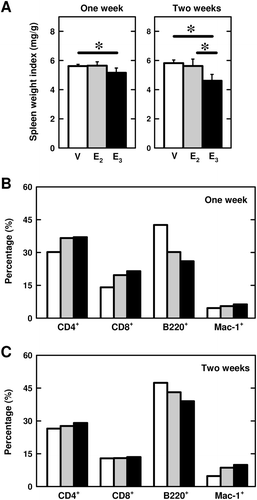
The effect of E3 and E2 on splenocyte populations in these animals was determined by staining for CD4+ and CD8+ (T-cells), B220 (B-cells), and Mac-1 (macrophages). In animals implanted with E3 or E2 for 1 week, the populations of CD4+, CD8+, and Mac-1+ cells were slightly increased, whereas the population of B220+ cells was decreased (). After 2 weeks of estrogen treatment, the population of macrophages remained elevated while the populations of other splenocytes (CD4+ and CD8+ cells) basically returned to their corresponding control levels ().
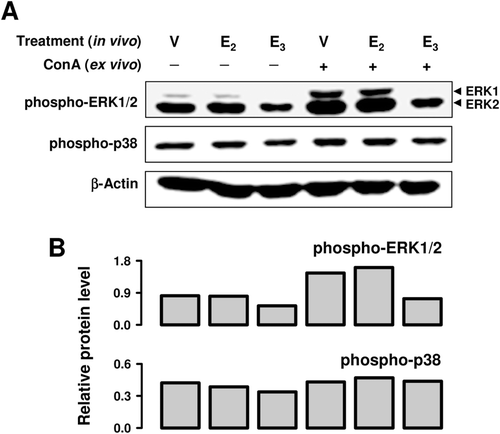
To further assess the splenocyte functions, we performed ex vivo experiments using cultured splenocytes prepared from animals that received E3 or E2 treatment in vivo. As shown in , in vivo treatment with E3 or E2 enhanced splenocyte proliferation in the absence of ConA by 3- or 2-fold, respectively. When splenocytes from vehicle-treated animals were cultured in the presence of ConA, their splenocyte proliferation ex vivo was stimulated 38-fold (compare and ). In vivo treatment with E3 or E2 further increased the ConA-stimulated splenocyte proliferation by 1.9- or 2.2-fold, respectively (). In addition, in vivo treatment with E3 and E2 also stimulated ConA-induced production of IFNγ by splenocytes ex vivo (). In contrast, treatment with these two estrogens in vivo had a considerably weaker stimulation of ConA-induced IL-2 production by splenocytes ex vivo, and E3 in this case was weaker than E2 (), which was different from their effect on the ex vivo production of IFNγ. In vivo treatment with E2 had no appreciable effect on the phospho-ERK1/2 protein levels in splenocytes either in the presence or absence of ConA, but E3 significantly decreased their levels. The levels of phospho-p38 remained unchanged in all groups ().
Figure 5. Effect of E3 and E2 on splenocyte proliferation and function in naïve mice. (a and b) Splenocyte proliferation in the absence and presence of ConA. (c and d) Production of cytokines (IFNγ and IL-2) in the presence of ConA. In these experiments, female BALB/c mice were first implanted with a pellet containing 10 mg E3 or 1 mg E2 for 2 weeks. The spleens from three animals per treatment group were then pooled for preparation of splenocytes. The splenocytes (at 5 × 106 cells/ml) were then cultured for 48 h in the absence (a) or presence (b) of ConA, respectively, for determining cell growth (based on [3H]-thymidine incorporation). For data shown in (c) and (d), the splenocytes were cultured for 24 h in the presence of ConA and then IFNγ and IL-2 levels in the medium were determined by ELISA. Data are reported as mean (± SD) of triplicates. Note that two separate experiments were performed and similar results were obtained; only one representative data set is shown. * p < 0.05 vs vehicle-treated animals. Group labels: open bar (V) = mice treated with vehicle only, light shaded bar (E2) = mice treated with E2, and black bar (E3) = mice treated with E3.
![Figure 5. Effect of E3 and E2 on splenocyte proliferation and function in naïve mice. (a and b) Splenocyte proliferation in the absence and presence of ConA. (c and d) Production of cytokines (IFNγ and IL-2) in the presence of ConA. In these experiments, female BALB/c mice were first implanted with a pellet containing 10 mg E3 or 1 mg E2 for 2 weeks. The spleens from three animals per treatment group were then pooled for preparation of splenocytes. The splenocytes (at 5 × 106 cells/ml) were then cultured for 48 h in the absence (a) or presence (b) of ConA, respectively, for determining cell growth (based on [3H]-thymidine incorporation). For data shown in (c) and (d), the splenocytes were cultured for 24 h in the presence of ConA and then IFNγ and IL-2 levels in the medium were determined by ELISA. Data are reported as mean (± SD) of triplicates. Note that two separate experiments were performed and similar results were obtained; only one representative data set is shown. * p < 0.05 vs vehicle-treated animals. Group labels: open bar (V) = mice treated with vehicle only, light shaded bar (E2) = mice treated with E2, and black bar (E3) = mice treated with E3.](/cms/asset/9da0f08e-2c5b-4e5d-b6d2-2328798a7fdf/iimt_a_617791_f0005_b.gif)
Figure 6. Effect of E3 and E2 on MAPK activation in splenocytes from naïve mice. Three spleens from vehicle- or estrogen-treated mice were pooled for the preparation of splenocyte suspension. The pooled splenocytes were then cultured in the presence or absence of ConA for 10 min. Cells from multiple in vitro treatment wells were pooled together for preparation of whole cell lysates, which were used for measurement of protein levels by Western blot analysis (a). The blots were re-probed with anti-β-actin antibody for confirmation of equal loading of proteins in each lane. Quantitative values shown in (b) were calculated according to laser densitometry scanning data (mean from two separate experiments). Since two bands were detected for ERK1/2, the quantitative data were based on their combined changes. The unit for the relative protein level is the densitometry ratio of the protein of interest to that of β-actin. Group labels: V = mice treated in vivo with vehicle only, E2 = mice treated with E2, and E3 for animals treated with E3.
Lastly, in order to determine whether the observed effect of E3 and E2 on splenocytes in vivo was attributable to their direct effect on the splenocytes, we determined the direct effect of E3 and E2 in vitro on viability and cytokine production by cultured splenocytes from untreated control female mice. Treatment of splenocytes with E3 and E2 in vitro at 1, 10, or 100 nM did not affect cell viability in the absence of ConA (). They also did not affect ConA-induced lymphocyte proliferation (). Whereas the two estrogens did not affect IL-2 production in vitro (), E3 slightly inhibited the production of IFNγ (). Altogether, these data suggest that the observed in vivo effects of E3 and E2 on splenocyte proliferation and function are not attributable to their direct actions in the splenocytes.
Figure 7. Effect of E3 and E2 on splenocyte proliferation and cytokine production in vitro. (a and b) Measurements of splenocyte viability (MTT assay) and proliferation ([3H]-thymidine incorporation assay) in the absence or presence of ConA. (c and d) Splenocyte cytokine production in presence of ConA. Pooled splenocytes were prepared from three untreated female BALB/c mice and cultured in 96-well plates (×106 cells/ml) with E2 or E3 in the absence (a) or presence (b–d) of ConA. For testing the effect of estrogens on cell viability and proliferation, cells were cultured for 48 h; for testing their effect on cytokine release, cells were only cultured for 24 h. Each value shown is the mean (± SD) of triplicate cultures. Three separate experiments were performed and similar results were obtained; one representative data set is shown here. * p < 0.05 vs control without estrogen.
![Figure 7. Effect of E3 and E2 on splenocyte proliferation and cytokine production in vitro. (a and b) Measurements of splenocyte viability (MTT assay) and proliferation ([3H]-thymidine incorporation assay) in the absence or presence of ConA. (c and d) Splenocyte cytokine production in presence of ConA. Pooled splenocytes were prepared from three untreated female BALB/c mice and cultured in 96-well plates (×106 cells/ml) with E2 or E3 in the absence (a) or presence (b–d) of ConA. For testing the effect of estrogens on cell viability and proliferation, cells were cultured for 48 h; for testing their effect on cytokine release, cells were only cultured for 24 h. Each value shown is the mean (± SD) of triplicate cultures. Three separate experiments were performed and similar results were obtained; one representative data set is shown here. * p < 0.05 vs control without estrogen.](/cms/asset/93ceb6ea-4821-4125-a92c-66df064d80dc/iimt_a_617791_f0007_b.gif)
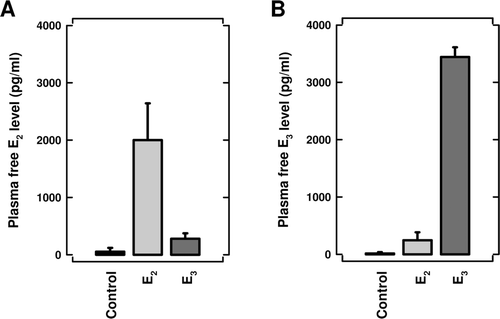
Plasma levels of unbound E3 and E2 in naïve mice implanted with pellet containing E3 or E2
For steroid hormones such as estrogens, it has been well documented that free (unbound) circulating hormone levels are a more useful parameter for assessing the magnitude of receptor activation in target cells than the total hormone levels (i.e., bound + free). Therefore, in order to provide an estimate of the magnitude of ER activation in animals implanted with 1 mg E2 or 10 mg E3, we determined plasma levels of unbound (free) E3 and E2 in naïve mice. As shown in , 5 days after SC implantation of a pellet containing 1 mg E2, plasma levels of free E2 were ~ 2 ng/ml (7.4 nM). In comparison, plasma levels of free E3 were ~ 3.6 ng/ml (12.5 nM) at 5 days after implantation of a pellet containing 10 mg E3 (). Surprisingly, the apparent plasma levels of free E2 or E3 in mice implanted with a pellet containing E3 or E2, respectively, were also slightly higher than the control group (implanted with a vehicle pellet). Although the metabolic conversion of E2 to E3 can occur in vivo (catalyzed by cytochrome P450 enzymes), there has been no evidence for the reverse conversion (i.e., E3 to E2). Accordingly, the apparent small increase in E3 plasma levels in mice implanted an E2 pellet most likely was because the E3 antibody used in the ELISA kit might have had certain degrees of cross-reactivity with E2.
Figure 8. Plasma levels of free (unbound) E3 and E2 in naïve mice implanted subcutaneously with a pellet containing 10 mg E3 or 1 mg E2. Control animals were implanted with a vehicle pellet only. Five days post-implantation, the mice were euthanized and their blood collected. Free plasma E3 and E2 levels were determined as described in the Materials and methods section. The data were expressed as mean (± SD) of 5–6 animals per treatment group.
Discussion
In humans, E3 is an endogenous estrogen that is predominantly produced, in very large quantities, during pregnancy (Fotsis, Citation1987). It is hypothesized that E3 may play a unique role in modulating the immune system functions during human pregnancy. In partial support of this possibility, a recent study showed that E3 administration significantly prolonged the survival of major histocompatibility complex (MHC)-compatible heart allografts in a rat model (Funeshima-Fuji et al., Citation2009). Moreover, E3 was also shown to have a rather different effect from E2 in modulating the production of antigen-induced specific antibodies (Ding and Zhu, Citation2008). In the present study, we investigated the modulating effect of E3 on splenocyte functions, and its effect was compared with that of E2. Our data showed that E3, in multiple instances, has distinctly different effects from E2 in modulating the functions of splenic lymphocytes in female BALB/c mice under inflammatory conditions, although the overall effect of E3 and E2 is very similar in immunologically-naïve mice.
It is known that estrogens can induce strong thymic atrophy in rodents (Staples et al., Citation1999). The results of our present study show that estrogens can also strongly suppress spleen weight gain under inflammatory immunological conditions, but not in naïve female mice. In naïve mice, the effect of E3 on the spleen weight change is modest (< 20%), and E2 does not show an appreciable effect. However, in mice treated with CFA, splenic weight was increased approximately 3-fold, and treatment with E3 almost completely inhibited splenomegaly development. The effect of E3 is significantly stronger than that of E2. This is the first report of a strong suppressive effect of E3 and E2 on splenic weight under inflammatory conditions. In addition, ex vivo experiments show that in vivo E3 treatment strongly suppresses proliferation of T-cells from CFA-treated animals and strongly inhibits IFNγ production (). While the effect of E2 on T-cell proliferation is far weaker than that of E3, its effect on splenocyte IFNγ production is completely opposite to that of E3 (). These data suggest that E3 has different effects than E2 in modulating splenocyte proliferation and function under different immunological and/or inflammatory conditions.
In naïve BALB/c mice, treatment with E3 or E2 increases the populations of splenic CD4+ and CD8+ T-cells, but decreases the population of B-cells. Additional ex vivo experiments showed that the rate of splenic T-cell proliferation is significantly elevated following in vivo estrogen treatment. In comparison, when splenocytes from naïve, untreated animals were cultured in vitro, E3 and E2 did not directly affect mitogen-induced splenic T-cell proliferation or TH1 cytokine production. These data suggest that the in vivo effects of E3 and E2 are not attributable to their direct effects on splenic lymphocyte functions. It is likely that estrogens may first act on other cells (non-splenocytes) to induce the production of regulatory factors, and these factors then act on the spleen to modulate its immunological functions. It will be of interest to test this hypothesis further in the future.
It is known that when cells of the immune system interact with the invading micro-organisms, their Toll-like receptors (TLRs) will be activated, which then leads to the activation of several intracellular signaling pathways, including the mitogen-activated protein (MAP) kinases such as ERK and p38 (Hitti and Kotlyarov, Citation2007). In recent years, the ERK and p38 signaling pathways have been recognized as important targets for anti-inflammatory therapy. In this study, we investigated the modulating effect of E3 and E2 on the activation of MAPKs in splenocytes and PEC of mice under different immunological conditions. In splenocytes, E3 treatment consistently blocks ERK1/2 activation (phosphorylation), whereas E2 has a milder effect on ERK1/2 activation. E3 also strongly suppresses p38 activation (phosphorylation), but the effect of E2 on p38 activation is significantly less efficacious. Similarly, we also showed that the effects of E3 and E2 are quite different in modulating the ERK1/2 and p38 signaling pathways in PEC. Since PEC are commonly used as an experimental model for studying the antigen-presenting cells (APC), the different effect of E3 and E2 on PEC functions suggests that they may have a different modulating effect on APC functions through differential modulation of the ERK1/2 and p38 pathways. Lastly, it is of interest to mention that the in vivo and in vitro effects of E3 and E2 on PEC functions are very similar, which suggests that estrogens may directly act on PEC to regulate their functions. In contrast, the in vitro and in vivo effects of these two estrogens on splenocyte functions are completely different, which suggests that the effects of E3 and E2 on splenocyte functions likely are indirect, i.e., they are likely mediated via their initial actions on other cells, such as the APCs, which then regulate the functions of splenocytes.
Several earlier studies showed that estrogens (particularly E3) could modulate antigen-specific immune response in autoimmune disease animal models, including those for experimental autoimmune encephalomyelitis (EAE) and collagen-induced arthritis (CIA) (Soldan et al., Citation2003; Palaszynski et al., Citation2004; Elloso et al., Citation2005). These autoimmune conditions are usually induced by injection of specific antigens that are emulsified with CFA. However, there are few studies examining the modulating effects of estrogens on CFA treatment alone. CFA contains various bacterial components that can effectively activate APC and may determine the immune response profiles. It is possible that the estrogen may exert a more predominant modulation of an immune response to CFA over its modulation of the immune response to the specific antigens used in inducing autoimmunity in these experimental disease models. The distinct differences in the immunomodulating effects of E3 vs E2 may be, in part, due to their differential ability to modulate APC functions in vivo during infection and inflammation. This possibility merits further investigation because the occurrence of infectious diseases and severe inflammation often are conditions that precede the onset of autoimmune diseases.
The mechanism underlying the distinctly different modulating effects of E3 and E2 on splenocyte functions is not clear at present, but a few speculative views are entertained below. It is known that both ER sub-types are expressed in cells of the mouse immune system (Smithson et al., Citation1995; Igarashi et al., Citation2001; Sakazaki et al., Citation2002), and that both E3 and E2 can activate ERα and ERβ signaling in these cells to modulate immune responses via their genomic actions (Salem et al., Citation2000; Maret et al., Citation2003; Polanczyk et al., Citation2003; Elloso et al., Citation2005; Baker et al., Citation2004). When E3 and E2 exert a similar modulating effect on certain immune system functions, it is likely that their actions are mediated through activation of the ERα and/or ERβ system(s), since both estrogens can bind to and activate these two receptor systems. However, when E3 exerts a very different effect from E2, the underlying mechanisms likely are more complex.
One possible explanation may lie in the possibility that E2 and E3 at different doses may activate ERα and/or ERβ differentially. However, under the experimental conditions used in this study, this possibility appears to be less likely. We found that the free plasma E2 concentration was ~ 2 ng/ml (7.4 nM), which could produce a near maximal activation of both ERα and ERβ, assuming that the Kd values of E2 for these two ERs are similar (at 0.1–0.2 nM). Notably, the free plasma concentration of E3, which was found to be at 3.6 ng/ml (12.5 nM), should also be able to produce a strong activation of both ERα and ERβ. Therefore, it is unlikely that a preferential activation of ERβ over ERα by E3 would contribute predominantly to the observed differences. Instead, it seems a possibility that, in addition to ERα and ERβ, E3 might activate additional signal transduction pathways in cells of the immune system that are refractory to E2. In recent years, there is growing support for the concept that certain biologically-active estrogen metabolites formed in the body under certain physiological or pathophysiological conditions may serve as new hormones or as local chemical mediators which exert a distinct profile of biological functions (discussed in Zhu and Conney, Citation1998). E3 has been suggested to be one of such bioactive estrogen metabolites in light of the fact that this endogenous estrogen derivative has unique biological functions during human pregnancy that are very different from E2 (Zhu and Conney, Citation1998; Zhu, Citation2008; Funeshima-Fuji et al., Citation2009).
In summary, the results of our present study show that E3 has distinctly different effects from E2 in modulating some of the splenocyte functions in female BALB/c mice under different immunological and/or inflammatory conditions. While E3 and E2 each promote splenic T-cell functions in immunologically-naïve mice, E3 suppresses splenic T-cell IFNγ release in CFA-treated mice, which is opposite to the effect of E2. It will be of considerable interest to better understand the cellular and molecular mechanisms by which E3 and E2 differentially modulate some of the splenocyte functions.
Acknowledgments
We thank Dr Xiao-Kang Li at the National Research Institute for Child Health and Development (Tokyo, Japan) for helpful discussion and suggestions regarding some of the experimental methods and assays. This study was supported, in part, by a grant from the National Institutes of Heath (ES015242). Some of the instruments employed in this study are part of the COBRE core facility that is supported by the NIH Grant P20RR021940 from the National Center for Research Resources.
Declaration of interest
The authors declare that there exists no conflict of interest that could be perceived as prejudicing the impartiality of the research reported here. The authors alone are responsible for the content and writing of the paper.
References
- Aluvihare V. R., KallikourdisM., and Betz A. G. 2004. Regulatory T-cells mediate maternal tolerance to the fetus. Nat. Immunol. 5:266–271.
- Amura C. R., KameiT., ItoN., Soares M. J., and Morrison D. C. 1998. Differential regulation of lipopolysaccharide (LPS) activation pathways in mouse macrophages by LPS-binding proteins. J. Immunol. 161:2552–2560.
- Baker A. E., Brautigam V. M., and Watters J. J. 2004. Estrogen modulates microglial inflammatory mediator production via interactions with estrogen receptor β. Endocrinology 145:5021–5532.
- Cargnello M, and Roux PP. 2011. Activation and function ofthe MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75:50–83.
- Clarke A. G., and Kendall M. D. 1989. Histological changes in the thymus during mouse pregnancy. Thymus 14:65–78.
- Clarke A. G., and Kendall M. D. 1994. The thymus in pregnancy: The interplay of neural, endocrine, and immune influences. Immunol. Today 15:545–551.
- DingJ., and Zhu B. T. 2008. Unique effect of the pregnancy hormone estriol on antigen-induced production of specific antibodies in female BALB/c mice. Steroids 73:289–298.
- Elloso M. M., PhielK., Henderson R. A., Harris H. A., and Adelman S. J. 2005. Suppression of experimental autoimmune encephalomyelitis using estrogen receptor-selective ligands. J. Endocrinol. 185:243–252.
- Fotsis T. 1987. The multicomponent analysis of estrogens in urine by ion exchange chromatography and GC-MS. II. Fractionation and quantitation of the main groups of estrogen conjugates. J. Steroid Biochem. 28:215–226.
- Funeshima-FujiN., FujinoM., XieL., KimuraH., TakaharaS., EzakiT., Zhu B. T., and Li X. K. 2009. Prolongation of rat major histocompatibility complex-compatible cardiac allograft survival during pregnancy. J. Heart Lung Transplant. 28:176–182.
- HittiE., and Kotlyarov A. 2007. The ERK and p38MAPK pathways as targets for anti-inflammatory therapy. Anti-inflamm. Anti-allergy Agents Med. Chem. 6:85–97.
- IgarashiH., KouroT., YokotaT., Comp P. C., and Kincade P. W. 2001. Age and stage dependency of estrogen receptor expression by lymphocyte precursors. Proc. Natl. Acad. Sci. USA 98:15131–15136.
- JanssonL., OlssonT., and Holmdahl R. 1994. Estrogen induces a potent suppression of experimental autoimmune encephalomyelitis and collagen-induced arthritis in mice. J. Neuroimmunol. 53:203–207.
- Johnston B. A., EisenH., and Fry D. 1991. An evaluation of several adjuvant emulsion regimens for the production of polyclonal antisera in rabbits. Lab. Anim. Sci. 41:15–21.
- Lang T. J. 2004. Estrogen as an immunomodulator. Clin. Immunol. 113:224–230.
- MaretA., Coudert J. D., GaridouL., FoucrasG., GourdyP., KrustA., Dupont S.. Chambon P., Druet P., BayardF., and Guery J. C. 2003. Estradiol enhances primary antigen-specific CD4 T-cell responses and TH1 development in vivo. Essential role of estrogen receptor-α expression in hematopoietic cells. Eur. J. Immunol. 33:512–521.
- Novotny E. A., Raveche E. S., SharrowS., OttingerM., and Steinberg A. D. 1983. Analysis of thymocyte subpopulations following treatment with sex hormones. Clin. Immunol. Immunopathol. 28:205–217.
- Palaszynski K. M., LiuH., Loo K. K., and Voskuhl R. R. 2004. Estriol treatment ameliorates disease in males with experimental autoimmune encephalomyelitis: Implications for multiple sclerosis. J. Neuroimmunol. 149:84–89.
- Pernis A. B. 2007. Estrogen and CD4+ T-cells. Curr. Opin. Rheumatol. 19:414–420.
- PolanczykM., ZamoraA., SubramanianS., MatejukA., Hess D. L., Blankenhorn E. P., TeuscherC., Vandenbark A. A., and Offner H. 2003. The protective effect of 17β- estradiol on experimental autoimmune encephalomyelitis is mediated through estrogen receptor-α. Am. J. Pathol. 163:1599–1605.
- Qin H. Y., Sadelain M. W., HitchonC., LauzonJ., and Singh B. 1993. Complete Freund’s adjuvant-induced T-cells prevent the development and adoptive transfer of diabetes in non- obese diabetic mice. J. Immunol. 150:2072–2080.
- SakazakiH., UenoH., and Nakamuro K. 2002. Estrogen receptor-α in mouse splenic lymphocytes: Possible involvement in immunity. Toxicol. Lett. 133:221–229.
- Salem M. L., Hossain M. S., and Nomoto K. 2000. Mediation of the immunomodulatory effect of β–estradiol on inflammatory responses by inhibition of recruitment and activation of inflammatory cells and their gene expression of TNFα and IFNγ. Int. Arch. Allergy Immunol. 121:235–245.
- SmithsonG., MedinaK., PontingI., and Kincade P. W. 1995. Estrogen suppresses stromal cell-dependent lymphopoiesis in culture. J. Immunol. 155:3409–3417.
- Soldan S. S., Retuerto A. I. A., Sicotte N. L., and Voskuhl R. R. 2003. Immune modulation in multiple sclerosis patients treated with the pregnancy hormone estriol. J. Immunol. 171:6267–6274.
- Staples J. E., Gasiewicz T. A., Fiore N. C., Lubahn D. B., Korach K. S., and Silverstone A. E. 1999. Estrogen receptor-α is necessary in thymic development and estradiol-induced thymic alterations. J. Immunol. 163:4168–4174.
- TangH., NishishitaT., FitzgeraldT., Landon E. J., Inagami T. 2000. Inhibition of AT1 receptor internalization by concanavalin A blocks angiotensin II-induced ERK activation in vascular smooth muscle cells. Involvement of epidermal growth factor receptor proteolysis but not AT1 receptor internalization. J. Biol. Chem. 275:13420–13426.
- UtsuyamaM., and Hirokawa K. 1989. Hypertrophy of the thymus and restoration of immune functions in mice and rats by gonadectomy. Mech. Ageing Dev. 47:175–185.
- Wang J. X, TangW., Yang Z. S., WanJ., Shi L. P., ZhangY., ZhouR., NiJ., Hou L. F., ZhouY., He P. L., Yang Y. F., LiY., Zuo J. P. 2007. Suppressive effect of a novel water-soluble artemisinin derivative SM905 on T cell activation and proliferation in vitro and in vivo. Eur. J. Pharmacol. 564: 211–218.
- ZhangY., Graubard B. I., Klebanoff M. A., RonckersC., Stanczyk F. Z. Longnecker M. P., and McGlynn K. A. 2005. Maternal hormone levels among populations at high and low risk of testicular germ cell cancer. Br. J. Cancer 92:1787–1793.
- ZhouR., Zheng S. X., TangW., He P. L., Li X. Y., Yang Y. F., Li Y. C., Geng J. G., and Zuo J. P. 2006. Inhibition of inducible nitric-oxide synthase expression by (5R)-5-hydroxy-triptolide (LLDT-8) in interferon-γ and bacterial lipopolysaccharide-stimulated macro- phages. J. Pharmacol. Exp. Ther. 316:121–128.
- Zhu B. T. 2008. Is it necessary to control the level of estrogen receptor-α and -β activation in post-menopausal hormone replacement therapy in order to achieve the optimal outcome? Mol. Med. Reports 1:15–20.
- Zhu B. T., and Conney A. H. 1998. Functional role of estrogen metabolism in target cells: Review and perspectives. Carcinogenesis 19:1–27.
- Zhu B. T., Han G. Z., Shim J. Y., WenY., and Jiang X. R. 2006. Quantitative structure- activity relationship of various endogenous estrogen metabolites for human estrogen receptor-α and -β subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology 147:4132–4150.