Abstract
The development and regulatory approval of immunomodulatory pharmaceuticals to treat many human diseases has increased significantly over the last two decades. As discussed by FDA and ICH guidelines, all human pharmaceuticals in development should be evaluated for potential adverse effects on the immune system. Developmental immunotoxicology (DIT) focuses on the concern that early-life (during pre-/post-natal development) exposure to agents which target the immune system may result in enhanced susceptibility to immune-related disease (e.g., infection, autoimmunity, and cancer, particularly leukemia) compared to adults, unique effects not observed in adults, or more persistent effects in comparison to those following adult exposure. This article provides a substantive review of the literature and presents detailed considerations for DIT testing strategies with a specific focus on pharmaceuticals and biopharmaceuticals. In this regard, differences between small molecule and large molecule therapeutics will be considered, along with recommendations for best practices in the assessment of DIT during drug development. In addition, gaps in the DIT knowledge base and current testing strategies are identified. Finally, a summary of an ILSI-HESI-ITC sponsored Workshop conducted in 2010, entitled ‘Developmental Immunotoxicity Testing of Pharmaceuticals’ will be presented. This Workshop consisted of participants from the pharmaceutical, biotechnology, academic, and regulatory sectors, where many of the issues relating to DIT outlined in this review were discussed, key points of consensus reached, and current gaps in the science identified.
| Abbreviations | ||
| ADA, | = | Anti-drug antibodies; |
| BSA, | = | Bovine serum albumin; |
| CMI, | = | Cell-mediated immunity; |
| CTL, | = | Cytotoxic T-lymphocyte; |
| DART, | = | Developmental and Reproductive Toxicology; |
| DIT, | = | Developmental Immunotoxicity; |
| DTH, | = | Delayed-type hypersensitivity; |
| EFD, | = | Embryo-fetal development; |
| EMA, | = | European Medicines Agency; |
| EOGRT, | = | Extended one-generation reproductive toxicity; |
| EPA, | = | Environmental Protection Agency; |
| ePPND, | = | Enhanced pre- and post-natal development; |
| Fc-bb, | = | Fc-bearing biopharmaceuticals (including mAbs); |
| FDA, | = | US Food and Drug Administration; |
| G-CSF, | = | Granulocyte-colony stimulating factor; |
| GD, | = | Gestation day; |
| HBsAg, | = | Hepatitis B Surface Antigen; |
| HESI, | = | Health and Environmental Sciences Institute; |
| HSC, | = | Hematopoeitic stem cell; |
| ICH, | = | International Conference on Harmonization; |
| Ig, | = | Immunoglobulin; |
| IHC, | = | Immunohistochemistry; |
| ILSI, | = | International Life Sciences Institute; |
| ITC, | = | Immunotoxicology Technical Committee; |
| KLH, | = | Keyhole limpet hemocyanin; |
| mAbs, | = | Monoclonal antibodies; |
| NHP, | = | Non-human Primate(s); |
| NK, | = | Natural Killer; |
| OECD, | = | Organization of Economic Cooperation and Development; |
| OPPTS, | = | Office of Prevention, Pesticides and Toxic Substances; |
| PND, | = | Post-natal day; |
| PPND, | = | Pre- and post-natal development; |
| PREA, | = | Pediatric Research Equity Act; |
| rh, | = | Recombinant human; |
| SCF, | = | Stem cell factor; |
| SME, | = | Small molecular entities; |
| SRBC, | = | Sheep red blood cells; |
| STS, | = | Standard Toxicity Study; |
| T- | = | Dependent antibody response (TDAR) |
| TK, | = | Toxicokinetics; |
| TT, | = | Tetanus toxoid; |
| WoE, | = | Weight of Evidence |
Introduction
Evaluation of developmental and reproductive toxicology (DART) has been an integral part of the safety assessment process for new chemicals for many years. During the past two decades, increased attention has been placed on the effects of pharmaceutical, agricultural, and industrial chemicals on the developing organ systems, including the immune system, of the fetus and newborn. The primary concern is that the developing offspring are inherently different in their basic biology and so their responses to environmental and/or pharmaceutical exposures may be different than those observed in adults. There is merit to this hypothesis given that some organ systems in humans continue to develop after birth, and this post-natal maturation appears to be different for various organ and biochemical systems. For example, most pulmonary alveolar maturation occurs in humans in the first 2 years after birth (Burri, Citation1997), while the skeletal system continues to mature well into adulthood (Zoetis and Walls, Citation2003). The consensus in the scientific and regulatory arenas is that the immune system of children continues to develop post-natally between 5–12 years-of-age (Miyawaki et al., Citation1981).
Developmental immunotoxicology (DIT) focuses on the concern that early-life (during pre- and/or post-natal development) exposure to agents which target the immune system may result in enhanced susceptibility to immune-related disease (e.g., infection, autoimmunity, and cancer, particularly leukemia) compared to adults, unique effects not observed in adults, or more persistent effects in comparison to those following adult exposure. Much of the recent literature focuses on the effects of environmental chemicals on the developing immune system, with relatively less regarding pharmaceutical agents; a more thorough discussion of these studies can be found elsewhere (Collinge et al., Citation2010) and in more recent publications (Tonk et al., Citation2010, 2011a).
There are a number of examples in the recent literature suggesting that the developing immune system is more sensitive than that of the adult for some immunotoxic alterations (Miller et al., Citation1998; Dietert et al., Citation2003; Dietert and Piepenbrink, Citation2006; Luebke et al., Citation2006). This apparent sensitivity may be due in part to novel immune maturation events including the need for rapid perinatal changes in functional equilibrium to restore the critical immune balance between T-helper (TH)-1 and TH2 cell populations through the enhancement of TH1 capacity in the newborn (Holt et al., Citation2005; Yun and Lee, Citation2005). While comparative interpretation across DART studies is, in most cases, impossible due to the varied dosing paradigms (in utero only, juvenile only, critical developmental windows, etc.), it is clear that a differential sensitivity can exist and should be considered during the safety evaluation of any drug or non-drug chemical. From a regulatory perspective there is concern, then, that toxicity studies conducted in young adult animals (typically rodents) would not detect this potential for greater sensitivity.
To date, no developmental immunotoxicants have been identified in animals that are not also immunomodulatory in the adult animal; however, this may be a function of the current state-of-the-science, the fact that most DIT studies to date have been conducted with known adult immunotoxicants, most of which are non-drug chemicals, and/or the availability of this information in published (e.g., public) forms. Examples of developmental immunotoxicity assessments with pharmaceutical compounds, including cyclosporine, dexamethasone, diazepam, aciclovir, and diethylstilbestrol (DES), have been described (Stahlmann et al., Citation1992; Schlumpf et al., Citation1989, Citation1994; Dietert et al., Citation2003; Fenaux et al., Citation2004; Hussain et al., Citation2005; Barrow et al., Citation2006; Luebke et al., Citation2006). Recently, it was suggested that the United States (US) Food and Drug Administration (FDA) has possibly observed a case of differential sensitivity where juvenile animals displayed effects not observed in toxicity studies conducted in the adult; however, due to confidentiality, no specific information could be shared.
In addition to examples of increased sensitivity of the developing immune system, observations of more persistent effects have also been described. Exposure of adults to cyclosporine produced acute immunotoxicity with minimum long-term effects (Hussain et al., Citation2005). However, in uteroexposure led to much more persistent effects, with TDAR responses reduced in offspring at 13 weeks-of-age, whereas adult TDAR responses returned to normal levels by 13 weeks. It is important to note that others have not shown similar effects (Barrow et al., Citation2006) and immunotoxicity endpoints may be differentially affected. However, it is clear that in some instances toxicity to the developing immune system may take longer to reverse, and the specific window of exposure may be important. Female mice dosed with DES from Post-natal day (PND) 1–5 demonstrated decreased mitogen-induced splenocyte proliferation several months after treatment (Kalland et al., Citation1979; Luebke et al., Citation2006), delated-type hypersensitivity (DTH) responses that were diminished 6 months after treatment (Kalland and Forsberg, Citation1978), and reduced TDAR at 16 weeks (Kalland, Citation1980).
This article will consider DIT testing in the context of pharmaceutical development. A number of prior publications have addressed the issue of DIT. However, this is the first substantive review that provides a detailed consideration of testing strategies and unique aspects of DIT that are of relevance solely to pharmaceuticals and biopharmaceuticals. Areas that will be addressed include strategic drivers that make pharmaceutical safety evaluation unique from that of industrial and environmental chemicals, causes for concern that might trigger the need for DIT testing, differences between small molecule and large molecule therapeutics that present unique challenges, and gaps in our knowledge and testing methodology. Finally, recommendations for assessing DIT in the drug development paradigm will be presented.
Regulatory drivers
Evaluations of DIT began to be required as part of the US Environmental Protection Agency (EPA) Office of Prevention, Pesticide and Toxic Substances (OPPTS) testing guideline for the two-generation reproductive toxicity study (USEPA, Citation1998), which requires collection of spleen and thymus weights in one pup/sex/litter in F1 and F2 weanlings. Within the pharmaceutical industry, the FDA was the first to put forward recommendations regarding DIT. In its 2002 guidance for industry (FDA, Citation2002), the Agency noted that all drugs that are immunotoxic in adult animals and are to be given to pregnant women should be evaluated for DIT. Endpoints recommended for assessment were hematology and lymphoid organ weights on F1 pups in standard DART studies. Other global regulatory authorities have not specifically addressed DIT; however, the topic has been indirectly considered in multiple guidance documents addressing juvenile toxicity testing.
In the wake of legislation changes aimed at children’s health assessments for non-drug chemicals in the late 1990s, and as a means to formally codify what had previously been known as the ‘Pediatric Rule’, the Pediatric Research Equity Act of 2003 (PREA) was enacted. PREA requires sponsors who submit drug applications for new active ingredients, indications, dosage forms, dosing regimen, or a new route of administration to include an evaluation for all relevant pediatric populations for which the sponsor is seeking an approved indication. These assessments must be adequate to assess the safety and effectiveness of the drug in each pediatric population and to support dosing instructions. The passage of PREA was followed closely by the development of a new FDA guidance document addressing the non-clinical safety evaluation of pediatric drug products (FDA, Citation2006). Although no specific recommendations for DIT testing are made, this guidance does include the age ranges through which multiple organ systems develop, with the understanding that Sponsors will consider these developmental milestones in the assessment of the need to conduct juvenile toxicity studies, as well as which endpoints should be included in that assessment. In the European Union, similar legislation and guidance for industry has also been passed. Regulation EC 1901/2006 (2006) requires pharmaceutical companies to submit their plans for pediatric investigation by the end of the Phase 1 trials supporting adult indications. As part of this, Sponsors must include any plans for non-clinical juvenile toxicity studies to support these trials. European regulatory guidance around juvenile toxicity testing (EMA, Citation2008) is similar to that from the FDA in that for DIT, the guidance notes that the European Medicines Agency (EMA) considers that the immune system continues to mature post-natally through the age of 12 and, thus, plans to treat children at age 12 or younger necessitates the consideration of whether a specific DIT evaluation is necessary in young animals.
Global regulatory agencies do not require evaluation of DIT as a routine assessment during the drug development process; rather, assessments are driven primarily by a cause-for-concern derived from a weight of evidence (WoE) review of the totality of the available data, the intended patient population, and the potential that the fetus, neonate, or juvenile might be exposed either intentionally or unintentionally. Notably, this approach is not unique for the immune system but applies to any organ system with continuing post-natal development. A number of novel therapeutics are emerging for which the immune system is a potential target organ, or for which a re-directed immune response is a pharmacologic end-point. Therefore, compliance with these regulations and guidelines, and regulatory agency or Sponsor concerns, may increase the need to conduct non-clinical and clinical pediatric immunotoxicity assessments in the future.
A paradigm difference: Drugs vs industrial chemicals
In a 2001 Workshop sponsored by the Immunotoxicology Technical Committee (ITC) of the International Life Sciences Institute (ILSI) Health and Environmental Sciences Institute (HESI) (Holsapple, Citation2002), several recommendations were made by the participants regarding assessment of DIT. Most notable was the assertion that the best approach to assess DIT may be to address all critical developmental windows (e.g., gestation, post-natal, weaning, juvenile) at once, ideally by adding DIT evaluations to DART studies wherever feasible. Although no regulatory guidance specifically addresses DIT from this perspective, the proposed change to the Organization of Economic Cooperation and Development (OECD) 415 test guideline (OECD, Citation2010) would extend the one-generation reproductive toxicity study to include exposure through all critical windows and an evaluation of both immunopathology and immune function (T-dependent antibody response; TDAR).
The main objective of the extended one-generation reproductive toxicity (EOGRT) study is to evaluate specific life stages not covered by other types of toxicity studies, and to test for toxicities that may occur as a result of pre- and post-natal exposure. The test substance is administered to the sexually-mature parental generation for a minimum of 2 weeks prior to mating, and for 2 weeks during the mating period. Parental females are dosed throughout pregnancy and lactation, and, following weaning, the F1 offspring continue to be dosed to adulthood. At weaning, offspring are randomly assigned to three cohorts: Cohort 1 is used to assess reproductive and developmental endpoints; Cohort 2 is used to assess the effect of chemical exposure on the developing nervous system; and Cohort 3 assesses the potential of chemical exposures on the developing immune system. Cohort 3 includes a minimum of 10 males and 10 females, with a primary TDAR conducted at PND 56. The significance of the TDAR is interpreted in the context of additional immunologic indicators, including bone marrow cellularity, weight and histopathology of lymphoid tissues, and lymphocyte subset distribution. There is no question that this represents the most robust assessment of the potential hazard of a chemical by utilizing in utero, lactational, and juvenile exposure, and has been used to assess the developmental immunotoxicity of, for example, di-n-octyltin dichloride (DOTC) (Tonk et al., 2011b).
However, there is a significant difference between chemicals used for therapeutic applications and those used for industrial/agricultural purposes. For industrial chemicals, exposure of humans is typically unintentional and there is often no direct benefit to humans by the exposure. In the case of pharmaceuticals, the intention is to treat a defined population for which there is clear benefit to intervention, often for a pre-defined period of time. Thus, there is a clear risk:benefit difference for human exposure between these two industrial sectors, and there is a clear intentional vs unintentional exposure paradigm. This is not to say that testing for pharmaceuticals should be any less rigorous than for industrial chemicals. Rather, it suggests that specialized testing (such as DIT evaluations) may be performed on a case-by-case basis rather than ‘across the board’. DIT testing for environmental chemicals by necessity requires full hazard identification and risk characterization since there is no upfront knowledge of when and for how long an individual may be unintentionally exposed.
On the other hand, in the case of drug development, exposure to the developing system is likely to occur only in two general scenarios: inadvertent exposure in utero or via breast milk as a result of the mother taking the drug, or direct dosing of children to treat a childhood disease. In either case, the goal in drug development is to assess risk of developmental effects resulting from these scenarios and not to generally assess a hazard, such as might be the desire when considering unintentional exposure to non-drug chemicals. For example, treating children aged 8 and older specifically addresses a defined population, and that population can be evaluated in animal studies with comparable developmental stages to ask the specific question, ‘what happens to organ system X in offspring of this age?’ This approach is reflected well in the FDA juvenile testing guidance (FDA, Citation2006). The ability to be flexible in determining both the need for and the study designs to assess DIT is a key, unique feature for pharmaceutical development.
Possible cause(s) for concern
The initial question for the pharmaceutical industry is whether there is a need to assess the impact of a new molecular entity on the developing immune system. Following the DIT and risk assessment workshop (Holsapple, Citation2002), the ILSI HESI ITC sponsored a Roundtable discussion in 2003 to reach agreement on the most appropriate methods to assess DIT, including under what conditions such testing might be needed (Holsapple et al., Citation2005). The following triggers, or possible cause(s)-for-concern, were identified: (1) findings in standard toxicity studies (e.g., increased susceptibility to disease, altered lymphoid organs weights, histologic changes in lymphoid tissue, etc.); (2) structural similarity, including structure-activity relationships or belonging to a class of compounds known to target the immune system (Dietert et al., 2001); (3) characteristics of the intended patient population, including the ability to improve the risk characterization by conducting a study; and (4) findings in clinical trials or Phase 4 follow-up (e.g., increased incidence of infection, particularly rare infections, or immune-related disease). Of particular interest, these cause(s)-for-concern also form the basis of the ‘additional information’ in the WoE review defined by the International Conference on Harmonization (ICH) S8 immunotoxicity guidance (ICH, Citation2006).
Differential challenges between small and large molecule biotherapeutics
Species selection
The selection of an appropriate species for DIT evaluation is a fundamental aspect of the assessment. The consensus in the scientific community has been that the rat is the best model for assessing DIT due to the large knowledge-base, availability of reagents/methods to assess immune function, and the ability to add these endpoints onto existing DART protocols (Holsapple, Citation2002; Holsapple et al., Citation2005; Ladics et al., Citation2005). While the authors of this review are in agreement with this statement, this consensus opinion reflects a broad generalization that, at the time it was being debated/discussed, specifically did not focus on possible differences between environmental chemicals and pharmaceuticals. For pharmaceuticals, the selection is critical and much more complex. Factors beyond the knowledge base and reagent availability must be considered, and there will be times when the rat is not the most appropriate species, for example where there is lack of rodent cross-reactivity with a biopharmaceutical drug.
For small molecule drugs, selection should reflect what is known about the absorption, distribution, metabolism, and elimination of the compound in both humans and non-clinical species. Biopharmaceuticals also have unique properties, including cross-reactivity, which can significantly influence the selection of appropriate species and the reader is referred to comprehensive reviews on the subject by Bussiere (Citation2008b) and de Haan et al. (Citation2011). In both cases the drug should be pharmacologically active; however, consideration must also be given to possible species differences in the biology of the target.
For some biopharmaceuticals, the rat (or sometimes mouse) is a pharmacologically relevant species. In this case, the rodent may be used for DIT testing, but other pharmacologically relevant species (e.g., non-human primates [NHP]) may be considered if DART evaluations are being conducted in the other species. For many biopharmaceuticals the only pharmacologically relevant species is the NHP, which compels DIT testing in this species. When no pharmacologically-relevant species exists, toxicity testing of the biopharmaceutical may be conducted in transgenic mice expressing the human target or the toxicity of a homologous protein (against the rodent orthologue of the human target) may be evaluated in rodents. In such cases, it may be possible to conduct DIT testing in these models. Whichever species is selected, it is important to understand the limitations of the animal model, which would include, but is not limited to (i) current understanding of the comparative age of the immune system in that species compared to the clinical population, (ii) availability of reagents/assays to test for DIT, (iii) differences in the biology of the target (and therefore possible differences in pharmacology) between humans and the selected species, and/or (iv) availability, or lack thereof, of historical data demonstrating translation of DIT findings from this species to the clinic.
Species differences in fetal drug exposure
NHP are frequently the only species in which a biopharmaceutical is active, and, therefore, NHP are also often the test species for any DART testing that may be required. For the developmental component, the assumption is that the fetus will be exposed to the test pharmaceutical during the sensitive period of organogenesis and immune system development. During organogenesis, placental transfer of small molecular entities (SME) is presumed to occur primarily by passive diffusion. Although placenta structural differences across species may impact the extent of passive diffusion, fetal exposure is anticipated. However, large molecular weight biopharmaceuticals are restricted in their ability to diffuse or cross the placenta and minimal transfer of Fc-bearing biopharmaceuticals (Fc-bb), which includes monoclonal antibodies (mAb) still containing their Fc-fragment, is anticipated during organogenesis. Hence, one of the fundamental concerns for biopharmaceutical testing is the degree of placental transfer of the large protein. This subject has been extensively reviewed (Pentsuk and van der Laan, Citation2009; Stewart, Citation2009), and only a general overview as it pertains to biopharmaceutical development is discussed here.
Timing of fetal exposure during pre-natal development is a key factor in determining what effect a drug may have on embryonic morphogenesis. Rodent gestation and development are relatively short compared to humans. Rodents initially develop a yolk sac placenta around Day 6–7 (inverted yolk sac placenta) followed by the establishment of a chorioallantoic placenta on day 11 of gestation (DeSesso, Citation2005). In contrast, in dog, monkey, and human, the chorioallantoic placenta is in place throughout gestation (de Rijk and van Esch, Citation2008). Nutrients and most SME drugs, and presumably large molecular weight biopharmaceuticals, more freely or quickly diffuse through an inverted yolk sac placenta than through the chorionic placenta (DeSesso, Citation2005; Stewart, Citation2009). SME (< 600 Da) and nutrients diffuse across the placenta by simple diffusion that depends on molecular weight, lipid solubility, degree of ionization, and plasma protein binding. Large molecular weight biopharmaceuticals (> 1000 Da) are presumed not to cross the placenta with the exception of Fc-bb. The Fc-bb is presumed to cross the placenta in parallel with the physiologic transfer of maternal IgG by Fc receptor-mediated endocytosis (Leach et al., Citation1996; Simister, Citation2003; DeSesso, Citation2005). The timing and transfer of maternal IgG is related to placental tissue expression and functionality of the neonatal Fc receptor (FcRn), and possibly other Fc receptors that mediate receptor-mediated transcytosis of IgG (Roopenian and Akilesh, Citation2007). As a consequence, very little IgG is transported during the first trimester when FcRn expression is low (Simister, Citation2003). In NHP, maternal IgG transfer gradually increases as gestation progresses, with a large increase in IgG transferred in the last month before birth (Fujimoto et al., Citation1983; Coe et al., Citation1993) ().
Figure 1. Fetal/maternal ratio of macaque IgG antibodies. Figure depicts estimated ratios of fetal-to-maternal IgG concentrations throughout gestation in macaques and the relationship of fetal antibody exposure relative to the period of organogenesis. Although fetal IgG exposure is represented as being at or below baseline prior to the second trimester, recent information suggests some IgG may be present during the 1st trimester at levels that could be pharmacologically or toxicologically active (Dybdal, Citation2010; Wang et al., Citation2011). (Reprinted with permission from Martin and Weinbauer, Citation2010).
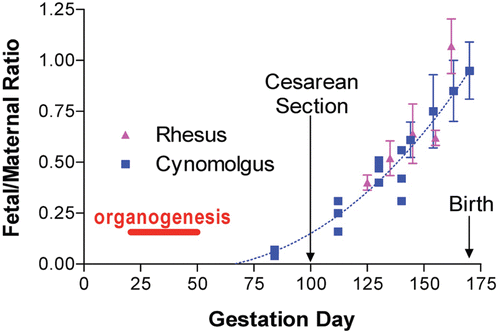
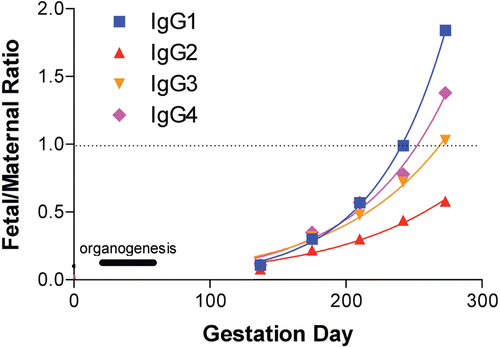
This pattern is very similar to that observed in humans, except that there is an exponential increase in maternal/fetal IgG transfer during the last 2 weeks of pregnancy (). As shown, there is preferential transport of IgG1 over other subclasses during this time (Fujimoto et al., Citation1983; Coe et al., Citation1993; Malek et al., Citation1996). This increase in pre-natal IgG transport during the latter part of gestation occurs in most animal species used in reproductive toxicity studies. Human Fc-bb cross the macaque placenta during the fetal period and Fc-bb detected in the blood of infants are derived from placental transfer. The presence of such Fc-bb (especially mAb) in a neonate/infant may continue for up to 3–6 months after in utero exposure. In contrast, for non-Fc-bb biopharmaceuticals it may be necessary to dose mothers through lactation in order to continue exposure to the infant. In rodents and dogs, FcRn-mediated IgG transport across the placenta is less efficient than in the neonatal period when FcRn transports maternally-derived IgG in ingested milk across the small intestine (Day, Citation2007; Roopenian and Akilesh, Citation2007; Pentsuk and van der Laan, Citation2009). In addition, fetal exposure in rodents may be the result of IgG transfer across the yolk sac (as mentioned above) or by ingestion of amniotic fluid. Measurable levels of a mouse surrogate of efalizumab (muM17;IgG2a) were detected in fetal mice following dosing of dams, indicating that exposure can occur prior to birth (Chao et al., Citation2009). While placental transfer of Fc-bb in rodents may be lower than post-natal transfer via the gut, and placental transfer of Fc-bb in NHP may be lower early in gestation compared to late in gestation, even low levels of transfer of a Fc-bb have potential pharmacologic or toxicologic relevance (Dybdal, Citation2010; Martin and Weinbauer, Citation2010; Wang et al., Citation2011). In the context of the clinical situation one should ask whether it is important if exposure occurs primarily by placental transfer as occurs in humans, or by other means, so long as exposure occurs.
Figure 2. Placental transport of IgG subtypes in humans. Figure shows the theoretical distribution of fetal serum immunoglobulin isotypes relative to maternal serum during the typical gestational period in humans. The relative order of transport capacity across the placenta is IgG1 > IgG4 > IgG3 > IgG2. (Schematic provided courtesy of Pauline L. Martin, PhD. For more detailed information, see Martin and Weinbauer, Citation2010).
Immunogenicity
In certain cases the human biopharmaceutical is pharmacologically active in several animal species. The default for DART screening in this scenario would be to use rodent or rabbit models rather than NHP. However, for DIT assessments the rabbit is not considered a viable model at this time due to the lack of: (1) knowledge of comparative immune ontogeny and effects of known immunotoxicants; (2) accepted methods to assess DIT; (3) understanding of when would be an appropriate time to assess DIT relative to developmental age; and (4) reagents. However, for the purposes of discussion of immunogenicity, conclusions from rabbit data are very relevant to the similar situation in the rodent.
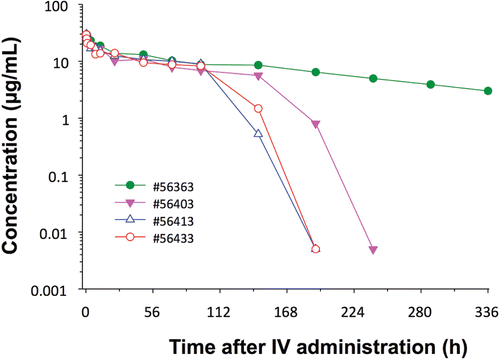
Aside from the issue of species differences in fetal exposure mentioned above, an important concern is that the human biopharmaceutical will generate an immunogenic response in species phylogenetically distant from humans (such as rodents or rabbits) that will either neutralize pharmacology of the biopharmaceutical and/or alter the fetal exposure. In the context of DIT, the formation of neutralizing anti-drug antibodies (ADA) in pregnant mothers may impact fetal exposure. Alternatively, ADA formation in the offspring will reduce exposure in juvenile studies. However, because it takes between 7–21 days to generate significant antibody titers against an antigen (in this case the biopharmaceutical), and because not all animals will mount an ADA response, one can design rodent DART studies to provide adequate safety information. For example, increasing the numbers of animals in each dose group could strengthen the study design, as the incidence of animals with ADA is generally not 100%. To illustrate, when rabbits were given a single dose of a human mAb and the pharmacokinetics of the mAb determined at the end of gestation, several rabbits had a loss of exposure during the study suggestive of an ADA response, but there were also rabbits that did not lose exposure (). In addition, taking into account the lag phase following initiation of dosing before significant titers of ADA occur, that gestation is relatively short in the rodent and rabbit, and that higher doses can be administered to overcome anticipated neutralizing antibodies, it is possible that a successful DART study and associated DIT endpoints can be conducted in most cases with a human biopharmaceutical in species more phylogenetically distant from humans than NHP (when those species have been demonstrated to be pharmacologically relevant).
Figure 3. Kinetics of Antibody X after intravenous administration to female NZW rabbits (1 mg/kg). Figure presents effect of a neutralizing anti-drug antibody on pharmacokinetics of a monoclonal antibody in New Zealand white rabbits that had been previously treated with a human monoclonal antibody. Serum half-life was reduced in three of four rabbits. In order to use this model for DART studies, consideration must be given to the 10–14 day lag-phase for the development of IgG anti-drug antibody and to the inclusion of more animals in each dose group as the incidence of ADA was not 100%.
Study designs and endpoint selection
General factors to consider
Dose selection is an important aspect of DIT evaluations, as it is known that exposures resulting in maternal toxicity, general target organ toxicity, or stress can impact immunotoxicology endpoints (Pruett, Citation2003). Effects resulting from doses causing secondary immunological effects may or may not be relevant to human risk evaluation for DIT. Therefore, testing to a maximum tolerated dose (MTD) is not desirable and should be avoided if possible. If unavoidable, this should be considered in data interpretation and risk evaluation. To the extent possible, an evaluation of systemic exposure during the various life stages is important to the overall characterization and interpretation of DIT. This also includes an understanding of placental transfer and possibly secretion into breast milk. Without an understanding of exposure, a negative DIT study is inadequate for assessing potential risk. If transfer of the compound through the breast milk is unknown, direct dosing of offspring should be considered. Because sexual dimorphisms are known to occur in the mammalian immune response, both sexes should be evaluated unless scientifically justified otherwise (e.g., sex-specific indications).
An evaluation of reversibility of any adverse effects should be included in planned assessments of DIT. While there is no consensus on the appropriate duration of a recovery phase on studies, 2–4 weeks is reasonable for most small molecules based on what is known about the maturation and natural turnover of immune elements from the primary lymphoid organs. Modifications of that duration based on other available information on the agent, including biotherapeutics with long half-lives (e.g., extend duration to 4–5 half-lives or more), would be appropriate.
Endpoint selection
Selection of the most appropriate endpoints to assess DIT, including the question around structure vs function, has been the subject of much debate. Any endpoint selected for use in human health risk evaluation should be predictive of underlying immune system status or capacity, sensitive, able to be performed with reliability, and well-understood biologically. The difficulty in selection lies primarily in the gaps and limitations of the knowledge base and currently available methodologies for assessing DIT. At this point in time, many fewer reliable endpoints are available for assessing the developing immune system compared with those for the adult. Generally speaking, there is consensus that immunopathology endpoints (e.g., hematology, lymphoid organ weights and histology, immunophenotyping) are an important part of the overall characterization of effects on the developing immune system. However, it is plausible that effects on immune function could be observed at drug doses at which there are no observable effects on immunopathology endpoints. Therefore, while immunopathology will likely be sufficiently sensitive to detect immunotoxicity in most cases, there may be instances where functional assessments may be needed. This is not to say that functional assessments should be conducted, irrespective of the data obtained from immunopathology endpoints that are indicative of immunotoxicity. Rather, immunopathology should be one of the primary drivers for conducting additional immunotoxicology assessments, but functional assays could be conducted in the absence of an immunopathology signal if there was a specific cause for concern based on, for example, the mechanism of action of the drug.
Similar methods, in general, can be used for evaluating DIT in rats and NHP. Some key considerations for inclusion of endpoints on DIT studies are: (i) that some functional endpoints have sub-optimal activity in the developing immune system; (ii) sample sizes and volumes may impact the age at which the endpoints can optimally be evaluated; and (iii) lack of historical data in developing animals. Measuring multiple immunotoxicity parameters can be important in interpreting the biological or clinical significance of observed changes (Holsapple et al., Citation2005; Dietert and Holsapple, Citation2007; Dietert and Burns-Naas, Citation2008). The most common endpoints used to evaluate the effects of drugs on the developing immune system are discussed below. Other immune endpoints such as cytokine measurements are less well characterized (Bussiere, Citation2008a; Dietert and Burns-Naas, Citation2008). The choice of DIT endpoints to be evaluated should be based on a scientific rationale.
Functional evaluations may often be combined with immunopathology evaluations in the assessments of DIT. For example, the TDAR can be performed in the NHP beginning between 2–4 weeks-of-age. However, the primary limitation of the TDAR in rodents relates directly to the post-natal maturation of immunocompetence (e.g., humoral immunity) in that species, and a robust TDAR cannot be measured in young rodents (Kimura et al., Citation1985; Ladics et al., Citation2000). Sub-optimal responses can be obtained on post-natal day (PND) 20–30, but responses of sufficient magnitude are only obtained at 40–56 days-of-age, suggesting that for sufficient sensitivity the TDAR should only be performed in adult rodents. While accepted and validated methods are desirable and preferred, there may be times when less well-validated methods or methods with unknown or limited sensitivity in younger systems may be necessary. These factors need to be considered in the overall data interpretation.
Immunopathology
The FDA guidance on immunotoxicology investigation of investigational new drugs recommends evaluating pathology endpoints in the F1 generation offspring when immunosuppression is seen in adult animals (FDA, Citation2002). Standard pathology endpoints (e.g., lymphoid organ weights, white blood cell counts and differentials, histology of lymphoid tissues) are believed to be sensitive enough to detect overt immunotoxicity in adult animals (FDA, Citation2002). Evidence of toxicity to the immune system would likely be evaluated with respect to the criteria outlined by Germolec (Citation2009), which include demonstration that an effect on one or more immune parameters is dose-related when the magnitude of the effect and the dose-response are also considered, and is not secondary to systemic toxic effects. However, the predictivity of pathology in DIT is far less established (Dietert and Burns-Naas, Citation2008). With this caution in mind, pathology endpoints can be used as an initial screen to assess DIT. Immunologically important organs for evaluation include the spleen, thymus, and draining and distant lymph nodes if available. Histopathologic evaluation of lymphoid tissues can easily be incorporated into DART studies, and can be sufficient to detect immune system alterations (Bussiere, Citation2008a). In rats, histopathologic evaluation of lymphoid tissues can be performed as early as PND 21 (CitationDietert and Holsapple, 2007). However, there is little potential for histopathologic detection of drug-related alterations in the developing immune system of laboratory rats prior to gestation day (GD) 15 (Burns-Naas et al., Citation2008). Substantial toxicity (e.g., failure of formation of immune organs) could be detected by GD20, but less significant effects could be missed. Finally, immunopathology may detect structural alterations associated with immunosuppression; however, histopathology is unlikely to detect misdirected or enhanced immune responses, a limitation not restricted to just the developing immune system.
Clinical pathology endpoints of particular interest to DIT include evaluation of total white blood cell counts and differential in the routine hematology evaluations. Determination of serum immunoglobulin levels may also be included, with the knowledge that, given the considerable variation between individual animals, only large changes may be detectable and small-to-moderate changes may be difficult to interpret. In rats, however, evaluation of these parameters may be limited by inadequate sample volume. Notably, when sample size is limited, evaluation of hematology parameters should be given priority (Burns-Naas et al., Citation2008).
Characterization of lymphocyte sub-populations can be determined by flow cytometric and immunohistochemistry (IHC) approaches. When changes in immune function are seen (e.g., alterations in TDAR), immunophenotyping can be used to investigate potential alterations in specific cell populations. Immunophenotyping of peripheral blood lymphocytes is also easily incorporated into clinical trials to monitor potential concerns. While it can provide supportive data and help elucidate potential mechanisms of toxicity, immunophenotyping alone is unlikely to be sufficient as an effective DIT screen in either the rat or NHP (CitationDietert and Holsapple, 2007). In addition to immunophenotyping of peripheral blood, flow cytometry and immunohistochemistry can be valuable in evaluating specific lymphocyte sub-populations in tissues following exposure to presumptive immunomodulatory agents (Gillett and Chan, Citation2000). In combination with flow cytometric analysis of peripheral lymphocyte sub-populations, evaluation of lymphocyte sub-populations in tissues can help determine if alterations detected in the peripheral blood are a result of trafficking or reflective of changes in tissue distribution. The need to collect tissue limits these evaluations, for all practical purposes, to the necropsy intervals.
Immune function tests
While some functional immunotoxicity assays have been developed and validated for use in rodents and NHP, few are currently accepted as sensitive and predictive in developing animals for a variety of reasons. For the most robust assessment of DIT, combining immunopathology with an examination of humoral immunity (e.g., TDAR) and an evaluation of cellular immunity such as the delayed-type hypersensitivity response (DTH), cytotoxic T-lymphocyte (CTL) response, or natural killer (NK) cell assay would appear to provide the broadest assessment of immunocompetence (Holsapple et al., Citation2004; Dietert and Holsapple, Citation2007; Burns-Naas et al., Citation2008, Dietert and Burns-Naas, Citation2008). However, it is not recommended that functional endpoints always be included in assessments of DIT. The determination as to whether to include functional tests should be made on a case-by-case basis following a WoE review. The relevant drivers to make the decision as to whether to include functional assessments may include previously observed immunologic changes in standard toxicity studies. The immune parameters included in STS studies are outlined in the ICH S8 guidance (ICH, Citation2006). Possible situations that may prompt functional immune assessments include: (i) hematological changes such as lymphopenia/lymphocytosis; (ii) alterations in immune organ (e.g., spleen, thymus, and lymph node) weights and/or histology; (iii) changes in serum immunoglobulins; or (iv) increased incidence of infections or tumors that may be due to immunosuppression. Additional drivers may also include changes in relevant cell phenotypes in developing animals, or may be based on observations from previous adult studies.
T-dependent antibody response (TDAR)
The TDAR assay is one of the most commonly used functional tests to evaluate the immunosuppressive potential of drug candidates in adult animals (FDA, Citation2002; ICH, Citation2006). This assay evaluates the participation of antigen-presenting cells, T-lymphocytes, and B-lymphocytes. Alterations in antigen-specific antibody production may result from effects on any of these cell populations, thus the TDAR remains the most holistic evaluation of immune functionality, although additional follow-up investigations may be needed to determine the affected cell type(s).
The TDAR assay may be valuable in evaluating the effects of drug candidates on the developing immune system (Dietert and Burns-Naas, Citation2008). In the rat, antibody responses to sheep red blood cells (SRBC) are sub-optimal at PND 21 (weaning). At this time point lymphoid tissues in rats are anatomically intact but relatively inactive. Thus, evaluation of the TDAR is more commonly included at a later timepoint (Kimura et al., Citation1985; Ladics et al., Citation2000) such as PND 42–45 when the TDAR is comparable to adult rats. Although SRBC are recommended for use in the TDAR when testing the immunotoxicity of environmental chemicals, keyhole limpet hemocyanin (KLH) is more frequently used when testing the immunotoxicity of pharmaceuticals.
TDAR assays can also be part of DIT assessments in NHP (Rasmussen et al., Citation2007) and can be performed as early as 2–4 weeks-of-age in this species. If the post-partum period is extended, the assay can be repeated if necessary with the same antigen to assess a memory response, or with a different antigen to assess recovery from test article-related effects. Consistent with immunization of human infants shortly after birth with a variety of antigens, non-clinical studies with immunomodulatory drugs may incorporate multiple antigenic challenges, either at different ages or with different antigens (e.g., KLH, tetanus toxoid [TT], or hepatitis B surface antigen [HBsAg]).
Several questions remain on the use of the TDAR assay in DIT testing. While not unique to DIT testing, concerns can be raised on which antigen(s) to use, antigen dose, the isotype to measure (IgM, IgG, or both), and the kinetics of the antibody response. While there is consensus that DIT endpoints should be incorporated into existing DART studies whenever possible, there is some debate as to whether a TDAR assay should be performed using the main study animals or performed with additional animals in satellite groups to the main study. Whether immunizing animals on general toxicology studies may affect the interpretation of standard toxicity endpoints has been previously discussed (Piccotti, Citation2008). Previous work (Ladics et al., Citation1995, Citation1998) has demonstrated that immunization of rats with SRBC does not affect hematology or clinical pathology parameters, lymphocyte subset numbers, nor lymphoid organs weight or histology, with the exception of anticipated effects on germinal centers of the spleen. However, less is published regarding the potential effects of KLH immunization on the interpretation of standard toxicity endpoints. There is evidence in NHP that KLH immunization does not alter these endpoints when evaluated in control animals (Piccotti et al., Citation2005). Until more information emerges regarding the impact of KLH immunization on standard toxicity endpoints, immunizing rats in DART studies should be considered on a case-by-case basis. It is possible to include a separate cohort(s) of rats for immunization on rat pre- and post-natal development (PPND) studies or to conduct a stand-alone DIT study in rats in parallel with or subsequent to DART studies. In contrast, a separate cohort or stand-alone DIT study in NHP is not practical, and in such cases the incorporation of antigen immunization into the main study group animals is justified. Thus, careful design of DIT assessments within DART studies in NHP is critical.
Cell-mediated immunity (CMI)
The DTH assay has been a part of the National Toxicology Program’s Tier 2 test panel for cellular immune responses (Luster et al., Citation1988), and can be performed using whole cells (xenogeneic erythrocytes), KLH, or bovine serum albumin (BSA) as antigens. The use of adjuvants, frequently required in DTH protocols, is concerning however (Holsapple, Citation2002). Another concern is the potential for some antigens to also produce antibody responses to the same antigens (e.g., KLH) that may contribute to the measured cellular immunity response, as part of a Type III hypersensitivity response.
Distinguishing the humoral (antibody) from the cellular response is critical, particularly in juvenile rodents where a CMI assay for PND 21 is highly desirable. It has been suggested, however, that when a protocol is truly optimized for a maximal cell-mediated response to an antigen such as KLH, no measureable antibody response to that antigen is observed, and vice-versa (Bretcher, Citation1994; Burleson et al., Citation2009). Thus, separate groups of animals should be considered when using KLH as an immunogen to evaluate both humoral and cellular immune responses, with each group treated following a differentially optimized protocol. There is significant scientific disagreement on the utility of the DTH in NHP, primarily because it has been very difficult to reliably reproduce or modulate in NHP. Although the DTH assay is yet to be considered adequately ‘validated’ for use in regulatory DIT assessment, and there is no general consensus on the best methodology, the DTH assay may have utility and could be used when appropriate or necessary.
As a result of the issues raised over the use of adjuvants and the potential for humoral responses to contribute to a DTH response, the CTL assay has also been proposed as a method for evaluation of cell-mediated immunity in DIT assessments (Dietert and Holsapple, Citation2007; Burns-Naas et al., Citation2008; Burns-Naas, Citation2011). The CTL assay has been widely used as an immunotoxicology screen in adult rodents, but its ability to be used in younger animals has generally not been explored until recently (Burleson et al., Citation2008) where it has shown promise. To date, however, no generalized CMI assay has been effectively evaluated in NHP that could be used as a DIT screen.
Natural killer (NK) cell assay
As a measure of innate immunity, the NK cell assay has been used effectively, along with the TDAR, in the assessment of DIT in rodents (Dietert and Holsapple, Citation2007). Additionally, it is also being investigated for use in immunologic assessment of NHP with limited success to date. NK cell activity can be measured in infant or juvenile monkeys, although NK cell activity is low in neonates of different species, and the ability to activate these cells with interleukin (IL)-2 is reduced (Hodge et al., Citation2001; Frings and Weinbauer, Citation2004). Taking this into account, along with the relatively high blood volume required, the NK cell assay is recommended in NHP at the age of 6-months and older. Like other assays of cellular immunity, the NK cell assay is not broadly considered ‘validated’ for application in regulatory DIT evaluations and its use should be driven by the needs of the project, the ongoing advances in the method development (particularly in the NHP), and its interpretation tempered by its limitations at the time.
Other evaluations
Serum immunoglobulinsrepresent an end-point that can be comparatively measured in NHP, rodents, and humans. Age-related changes in serum immunoglobulin levels have been evaluated in rhesus (Voormolen-Kalova et al., Citation1974; Hendrickx et al., Citation2005) and cynomolgus (Terao, Citation1981) monkeys, and patterns typical of those seen in humans are observed. However, there is considerable lack of sensitivity of this end-point by itself in predicting immunotoxicity associated with drug or non-drug chemical exposure. Only profound changes in serum immunoglobulins or significant shifts in isotypes would be considered potentially relevant findings for immunotoxicity assessment, whether in adults or in juveniles.
Cytokine measurementsin peripheral blood following drug administration can be performed as a means to assess immunomodulation or determining disease susceptibility and do represent a potentially translatable clinical biomarker. Currently though, cytokine measurements as screening biomarkers of DIT should not be used (Dietert and Holsapple, Citation2007; Burns-Naas et al., Citation2008) because of a lack of: (1) baseline data on normal levels of cytokines occurring within the immune system during the various windows of immune system development; (2) understanding of the time- and dose-dependent kinetics of cytokine levels to known developmental immunotoxicants in peripheral blood and immune organs; and (3) standardization of methods.
Macrophage functionhas been deemed to require further evaluation and validation before its utility in assessing DIT can be determined.
Host resistance assaysare not generally considered appropriate for a DIT screen (Holsapple, Citation2002) as they are considered to be a final tier of testing and are typically conducted only when data from a primary screen suggest alterations in immune parameters and affected cell types are known.
Study designs
Rat
Nearly all workshops to date recommend the inclusion of DIT endpoints onto existing DART studies where feasible, rather than conducting stand-alone DIT assessments. Schemes for DIT testing in rodents have been discussed extensively elsewhere (Holsapple, Citation2003; Holsapple et al., Citation2005; Burns-Naas et al., Citation2008; Dietert and Burns-Naas, Citation2008). This section will provide only a brief overview of some factors to consider in designing a DIT study in rats.
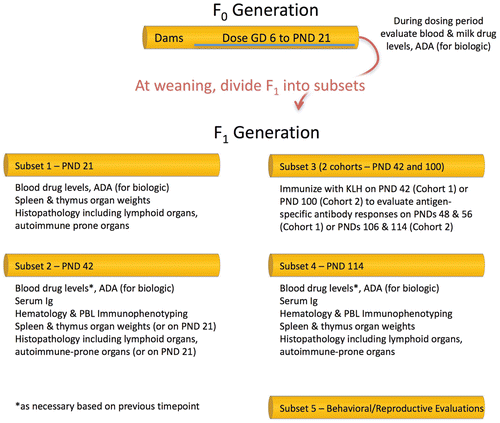
depicts a robust DIT evaluation (all possible evaluations) that could be included in a PPND study design in rodent. The detailed analyses represent options and are not advocated as ‘across the board’ evaluations for all studies. In this example, maternal dosing occurs from GD6 through PND 21. If transfer of the agent through breast milk is unknown, or not expected, direct dosing of pups can begin as early as PND 4, depending on the route of administration. Regardless of the experimental design, weanlings may be evaluated on PND 21 for immunopathology endpoints, as well as drug levels and ADA (for biotherapeutics). Functional immunotoxicity assays could also be performed on PND 21 (with the caveats discussed above). Direct dosing of pups can be continued from PND 21 through PND 42–56 when multiple structural (immunopathology) and functional endpoints can be evaluated. There is consensus that recovery should be included for all DIT evaluations wherever possible, and it would be desirable to include the same evaluations in recovery as performed during the dosing phase when exposure and pharmacodynamics are expected to have dissipated. It is important to note that if DIT assessments are not performed during the exposure period, and only performed at the end of the recovery period, transient effects on the developing immune system may be missed.
Figure 4. Possible study design for PPND study in rats incorporating DIT endpoints. The choice and timing of DIT to be included on a PPND study should be based on scientific rationale. Endpoints should include hematology (to evaluate changes in peripheral mononuclear cells), histopathology of lymphoid organs, and immunotoxicology endpoints that were affected in adult animals. The timing of evaluations may depend on the dosing interval. GD = Gestation Day; PND = Post-natal day; ADA = anti-drug antibodies (immunogenicity); Ig = Immunoglobulin; PBL = Peripheral Blood Lymphocyte.
represents a robust evaluation of DIT in the context of a juvenile toxicology study in rats. The F1 generation is continuously exposed to drug by direct dosing following weaning, and during lactation if sufficient secretion in the milk is unknown or does not occur. Many of the same principles discussed in relation to the inclusion and timing of DIT evaluations in PPND studies apply to juvenile studies.
Figure 5. Possible study design for a juvenile toxicology study in rats incorporating DIT endpoints.
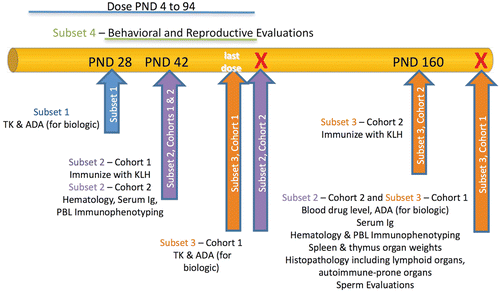
NHP
Evaluations of DIT in NHP can be conducted as part of embryo-fetal development (EFD), PPND, or juvenile studies. With the desire to use fewer animals and gain greater information to inform clinical risk, modifications to more standard study designs have begun to be adopted which expand the scope of the EFD and PPND studies, i.e., enhanced (e)EFD or ePPND studies. These general schemes, and the potential of incorporation of DIT endpoints, are considered in detail in Burns-Naas (Citation2011). In the standard EFD studies, dosing typically occurs from the confirmation of pregnancy (around GD20) through the remainder of the first trimester (around GD50) to cover the major period of organogenesis (Hendrickx et al., Citation2005). The only end-point that can be evaluated at this time is immunopathology of the lymphoid tissues. Since the fetal immune system continues to develop until later in gestation (Buse, Citation2005), dosing may be extended into the second or third trimester for test articles, with known effects on the immune system. For Fc-bb dosing is best extended to GD90 or GD100 (end of second trimester) or even later (late third trimester) to ensure fetal exposure (Fujimoto et al., Citation1983; Coe et al., Citation1993; Leach et al., Citation1996). Extending dosing allows increased fetal exposure due to the increased expression of the FcRn receptor during this time (Henck et al., Citation1996; Weinbauer et al., Citation2008). As with the standard EFD study, immunopathology of lymphoid tissues is the DIT end-point evaluated at the termination of the study. While it is always desirable to generate exposure data, and toxicokinetic (TK) evaluations are recommended wherever possible, there are frequently times where obtaining samples may not be feasible. For example, in young animals, it may not be possible to obtain sufficient blood volume from serial collections in order to perform TK analysis, particularly if there are other endpoints requiring the collection of peripheral blood. In other instances, the analytical assay may either not be available or is not sufficiently sensitive.
Since EFD studies allow only limited assessment of the developing immune system of NHP, DIT testing is most commonly incorporated into PPND studies, including the ePPND design (Chellman et al., Citation2009; Stewart, Citation2009). Dosing in PPND studies may be limited to that portion of gestation after the range covered by any previous EFD study. More frequently, however, dosing for PPND studies in NHP will cover the entire gestation period beginning at ~ GD20 to the time of delivery, ensuring exposure during all developmental phases (Holsapple et al., Citation2005; Burns-Naas et al., Citation2008). Extending dosing post-partum is discussed below.
The PPND or ePPND designs afford the most options for adding DIT endpoints, and allow tracking over a longer developmental period than other designs. EFD-PPND combination studies are also possible (discussed below). In both dosing schemes multiple DIT endpoints can be evaluated in the infant, including immune function tests, lymphoid histopathology, and/or immunohistochemistry, immunophenotyping of peripheral blood, and clinical pathology. Infants can be retained for periods of 1 month to 1 year, depending on the needs of the program, allowing the opportunity for serial DIT assessments in the same animals within a study. All of these designs are long-duration studies; the gestation period is typically 160 [± 10] days in cynomolgus monkeys, the NHP species most commonly used for DART testing. For biopharmaceuticals, it is common to use two to three experimental groups (control + 1 or 2 dose groups), compared to the conventional four groups for standard toxicology (control + 3 dose groups) (Chapman et al., Citation2009). Multiple DIT endpoints can be evaluated in the mothers and/or offspring of a PPND study that offers an advantage over EFD designs.
Dosing mothers post-partum could be considered as a means to extend infant exposure via continued secretion of the test article into milk, but it is not generally necessary to dose NHP post-natally in PPND studies to mimic human neonatal/fetal exposure. If the test article is an Fc-bb, placental transfer from mother to fetus in NHP will be similar to humans. There is risk to continuing dosing post-partum since the procedures involved can irreversibly disrupt mother–infant bonding, especially during the first 2 weeks post-natal. Also, data currently available for some Fc-bb administered to cynomolgus monkeys indicate that the concentrations in milk are very low compared to maternal serum (Martin et al., Citation2007; Auyeung-Kim et al., Citation2009); therefore, the additional exposure gained in infants by post-partum dosing of mothers is relatively small. IgG antibodies are transferred from mother to offspring, primarily across the placenta rather than in the milk; the predominant immunoglobulin secreted in milk is IgA (Cole and Bowen, Citation1976; Malek et al., Citation1994; van de Perre, Citation2003; Martin et al., Citation2007).
A combined EFD/PPND study can also be conducted. In this design, half of the pregnant females are Caesarian-sectioned during gestation, whereas the other half is allowed to deliver. This enables DIT to be tracked across the continuum of maternal/fetal/infant stages of development. DIT testing in NHP can also be conducted as part of a juvenile toxicity study (Chellman et al., Citation2009). For juvenile studies, dosing can span whatever range is determined appropriate to address DIT, while simultaneously accomplishing the overall objectives of the study.
Consideration of the gaps in the science and recommendations for future interactions and research
Despite progress in recent years, major gaps in our current understanding of the developing immune system and in the state of the science of DIT still exist, and affect our ability to (1) select the most relevant species for non-clinical DIT testing, (2) develop appropriate dosing and exposure regimens, and (3) conduct the most appropriate assays for evaluation of immune endpoints. Some areas where there is a need for more research are indicated in . Evaluating possible effects on the developing immune system therefore introduces significant challenges. In the following section, gaps in the science and methods for DIT testing, the impact of those gaps, and possible pathways to resolution are discussed.
Table 1. Areas for Continued Research and Pathways to Resolution.
Incomplete knowledge of comparative immune system development across species
Incomplete knowledge of comparative immune system development across species is the biggest data gap impacting DIT testing and data interpretation. Historically, immunotoxicity testing has been conducted primarily in rats (Holsapple et al., Citation2005), but with the rise of species-specific biopharmaceuticals, testing in NHP is increasingly needed. Testing in rodents with homologous proteins or in transgenic mice expressing the human target is also used in some cases. For SME, dogs may also be used for juvenile immunotoxicology testing (Smith et al., Citation2002). Understanding species differences in immune system development and how DIT findings in non-clinical species relate to developing humans is critical for study design and data interpretation.
Although significant progress has been made to characterize immune system development across species (reviewed in Holsapple et al., Citation2003; Dietert and Piepenbrink, Citation2006; Burns-Naas et al., Citation2008), many gaps in understanding remain. Further evaluations of age-related changes in immune system structure, phenotype, and function must be conducted both within a species and across species. There is a clear need to compile current data, particularly from NHP and rat, which is dispersed throughout the literature, into one comprehensive source. This information should not only include structural and functional data, but also historical data on baseline values for a number of immunologically relevant phenotypic parameters (i.e., cytokines, immunoglobulins, lymphocyte subsets, etc.).
Incomplete knowledge of placental transfer of SME and biopharmaceuticals
A central question when designing DIT studies is: ‘Will exposure occur during the developmental period under evaluation?’ With respect to evaluation of the fetus/neonate following maternal dosing, understanding placental transfer of the investigative compound is required. However, placental transfer of SME and biopharmaceuticals is incompletely understood. While the following discussion primarily focuses on biopharmaceuticals, placental transfer of SME and subsequent fetal exposure are also important considerations for DIT studies. In the context of DIT studies, fetal exposure to SME is not routinely measured. However, this is something that should be given serious consideration when designing such studies. It should not be assumed that all SME are equivalent in their ability to cross the placenta, and fetal exposure cannot necessarily be determined from maternal dosing and exposure levels.
For Fc-bb, the relative timing and efficiency of FcRn-mediated placental transport for each immunoglobulin isotype commonly used for Fc-bb across all non-clinical species needs to be fully evaluated. Whether placental transfer of Fc-modified Fc-bb molecules or Fc-devoid biopharmaceuticals can be mediated by binding to FcRn, by receptors other than FcRn, or by limited passive diffusion, is unknown. Some studies exist in the literature, demonstrating limited placental transfer of Fc-devoid biopharmaceuticals such as those that are fragment antigen binding (Fab)-based (see Miller et al. (Citation2003) for the example of ReoPro [abciximab; ~ 15 kDa]). In the case of recombinant human cytokines, neither recombinant human stem cell factor nor granulocyte-colony stimulating factor (G-CSF) readily crossed the placenta of rhesus monkeys (Tarantal and Cowan, Citation1999). However, in rats placental transfer of G-CSF was shown to occur, albeit at very low levels, but this small amount was sufficient to evoke a pharmacologic effect in the fetus and neonate (Medlock et al., Citation1993). It has been speculated that placental transfer of small amounts of cytokines may be mediated by binding to the respective cytokine receptors when expressed by the placenta (Tarantal and Cowan, Citation1999). It has also been suggested that the peptide, bovine insulin, can cross the placenta by piggybacking on anti-insulin antibodies (Bauman and Yalow, Citation1981); however, this theory has not been universally accepted and has not been demonstrated for other protein or peptide pharmaceuticals.
Lack of robust understanding of when the human immune system reaches maturity
It is understood that the rodent immune system matures predominantly post-natally, in contrast to that of the human that matures predominantly prenatally. However, the age at which the immune system reaches ‘adulthood’ is not easily defined. Although functional at birth, maturation of the human immune system continues post-natally due to the naïveté of the neonatal immune system. Different immune system components and functions mature at different rates (Holsapple et al., Citation2003; Burns-Naas et al., Citation2008; Collinge et al., Citation2010). However, it is unclear which parameter is the last to attain adult levels and as such at what age the immune system should be considered fully mature.
An important trigger for conducting juvenile toxicology studies is whether a potentially sensitive organ system is considered fully functionally mature in the targeted pediatric population; functional maturity at the time of dose-initiation may obviate the need for testing (FDA, Citation2006). Current regulatory guidances (FDA, Citation2006; EMA, Citation2008), based on data from Miyawaki et al. (Citation1981), assert that the human immune system continues to mature until 5 and 12 years-of-age, when adult levels of IgG and IgA, respectively, are achieved. However, it is important to recognize that post-natal immune system development mainly occurs in the very early years after birth, and only to a lesser extent in older children. Based on these guidances, for pediatric indications in children > 12 years-of-age, DIT testing may not be warranted. Moreover, if a drug is not expected to have any impact on an immune parameter that is fully mature before the age of 12, then the question could be raised as to whether DIT testing is warranted for use in pediatrics older than the age at which the parameter reaches maturity.
Impact on age for dose-initiation and duration of dosing/exposure
The relative immune system age across species should be considered in order to determine when dosing should be initiated and terminated. The development of the immune system of the NHP is largely similar to that of humans as a proportion of the total gestation period, although development is temporally compressed, and NHP are therefore considered useful models for DIT testing. However, while in humans and NHP the majority of immune functions appear before the end of the first trimester, some rodent immune functions do not appear until, on, or after birth (Daubener et al., Citation1985). Thus, to evaluate possible immunotoxicology effects on the human fetus following exposure to a drug, initiating dosing of neonatal rats instead of initiating dosing of dams may be sufficient. Similarly, the duration of treatment should include, at a minimum, the significant periods of relevant post-natal development for the selected species (FDA, 2006).
For DIT studies, continued drug exposure until immune system adulthood, addressing all developmental windows, is sufficient in the absence of an immunotoxicology finding. However, in the presence of an adverse finding with potential relevance to humans, a window of susceptibility may need to be defined to guide the age at which it is acceptable to expose humans. In this regard, the age at which the immune system of the test species surpasses particular critical windows of maturation relative to humans would be informative. Five critical windows of vulnerability in the development of the immune system have been described, and the timing of these critical windows compared in rodents and humans (Dietert et al., Citation2000), but it is unclear how the timing of these critical windows compares across other non-clinical species. In addition, seven events that are either completely unique to the developing immune system (i.e., they lack equivalents in the adult immune system) or play a unique role in the developing immune system have been described (Dietert and Piepenbrink, Citation2006), but it is not entirely transparent within which critical window(s) each of these seven events fall. Our lack of understanding challenges our ability to rationally design studies that will identify risks to specific stages of human immune system development. Regardless, data interpretation and translation to humans must be based on the relative maturity of the immune system, rather than age, with the recognition that different aspects of the immune system develop at different times.
Impact on interpreting differential findings across non-clinical species and relevance to humans
In some cases, DIT testing is conducted in two species or DIT signals are observed in one species but not another during DART studies. Reconciling differential observations across species and the relevance to humans is hampered by the current state of the science. To illustrate these difficulties, the effect of neonatal thymectomy and/or cyclosporine treatment in mouse, rat, and human will be discussed. Neonatal thymectomy in mice at PND 2–4 leads to multi-organ autoimmunity (Sakaguchi et al., Citation1982a, Citationb; Bonomo et al., Citation1995), whereas thymectomy after the first week-of-age does not. Similarly, treatment of newborn mice with cyclosporine leads to autoantibody production when given during the first week of life (Classen, Citation1998). These data indicate that the first week after birth is a critical window where alterations in thymocyte development lead to autoimmunity in mice. On the other hand, to the authors’ knowledge, thymectomy at birth in rats has not been reported to result in immune dysregulation. Although cyclosporine treatment of newborn rats starting on PND 4 did have effects on the immune system, autoantibodies were not produced (Barrow et al., Citation2006). Clearly, even species presumed to follow similar immune system development can have different responses to immunotoxicants. Removal of the human thymus at birth (due to cardiac transplantation) does not result in autoimmunity or abnormal immune function in children evaluated up to 16 years post-transplantation (Eysteindottir et al., Citation2004). Hence, the rat may be more predictive of the outcome of neonatal thymectomy in humans than the mouse, but why this is the case is unknown. Current understanding of the development of the rodent immune system is primarily based on data from mice (Holsapple et al., Citation2003); much less is known about the maturation of the rat immune system. It is important that the development of the immune system in rats be better characterized and not simply assumed to be ‘mouse-ish’. Interpreting contradictory and/or differential results across species will remain challenging until a further understanding of immune system development across species is obtained.
Translatability of DIT findings in non-clinical species to humans
It is fair to say that there is a very limited understanding of translatability of DIT findings in non-clinical species to humans. Previous workshops concluded that the rat is the most appropriate species for DIT testing based on practical as well as scientific considerations (Luster et al., Citation2003; Holsapple et al., Citation2005). However, validation of either rat or NHP in regards to translation of findings to humans is generally lacking. It has been proposed (Neubert et al., Citation2002) that immunotoxicity testing in non-clinical species be focused on tests that are directly applicable to human clinical testing to help with translation between species. Endpoints based on peripheral blood testing would thus be advocated. Clinical trials, however, generally exclude pregnant women and, thus, possible effects on the vast majority of human immune system development, which occurs in utero, would be missed. Notwithstanding, even if pregnant women were included in clinical trials, only the pregnant women and not the developing offspring could be tested. Instead, direct comparisons are limited to the effects of drugs on the developing immune systems of non-clinical species and pediatrics limiting comparative evaluation to human post-natal development.
Due to the limited possibilities to evaluate clinically the effects of drugs on the developing immune system in utero, sensitive in vitro assays that are translatable across species could be of great value. However, the utility of in vitro assays can only be validated by careful monitoring and reporting of any effects on the developing immune system observed clinically (e.g., publishing of case studies when clinical trial participants inadvertently become pregnant and choose to continue the pregnancy). As our understanding of comparative immune system development across species increases, immunotoxicity assays that are currently limited to use in non-clinical species will have increasing value, as translatability to humans may become better understood.
Which tests should be employed in DIT?
In vitro screening for DIT
No routine in vitro assays are currently available for DIT screening. Due to the complexity of the immune system, a battery of in vitro tests will have to be developed to evaluate DIT, and possiblyin vitro tests alone will never be sufficient. Only isolated examples of in vitro tests for DIT have been published (Dietert and Holsapple, Citation2007). If data from an in vivo non-clinical study suggests that a specific developmental pathway is targeted, then a validated in vitro test for that specific pathway and species would be useful. If in vivo findings were recapitulated in vitro, then a similar human in vitro assay may allow translation to humans. Assays using fetal thymus or immunologically-relevant cells isolated from the umbilical cord are some ideas that have been investigated (Neubert et al., Citation2002).
In vivo screening for DIT
One of the challenges forin vivo DIT assessments is the limited historical data and normal ranges for many immune parameters in developing animals. Until more data becomes available in this regard, interpretation of DIT data will be limited to comparison between dosed and control animals, since pre-test values are usually not available from developmental toxicology studies due to exposure initiating in utero or prior to feasible testing.
A species comparison of anatomical immune system development published in 2003 identified many data gaps, especially in regard to rats, dogs, and NHP (Holsapple et al., Citation2003). Since that time, the histology of the developing immune system of normal rats from GD15 to PND 22 has been extensively characterized (Burns-Naas et al., Citation2008), with intentions to follow this through until at least PND 42. Additionally, further evaluation of the fetal development of lymphoid organs in NHP (specifically the cynomolgus monkey) has been conducted (Buse, Citation2005; Buse et al., Citation2006). However, no new data on lymphoid organ development in dogs have been published.
Peripheral-blood lymphocyte phenotypes in humans and marmosets, in both newborns and adults, have been evaluated, and significant age-dependent changes and differences between marmosets and humans identified (Neubert et al., Citation2002). Age-dependent and species-related changes in serum immunoglobulins, anti-nuclear antibodies, cytokines, and complement is generally lacking, although IgG and IgM concentrations from ~ GD100 to 1 year-of-age in the cynomolgus monkey have been evaluated (Buse, Citation2005). The lack of age-dependent historical data for peripheral-blood evaluations, at least in rodents, is in part related to the difficulties in bleeding small animals and the paucity of blood available for evaluation. A study of peripheral blood-based, phenotypic, immunotoxicity endpoints in multiple species from soon after birth to adulthood would provide useful comparative data on immune system development and also context to differentiate normal age-related changes from possible drug-related effects.
Functional tests for DIT
Practical considerations, limited signal-to-noise, or uncontrollable confounding factors may preclude the use of functional immunotoxicity assays designed for adult animals in developing animals. Further age-related comparisons of functional assays optimized for use in adult animals are needed in all non-clinical species. If functional tests optimized in adult animals are not ideal for use in developing animals then the following questions need to be considered, and will be addressed in further detail below: (1) should further attempts to optimize these assays for use in developing animals be undertaken, (2) should tests specific to developing animals be developed, or (3) should immunotoxicology testing be limited to evaluation of irreversible effects, i.e., immunotoxicity only evaluated in adult animals following developmental exposures?
Should the impact of developmental exposure be limited to evaluation of outcomes in adult animals?
Most studies have not assessed until adulthood possible adverse effects on the immune system following developmental exposure, in part to address the potential for long-lasting, or permanent, changes to the immune system (Holsapple, Citation2003). The risk of limiting evaluations to adult animals following developmental exposure is that it is possible that transient effects could be missed. The significance of transient effects on the health of the animal or for human risk assessment is unclear. Studies to address whether immunotoxic effects on critical windows of immune development could be missed if data were only collected from adults has been identified as the most pressing need from workshops since 1995 (Holsapple, Citation2003).
From an experimental standpoint, limiting evaluations to adult animals is also in part due to the fact that the current battery of immunotoxicology tests were generally optimized and validated in adult animals when the immune system is mature. Historical data is also generally only available for adult animals. Thus, when it comes to DIT, it is important to consider the age of the animals used to experimentally validate the assay and to generate reference ranges. Ideally, experimental validations should include a comparison of various age groups to support DIT evaluations on developing animals.
Is it appropriate to use data from adult animals to drive the need for DIT testing?
Is it reasonable to use findings in a possibly less-sensitive population (the adult) to decide the need for DIT? At least seven occurrences that are either completely unique to the developing immune system (i.e., they lack equivalents in the adult immune system) or play a unique role in the developing immune system have been described (Dietert and Piepenbrink, Citation2006). While there is at least a theoretical concern that uniquely developmental immunotoxicants exist, to date, there are no published reports of any drug (or chemical) that is uniquely toxic to the developing immune system in animals or humans. However, drugs which present no cause for concern in standard toxicity testing in adult animals would unlikely be tested specifically for DIT. Thus, an absence of data should not be interpreted as an absence of risk. Data do exist on differential sensitivity to an immunotoxic drug (or chemical) depending on the stage of immune system development (Dietert et al., Citation2000; Luebke et al., Citation2006); however, the current data set is quite limited.
What is the risk of not identifying a unique developmental immunotoxicant?
Although increased sensitivity following developmental exposure to a potentially immunotoxic pharmaceutical is an established risk based on animal studies, there currently is very limited evidence of an increased risk of immunotoxicity following developmental exposures in humans. Potential developmental immune disruptions that may pre-dispose children to developing childhood diseases have been described (Dietert and Burns-Naas, Citation2008); however, links to specific drugs are lacking. Although the possibility for increased sensitivity of developing humans to immunotoxicants is not disputed, an industry-wide survey would be useful to determine: (1) The prevalence of increased sensitivity in animals following developmental exposures (compared to adult exposures); (2) If increased sensitivity was observed in the developing animals, how did the company proceed?; and (3) If increased sensitivity was observed in juvenile animal studies, was increased sensitivity observed in pediatric clinical studies? Until a larger database of information is available, it is prudent to give rigorous scientific consideration to the need for and design of DIT evaluations. However, as further human information is collected it makes sense to revisit this topic to ensure that the depth of research focus is in line with the actual identified risk.
Summary of the ILSI-HESI-ITC Workshop–2010
In May, 2010 a 2-day Workshop entitled ‘Developmental Immunotoxicity Testing of Pharmaceuticals’, organized by ILSI HESI ITC, was held in Washington DC. The Workshop (hereafter called the ITC-2010 Workshop) brought together representatives from the pharmaceutical, biotechnology, academic, and regulatory sectors (Food and Drug Administration [FDA] and European Medicines Agency [EMA]) to discuss the current state of the science of DIT as it pertains to the testing of pharmaceuticals. Much of the focus of the workshop was on biopharmaceuticals, primarily because much of the DIT performed in the pharmaceutical sector has been performed with biologic molecules, as the majority of intentional immunomodulatory drugs are also biologics. However, historically DIT evaluations with pharmaceuticals have focused on SME that have immune targets and, therefore, SME were also considered. A preliminary draft of this state of the science manuscript was circulated prior to the Workshop in order to obtain additional input from all attendees, and also as a basis for discussion. The Workshop provided an opportunity for Pharma to interact directly with representatives from both the FDA and EMA. Key points of discussion and consensus from the Workshop have been summarized in .
Table 2. Key Points of Consensus from the ITC 2010 DIT Workshop.
There was consensus that DIT should be performed on a case-by-case basis, and that there should be no routine DIT testing for all compounds. The inclusion of what (if any) DIT assessments to incorporate into pre-clinical studies should be based on sound scientific rationale, including specific cause for concern, such as findings from repeat dose studies (adult), the mode of action of the drug (target and pharmacology), the therapeutic indication (including whether the drug will be administered to women of childbearing potential [WOCBP]), and the target pediatric age. Consideration should also be given to how these non-clinical studies will impact the clinical development plan. There was also consensus that, while regulatory guidance documents are a good starting point for considering DIT assessments, they should not be the ultimate drivers for performing DIT. It was also agreed that, in the future, in the case of some immunomodulatory drugs, it might be possible that no juvenile studies would be needed if the effects of those drugs is expected and predictable. However, these decisions would be the result of discussions between both the Sponsor and the Regulatory Agencies.
The recommendation from this Workshop was that if DIT assessments are to be conducted then these should be incorporated into PPND studies wherever possible, and that a PPND study design that incorporates DIT endpoints should be discussed with the regulatory agencies prior to conducting the study. An approach where parallel advice is requested from both the FDA and the EMA is recommended, and it was indicated that regulatory agencies were better able to respond to questions the more focused they are on study design. Such discussions are best included into pivotal regulatory meetings that have already been planned (pre-IND or Phase II). There was consensus that DIT studies should be performed on a case-by-case basis, and that a reversibility assessment is expected in such studies. It was clear from discussion at the Workshop that companies have typically been pro-active in performing DIT, and that no company at the ITC-2010 Workshop had been specifically requested to perform a DIT study by the Regulatory Agencies.
There was consensus, consistent with previous workshops, that the rat remains the preferred species for DIT testing unless there is a specific reason, such as lack of pharmacological relevance, that this species cannot be used. The use of NHP is only advocated if there is no alternative. Thus, the use of rats in DIT studies with biopharmaceuticals should not be disregarded: the NHP should not be the default species for all biopharmaceuticals. There was also clear consensus that, irrespective of whether the drug under consideration is an SME or a biopharmaceutical, only species that are pharmacologically responsive should be used for DIT testing of intentional immunomodulators.
Participants at the Workshop acknowledged that incomplete knowledge of comparative immune system development across species is perhaps the biggest data gap impacting DIT testing and data interpretation. Indeed, one of the key challenges identified was how to interpret non-clinical DIT results to be predictive for humans and how to advise on clinical use.
The use of one species for DIT studies was considered in most circumstances to be adequate. If immunotoxicity is observed in two adult species, then it would only be necessary to perform DIT testing in one of those species. If immunotoxicity is observed in one adult species then that species should be used for DIT testing.
The lack of translational biomarkers from non-clinical DIT assessments that can be applied to the clinical situation was identified as a key gap at the Workshop. It was suggested that some effort should be invested into discovering biomarkers for human immunotoxicity testing that can be applied to non-clinical studies.
The ITC-2010 Workshop participants agreed that there is a need for a better understanding of the relative sensitivity to immunomodulatory drugs between adults and developing animals. None of the Workshop participants were aware of any instances of drugs that display unique sensitivity to the developing immune system. However, it was recognized that there was a lack of human data on DIT, and there was a clear need to engage clinicians regarding assessment and interpretation of DIT studies in the future.
With respect to development of methods specifically for DIT testing, Workshop participants identified the development of a robust CMI assay that could be applied to DIT assessments as a high priority. In addition, development of methods to assess autoimmunity was also viewed to be a high priority. Development of assays to measure immune enhancement have generally been neglected in favor of immune suppression. For example, studies may be needed to determine if pharmaceuticals intended to enhance the immune response have the capacity to induce autoimmune disease. However, currently validated assays for this purpose are limited in adults and, therefore, difficult to apply to assessments of DIT. Similarly, methods to assess hypersensitivity or drug allergy with systemic exposure have not been adequately validated for adult use. Once such methods are available they should be validated for use as DIT endpoints.
Consensus from the ITC-2010 Workshop was that alternative models could be useful under certain circumstances. It was concluded that in some cases a mouse homolog might be a more relevant model than NHP for biologics, but that science needs to drive the studies. However, when considering such an approach the following must be borne in mind: (i) co-development of a full homolog program is needed in parallel with the therapeutic program (cost); (ii) questions of how similar, with respect to pharmacologic and physiologic consequences, the homolog is to the human therapeutic need to be addressed; and (iii) if toxicities are observed, then follow-up studies in a second species may be required. In general it was agreed that transgenic animals expressing the human protein might be used if the physiologic consequences are the same as expected in humans. However, in each case it is important to understand the limitations of such models.
Conclusions
Global Regulatory Agencies do not routinely require assessment of DIT during the drug development process, nor has any company represented at the ITC-2010 Workshop been asked by an Agency to perform this testing. Rather, pharmaceutical companies have tended to be pro-active with respect to conducting DIT assessments, although the scope of these assessments varies between companies. DIT assessments are driven primarily by a cause-for-concern derived from a WoE review of the totality of the available data, immunotoxicity observed in adult animals, the intended patient population, and potential that the fetus, neonate, or juvenile might be exposed. To date, DIT evaluations in the pharmaceutical industry have focused on agents that target the immune system, often biopharmaceuticals. In general, the data in the offspring has recapitulated the data in the adults, although there may be an instance where a drug has demonstrated some sensitivity differences. Any decision to conduct an assessment of DIT should be based on sound science and should aid in the overall risk evaluation for the proposed new drug. An essential characteristic somewhat unique to the drug development paradigm is that flexibility to alter existing DART and juvenile testing paradigms to meet the needs for the test material in a scientifically justifiable manner is paramount to a value-added study. Studies should not be performed, particularly in non-rodents, simply to minimize regulatory risk for registration. DIT assessments should only be performed where there is scientific justification for improving the evaluation of safety risks of the drug to the pediatric patient population. DIT studies should not be performed simply to ensure against delays in drug approval due to the assumption of a hypothetical request by regulatory agencies to perform additional studies prior to the approval of clinical trials or registration.
It is recognized that while immunopathology and immunophenotyping have a role in DIT evaluations, in some cases immunotoxicity may be most appropriately addressed with a functional evaluation such as the TDAR. The TDAR can be used in NHP shortly after birth, but a near adult-level response cannot be achieved in rodents until they are nearly 6 weeks-of-age due to post-natal maturation of humoral immunity in this species. Additional work is needed to fully validate assays of innate (e.g., NK cell assay) and cellular immunity (e.g., DTH, CTL) in the rodent and in the NHP in general. Suppression of immune function has received the greatest attention with respect to the developing immune system as opposed to enhancement of immune function. Immunity, though, is a continuum, with suppression being at one end and stimulation (e.g., hypersensitivity or autoimmunity) at the other. Methods and models to improve our understanding of the potential impact of chemical exposures during immune system development on the ability to exacerbate hypersensitivity responses or influence the development of autoimmune disease remains a significant data gap.
Finally, as noted by others, research is still needed in the area of translational immunotoxicology (Holsapple et al., Citation2005; Burns-Naas et al., Citation2008). We need to be able to extrapolate results from DIT screening studies to potential health effects in children. Detailed data on expression of immune endpoints in juveniles and an understanding of whether an effect on an immune end-point from a DIT study in adult animals can be applied to set safe levels in juveniles is also needed. Although animal data support the contention for some environmental chemicals, it remains largely unknown whether juvenile humans are truly more sensitive to immune system perturbations by exogenous molecules than adults.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Notice of correction:
This paper published early online on 19 March 2012 contained an error in the affiliations which has subsequently been corrected in this version
References
- Adkins, B., Leclerc, C. and Marshall-Clarke, S. 2004. Neonatal adaptive immunity comes of age. Nat Rev Immunol. 4:553–564.
- Albrecht, P., Ennis, F. A., Saltzman, E. J. and Krugman, S. 1977. Persistence of maternal antibody in infants beyond 12 months: Mechanism of measles vaccine failure. J Pediatr. 91:715–718.
- Auyeung-Kim, D. J., Devalaraja, M. N., Migone, T. S., Cai, W. and Chellman, G. J. 2009. Developmental and peri-postnatal study in cynomolgus monkeys with belimumab, a monoclonal antibody directed against B-lymphocyte stimulator. Reprod Toxicol. 28:443–455.
- Barrow, P. C., Horand, F. and Ravel, G. 2006. Developmental immunotoxicity investigations in the SD rat following pre- and post-natal exposure to cyclosporin. Birth Defects Res B Dev Reprod Toxicol. 77:430–437.
- Bauman, W. A. and Yalow, R. S. 1981. Transplacental passage of insulin complexed to antibody. Proc Natl Acad Sci USA. 78:4588–4590.
- Bonomo, A., Kehn, P. J. and Shevach, E. M. 1995. Post-thymectomy autoimmunity: Abnormal T-cell homeostasis. Immunol Today. 16:61–67.
- Bretcher, P. A. 1994. Requirements and basis for efficacious vaccination by a low antigen dose regimen against intracellular pathogens uniquely susceptible to cell-mediated attack. In: Strategies in Vaccine Design. (Ada, G. L. Ed.).Austin, TX: RG Landes, pp.99–112.
- Burleson, G. R., Burleson, F. G. and Dietert, R. R. 2008. Juvenile immunotoxicity (DIT) testing in SD rats: Effect of dexamethasone on influenza viral clearance, NK activity, CTL activity, and T-dependent viral-specific IgM and IgG antibody production (TDAR). The Toxicologist. 102:12.
- Burleson, G. R., Burleson, F. G. and Dietert, R. R. 2009. The cytotoxic T lymphocyte (CTL) assay for evaluating cell-mediated immune function. In: Immunotoxicity Testing. Series: Methods in Molecular Biology. (Dietert, R. R, Ed.). Totowa, NJ: Humana Press, Inc., pp. 195–205.
- Burns-Naas, L. A. 2011. Developmental immunotoxicity testing. In: Reproductive and Developmental Toxicity Testing. A Practical Approach.. 3rd Edition (Hood, R. D., Ed.). London, UK: Informa pp. 491–508
- Burns-Naas, L. A., Hastings, K. L., Ladics, G. S., Makris, S. L., Parker, G. A. and Holsapple, M. P. 2008. What’s so special about the developing immune system? Int J Toxicol. 27:223–254.
- Burri, P. 1997. Structural aspects of prenatal and postnatal development and growth of the lung. In: Lung Growth and Development. McNoald, J. A., Ed.). New York: Marcel Dekker, Inc., pp. 1–35.
- Buse, E. 2005. Development of the immune system in the cynomolgus monkey: The appropriate model in human targeted toxicology? J Immunotoxicol. 2:211–216.
- Buse, E., Habermann, G. and Vogel, F. 2006. Thymus development in Macaca fascicularis (Cynomolgus monkey): An approach for toxicology and embryology. J Mol Histol. 37:161–170.
- Bussiere, J. L., 2008a. General toxicity testing and immunotoxicity testing for biopharmaceuticals. In: Preclinical Safety Evaluation of Biopharmaceuticals. A Science-Based Approach to Facilitating Clinical Trials. (Cavagnaro, J. A., Ed.). Hoboken, NJ: John Wiley and Sons, Inc., pp. 343–356.
- Bussiere, J. L. 2008b. Species selection considerations for preclinical toxicology studies for biotherapeutics. Expert Opin Drug Metab Toxicol. 4:871–877.
- Chao, D. T., Ma, X., Li, O., Park, H. and Law, D. 2009. Functional characterization of N297A, a murine surrogate for low-Fc binding anti-human CD3 antibodies. Immunol Invest. 38:76–92.
- Chapman, K., Pullen, N., Coney, L., Dempster, M., Andrews, L., Bajramovic, J., Baldrick, P., Buckley, L., Jacobs, A., Hale, G., Green, C., Ragan, I. and Robinson, V. 2009. Preclinical development of monoclonal antibodies: Considerations for the use of non-human primates. MAbs. 1:505–516.
- Chellman, G. J., Bussiere, J. L., Makori, N., Martin, P. L., Ooshima, Y. and Weinbauer, G. F. 2009. Developmental and reproductive toxicology studies in non-human primates. Birth Defects Res B Dev Reprod Toxicol. 86:446–462.
- Classen, J. B. 1998. Cyclosporine induced autoimmunity in newborns prevented by early immunization. Autoimmunity. 27:135–139.
- Coe, C. L., Kemnitz, J. W. and Schneider, M. L. 1993. Vulnerability of placental antibody transfer and fetal complement synthesis to disturbance of the pregnant monkey. J Med Primatol. 22:294–300.
- Cole, M. F. and Bowen, W. H. 1976. Immunoglobulins A, G, and M in serum and in some secretions of monkeys (Macaca fascicularis syn. irus). Infect Immun. 13:1354–1359.
- Collinge, M., Burns-Naas, L. A. and Kawabata, T. T. 2010. Developmental immunotoxicology. In: Reproductive Toxicology,.3rd Edition (Kapp, R. W. Jr. and Tyl, R.W., Eds.). London: Informa Healthcare, pp. 134–156.
- Däubener, W., von Steldern, D., Maxeiner, B., Scheurich, P. and Pfizenmaier, K. 1985. Postnatal development of functional T-cell subsets in the mouse: A frequency analysis of mitogen reactive precursors of proliferating, of cytotoxic and of IL 2 producing T cells. Immunobiology. 169:472–485.
- Day, M. J. 2007. Immune system development in the dog and cat. J Comp Pathol. 137 Suppl 1:S10–S15.
- de Haan, L., Henderson, S., McFarlane, M., Scott, A. and Shenton, J. 2011. Species selection. Eur Biopharm Rev.
- de Rijk, E. P. and van Esch, E.2008. The macaque placenta - a mini-review. Toxicol Pathol. 36:108S–118S
- DeSesso, J. M. 2005. Comparative features of vertebrate embryology. In: Developmental and Reproductive Toxicology. A Practical Approach. 2nd Edition (Good, R. D., Ed.). Boca Raton, FL: CRC Press, pp. 147–198.
- Dietert, R. R. 2008. Developmental immunotoxicity (DIT) in drug safety testing: Matching DIT testing to adverse outcomes and childhood disease risk. Curr Drug Saf. 3:216–226.
- Dietert, R. R. and Burns-Naas, L. A.. 2008. Developmental immunotoxicity in rodents. In: Immunotoxicology Strategies for Pharmaceutical Safety Assessment. (Herzyk, D. J. and Bussiere, J. L., Eds.). Hoboken, NJ: John Wiley and Sons, Inc., pp. 273–297.
- Dietert, R. R. and Holsapple, M. P. 2007. Methodologies for developmental immunotoxicity (DIT) testing. Methods. 41:123–131.
- Dietert, R. R. and Piepenbrink, M. S. 2006. Perinatal immunotoxicity: Why adult exposure assessment fails to predict risk. Environ Health Perspect. 114:477–483.
- Dietert, R. R., Etzel, R. A., Chen, D., Halonen, M., Holladay, S. D., Jarabek, A. M., Landreth, K., Peden, D. B., Pinkerton, K., Smialowicz, R. J. and Zoetis, T. 2000. Workshop to identify critical windows of exposure for children’s health: Immune and respiratory systems work group summary. Environ Health Perspect. 108 Suppl 3:483–490.
- Dietert, R. R., Lee, J. E. and Bunn, T. L. 2002. Developmental immunotoxicology: Emerging issues. Hum Exp Toxicol. 21:479–485.
- Dietert, R. R., Lee, J. E., Olsen, J., Fitch, K. and Marsh, J. A. 2003. Developmental immunotoxicity of dexamethasone: Comparison of fetal versus adult exposures. Toxicology. 194:163–176.
- Dybdal, N. 2010. Impact of monoclonal antibody administration to pregnant cynomolgus monkeys on early fetal health and development. In: 16th Annual Charles River Biotech Symposium - Biotechnology Derived Therapeutics, Perspectives on Non-clinical Development, 2010., San Diego, CA
- EMA. 2008. European Medicines Agency (EMA) Committee for Human Medicinal Products (CHMP). Guideline on the Need for Non-clinical Testing in Juvenile Animals of Pharma-ceuticals for Paediatric Indications.
- Eysteindottir, J. H., Freysdottir, J. and Al, E. 2004. The influence of partial or total thymectomy during open-heart surgery in infants on immune function later in life. Clin Exp Immunol. 136:349–355.
- FDA. 2002. FDA (United States Food and Drug Administration). 2002. Guidance for Industry-Immunotoxicology Evaluation of Investigational New Drugs. Washington DC: Office of Training and Communication. Division of Drug Information, Center for Drug Evaluation and Research, U.S. Department of Health and Human Services, 2002.
- FDA. 2006. Food and Drug Administration (FDA). Guidance for Industry - Non-clinical Safety Evaluation of Pediatric Drug Products. Washington DC: Office of Training and Communication, Division of Drug Information, Center for Drug Evaluation and Research, U.S. Department of Health and Human Services, 2006.
- Fenaux, J. B., Gogal, R. M. Jr and Ahmed, S. A. 2004. Diethylstilbestrol exposure during fetal development affects thymus: Studies in fourteen-month-old mice. J Reprod Immunol. 64:75–90.
- Frings, W. and Weinbauer, G. 2004. Flow cytometry-based evaluation of lymphocyte subsets and natural killer cell activity in developing and adult cynomolgus monkeys. The Toxicologist. 71:430.
- Fujimoto, K., Terao, K., Cho, F. and Honjo, S. 1983. The placental transfer of IgG in the cynomolgus monkey. Jpn J Med Sci Biol. 36:171–176.
- Germolec, D. 2009. Explanation of levels of evidence for immune system toxicity. Available online at: http://ntp.niehs.nih.gov/go/9399.
- Gillett, N. A. and Chan, C. 2000. Applications of immunohistochemistry in the evaluation of immunosuppressive agents. Hum Exp Toxicol. 19:251–254.
- Hastings, K. L. 2005. Prospects for developmental immunotoxicity guidance and an update on ICH S8. J Immunotoxicol. 2:217–220.
- Henck, J. W., Hilbish, K. G., Serabian, M. A., Cavagnaro, J. A., Hendrickx, A. G., Agnish, N. D., Kung, A. H. and Mordenti, J. 1996. Reproductive toxicity testing of therapeutic biotechnology agents. Teratology. 53:185–195.
- Hendrickx, A. G., Peterson, P. E. and Makori, N. M. 2005. The non-human primate as a model of developmental immunotoxicity. In: Developmental Immunotoxicology. (Holladay, S. D., Ed.). Boca Raton, FL: CRC Press, pp. 117–136.
- Hodge, S., Hodge, G., Flower, R. and Han, P. 2001. Cord blood leucocyte expression of functionally significant molecules involved in the regulation of cellular immunity. Scand J Immunol. 53:72–78.
- Holsapple, M. P. 2002. Developmental immunotoxicology and risk assessment: A workshop summary. Hum Exp Toxicol. 21:473–478.
- Holsapple, M. P. 2003. Developmental immunotoxicity testing: A review. Toxicology. 185:193–203.
- Holsapple, M. P., Burns-Naas, L. A., Hastings, K. L., Ladics, G. S., Lavin, A. L., Makris, S. L., Yang, Y. and Luster, M. I. 2005. A proposed testing framework for developmental immunotoxicology (DIT). Toxicol Sci. 83:18–24.
- Holsapple, M. P., Paustenbach, D. J., Charnley, G., West, L. J., Luster, M. I., Dietert, R. R. and Burns-Naas, L. A. 2004. Symposium summary: Children’s health risk – what’s so special about the developing immune system? Toxicol Appl Pharmacol. 199:61–70.
- Holsapple, M. P., West, L. J. and Landreth, K. S. 2003. Species comparison of anatomical and functional immune system development. Birth Defects Res B Dev Reprod Toxicol. 68:321–334.
- Holt, P. G., Upham, J. W. and Sly, P. D. 2005. Contemporaneous maturation of immunologic and respiratory functions during early childhood: Implications for development of asthma prevention strategies. J Allergy Clin Immunol. 116:16–24; quiz 25.
- Hussain, I., Piepenbrink, M. S., Fitch, K. J., Marsh, J. A. and Dietert, R. R. 2005. Developmental immunotoxicity of cyclosporin-A in rats: Age-associated differential effects. Toxicology. 206:273–284.
- ICH. 2006. International Conference on Harmonization (ICH) Harmonized Tripartite Guideline. ICH S8. Immunotoxicity studies for human pharmaceuticals. 2006. Available online at: http://www.ich.org/products/guidelines/safety/article/safety-guidelines.html.
- Kalland, T. 1980. Alterations of antibody response in female mice after neonatal exposure to diethylstilbestrol. J Immunol. 124:194–198.
- Kalland, T. and Forsberg, J. G. 1978. Delayed hypersensitivity response to oxazolone in neonatally estrogenized mice. Cancer Lett. 4:141–146.
- Kalland, T., Strand, O. and Forsberg, J. G. 1979. Long-term effects of neonatal estrogen treatment on mitogen responsiveness of mouse spleen lymphocytes. J Natl Cancer Inst. 63:413–421.
- Kimura, S., Eldridge, J. H., Michalek, S. M., Morisaki, I., Hamada, S. and McGhee, J. R. 1985. Immunoregulation in the rat: Ontogeny of B-cell responses to Types 1, 2, and T-dependent antigens. J Immunol. 134:2839–2846.
- Ladics, G. S., Chapin, R. E., Hastings, K. L., Holsapple, M. P., Makris, S. L., Sheets, L. P., Woolhiser, M. R. and Burns-Naas, L. A. 2005. Developmental toxicology evaluations–issues with including neurotoxicology and immunotoxicology assessments in reproductive toxicology studies. Toxicol Sci. 88:24–29.
- Ladics, G. S., Smith, C., Bunn, T. L., Dietert, R. R., Anderson, P. K., Wiescinski, C. N. and Holsapple, M. P. 2000. Characterization of an approach to developmental immunotoxicity assessments in rats using SRBC as the antigen. Toxicol Methods. 10:283–311.
- Ladics, G. S., Smith, C., Elliott, G. S., Slone, T. W. and Loveless, S. E. 1998. Further evaluation of the incorporation of an immunotoxicological functional assay for assessing humoral immunity for hazard identification purposes in rats in a standard toxicology study. Toxicology. 126:137–152.
- Ladics, G. S., Smith, C., Heaps, K., Elliott, G. S., Slone, T. W. and Loveless, S. E. 1995. Possible incorporation of an immunotoxicological functional assay for assessing humoral immunity for hazard identification purposes in rats on standard toxicology study. Toxicology. 96:225–238.
- Leach, J. L., Sedmak, D. D., Osborne, J. M., Rahill, B., Lairmore, M. D. and Anderson, C. L.1996. Isolation from human placenta of the IgG transporter, FcRn, and localization to the syncytiotrophoblast: Implications for maternal-fetal antibody transport. J Immunol. 157:3317–3322.
- Luebke, R. W., Chen, D. H., Dietert, R., Yang, Y., King, M. and Luster, M. I.; Immunotoxicology Workgroup. 2006. The comparative immunotoxicity of five selected compounds following developmental or adult exposure. J Toxicol Environ Health B Crit Rev. 9:1–26.
- Luster, M. I., Dean, J. H. and Germolec, D. R. 2003. Consensus workshop on methods to evaluate developmental immunotoxicity. Environ Health Perspect. 111:579–583.
- Luster, M. I., Munson, A. E., Thomas, P. T., Holsapple, M. P., Fenters, J. D., White, K. L.Jr, Lauer, L. D., Germolec, D. R., Rosenthal, G. J. and Dean, J. H. 1988. Development of a testing battery to assess chemical-induced immunotoxicity: National Toxicology Program’s guidelines for immunotoxicity evaluation in mice. Fundam Appl Toxicol. 10:2–19.
- Malek, A., Sager, R., Kuhn, P., Nicolaides, K. H. and Schneider, H. 1996. Evolution of maternofetal transport of immunoglobulins during human pregnancy. Am J Reprod Immunol. 36:248–255.
- Malek, A., Sager, R. and Schneider, H. 1994. Maternal-fetal transport of immunoglobulin G and its subclasses during the third trimester of human pregnancy. Am J Reprod Immunol. 32:8–14.
- Martin, P. L. and Weinbauer, G. F. 2010. Developmental toxicity testing of biopharmaceuticals in nonhuman primates: Previous experience and future directions. Int J Toxicol. 29:552–568.
- Martin, P. L., Oneda, S. and Treacy, G. 2007. Effects of an anti-TNF-alpha monoclonal antibody, administered throughout pregnancy and lactation, on the development of the macaque immune system. Am J Reprod Immunol. 58:138–149.
- Medlock, E. S., Kaplan, D. L., Cecchini, M., Ulich, T. R., del Castillo, J. and Andresen, J. 1993. Granulocyte colony-stimulating factor crosses the placenta and stimulates fetal rat granulopoiesis. Blood. 81:916–922.
- Miller, T. E., Golemboski, K. A., Ha, R. S., Bunn, T., Sanders, F. S. and Dietert, R. R. 1998. Developmental exposure to lead causes persistent immunotoxicity in Fischer 344 rats. Toxicol Sci. 42:129–135.
- Miller, R. K., Mace, K., Polliotti, B., DeRita, R., Hall, W. and Treacy, G. 2003. Marginal transfer of ReoPro (Abciximab) compared with immunoglobulin G (F105), inulin and water in the perfused human placenta in vitro. Placenta. 24:727–738.
- Miyawaki, T., Moriya, N., Nagaoki, T. and Taniguchi, N. 1981. Maturation of B-cell differentiation ability and T-cell regulatory function in infancy and childhood. Immunol Rev. 57:61–87.
- Neubert, R. T., Webb, J. R. and Neubert, D. 2002. Feasibility of human trials to assess developmental immunotoxicity, and some comparison with data on New World monkeys. Hum Exp Toxicol. 21:543–567.
- OECD. 2010. Organization of Economic Cooperation and Development (OECD). OECD Guideline for the Testing of Chemicals, Extended One-Generation Reproductive Toxicity Study (DRAFT). November 17, 2010. Available online at: http://www.oecd.org/dataoecd/23/10/46466062.pdf.
- Pentsuk, N. and van der Laan, J. W. 2009. An interspecies comparison of placental antibody transfer: New insights into developmental toxicity testing of monoclonal antibodies. Birth Defects Res B Dev Reprod Toxicol. 86:328–344.
- Piccotti, J. R.. 2008. T-Cell-dependent antibody response tests. In: Immunotoxicology Strategies for Pharmaceutical Safety Assessment. (Herzykm, D. J. and Bussiere, J. L., Eds.). Hoboken, NJ: John Wiley and Sons Inc., pp. 67–76.
- Piccotti, J. R., Alvey, J. D., Reindel, J. F. and Guzman, R. E. 2005. T-cell-dependent antibody response: Assay development in cynomolgus monkeys. J Immunotoxicol. 2:191–196.
- Pruett, S. B. 2003. Stress and the immune system. Pathophysiology. 9:133–153.
- Rasmussen, A. D., Nelson, J. K., Chellman, G. J., Golub, M. and McAnulty, P. A. 2007. Use of barusiban in a novel study design for evaluation of tocolytic agents in pregnant and neonatal monkeys, including behavioural and immunological endpoints. Reprod Toxicol. 23:471–479.
- Regulation-(EC)-1901/2006. 2006. European Parliament. 2006. Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006 on medicinal products for pediatric use and amending Regulation (EEC) No 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC) No 726/2004.
- Roopenian, D. C. and Akilesh, S. 2007. FcRn: The neonatal Fc receptor comes of age. Nat Rev Immunol. 7:715–725.
- Sakaguchi, S., Takahashi, T. and Nishizuka, Y. 1982. Study on cellular events in post-thymectomy autoimmune oophoritis in mice. II. Requirement of Lyt-1 cells in normal female mice for the prevention of oophoritis. J Exp Med. 156:1577–1586.
- Sakaguchi, S., Takahashi, T. and Nishizuka, Y. 1982. Study on cellular events in postthymectomy autoimmune oophoritis in mice. I. Requirement of Lyt-1 effector cells for oocytes damage after adoptive transfer. J Exp Med. 156:1565–1576.
- Schlumpf, M., Lichtensteiger, W. and van Loveren, H. 1994. Impaired host resistance to Trichinella spiralis as a consequence of prenatal treatment of rats with diazepam. Toxicology. 94:223–230.
- Schlumpf, M., Ramseier, H. and Lichtensteiger, W. 1989. Prenatal diazepam induced persisting depression of cellular immune responses. Life Sci. 44:493–501.
- Smister, N. E. 2003. Placental transport of immunoglobulin G. Vaccine. 21:3365–3369.
- Smith, D., Broadhead, C., Descotes, G., Fosse, R., Hack, R., Krauser, K., Pfister, R., Phillips, B., Rabemampianina, Y., Sanders, J., Sparrow, S., Stephan-Gueldner, M. and Jacobsen, S. D. 2002. Preclinical safety evaluation using nonrodent species: An industry/welfare project to minimize dog use. ILAR J. 43 Suppl:S39–S42.
- Stahlmann, R., Korte, M., Van Loveren, H., Vos, J. G., Thiel, R. and Neubert, D. 1992. Abnormal thymus development and impaired function of the immune system in rats after prenatal exposure to aciclovir. Arch Toxicol. 66:551–559.
- Stewart, J. 2009. Developmental toxicity testing of monoclonal antibodies: An enhanced pre- and postnatal study design option. Reprod Toxicol. 28:220–225.
- Tarantal, A. F. and Cowan, M. J. 1999. Administration of recombinant human stem cell factor (rhSCF) and granulocyte-colony stimulating factor (rhG-CSF) to maternal and fetal rhesus monkeys (Macaca mulatta). Cytokine. 11:290–300.
- Terao, K. 1981. Serum immunoglobulin levels in relation to age in the Cynomolgus monkey. Jpn J Med Sci Biol. 34:246–249.
- Tonk, E. C., de Groot, D. M., Penninks, A. H., Waalkens-Berendsen, I. D., Wolterbeek, A. P., Piersma, A. H. and van Loveren, H. 2011. Developmental immunotoxicity of di-n-octyltin dichloride (DOTC) in an extended one-generation reproductive toxicity study. Toxicol Lett. 204:156–163.
- Tonk, E. C., de Groot, D. M., Penninks, A. H., Waalkens-Berendsen, I. D., Wolterbeek, A. P., Slob, W., Piersma, A. H. and van Loveren, H. 2010. Developmental immunotoxicity of methylmercury: The relative sensitivity of developmental and immune parameters. Toxicol Sci. 117:325–335.
- Tonk, E. C., Verhoef, A., de la Fonteyne, L. J., Waalkens-Berendsen, I. D., Wolterbeek, A. P., van Loveren, H. and Piersma, A. H. 2011. Developmental immunotoxicity in male rats after juvenile exposure to di-n-octyltin dichloride (DOTC). Reprod Toxicol. 32:341–348.
- USEPA. 1998. U.S. Environmental Protection Agency. Health Effects Test Guidelines, OPPTS 870.3800, Reproduction and Fertility Effects. Washington, DC: Office of Prevention, Pesticides and Toxic Substances; EPA 712-C-98–208. 1998. Available online at: http://www.epa.gov/opptsfrs/OPPTS_Harmonized/870_Health_Effects_Test_Guidelines/Series/
- van de Perre, P. 2003.Transfer of antibody via mother’s milk. Vaccine. 21:3374–3376.
- Voormolen-Kalova, M., van den Berg, P. and Rádl, J. 1974. Immunoglobulin levels as related to age in non-human primates in captivity. II. Rhesus monkeys. J Med Primatol. 3:343–350.
- Wang, H., Schuetz, C., Arima, A., Woods, C., Xiao, J., Iyer, S., Gelzleichter, T. and Cain, G. 2011. Assessment of placental transfer and the effect on embryo-fetal development of a humanized monoclonal antibody targeting lymphotoxin-α in non-human primates. The Toxicologist. Abstract No. 2772.
- Weinbauer, G. F., Frings, W., Fuchs, A. N. and Osterburg, I. 2008. Reproductive/developmental toxicity assessment of biopharmaceuticals in non-human primates. In: Preclinical Safety Evaluation of Biopharmaceuticals (Cavagnaro, J. A. Ed.). Hoboken, NJ: John Wiley and Sons, Inc., pp. 379–397.
- Yun, A. J. and Lee, P. Y. 2005. The link between T-helper balance and lymphoproliferative disease. Med Hypotheses. 65:587–590.
- Zoetis, T. and Walls, I. (Eds.). 2003. Principles and Practices for Direct Dosing of Pre-weaning Mammals in Toxicity Testing and Research. Washington DC: ILSI Press.