Abstract
Mercuric chloride (HgCl2), which induces kidney toxicity, constitutes a potential threat to human health. In addition to direct toxic effects, kidney inflammatory events take place during the HgCl2-induced nephropathy. There is no information currently available about the role of angiotensin II (Ang II) in this inflammatory process. Accordingly, the aim of this study was to determine the expression of Ang II and Ang II-associated inflammatory molecules, i.e. intercellular adhesion molecule-1 (ICAM-1), inducible nitric oxide synthase (iNOS), and mono-cyte/macrophage infiltration (ED-1), in HgCl2-induced nephropathy. Three groups of Sprague Dawley rats that were to receive HgCl2 (2.5 mg HgCl2/kg BW, by gavage) were utilized: one had received Losartan at 30 mg/kg BW; one had received Enalapril at 30 mg/kg BW; and one had received distilled water, in each case daily for 3 days prior to the HgCl2 exposure. For these studies, an extra set of controls treated with saline solution in place of HgCl2 and water in place of the test drugs was employed. Renal biopsies were obtained 96 h after HgCl2 injection and the expressions of Ang II, ICAM-1, iNOS, and ED-1 were analyzed by indirect immunoflourescence while tubular damage was assessed via histopathology. An increased expression of Ang II, ICAM-1, iNOS, and ED-1 as well as increases in tubular necrosis were observed in all HgCl2-animals. Treatments with Losartan or Enalapril diminished the induced expressions as well as the extent of tubular damage. The data here suggest that Ang II is involved in the pro-inflammatory events during HgCl2-induced nephropathy, and that this is probably mediated, in part, by Ang II receptors Type 1 (AT-1).
Introduction
Heavy metals, such as cadmium, lead, and mercury, constitute potential threats to human health in both occupational and environmental settings (Hu, Citation2000). Metals are chemically stable and tend to persist in human tissue as well as in the environment. Most metals are excreted almost exclusively via the kidney, thus renal exposure to metals is unavoidable and metal toxicity commonly involves the kidney (Madden and Fowler, Citation2000). Besides the direct toxic effect of mercuric chloride (HgCl2) in the kidney, inflammatory events may occur (Ghielli et al., Citation2000; de Greef et al., Citation2003).
Angiotensin II (Ang II) has been involved in renal pathophysiology by its hemodynamic and pro-inflammatory actions (Ruster and Wolf, Citation2006; Marchesi et al., Citation2008; Hayashi et al., Citation2010). There is information about the vasopressor role of glomerular Ang II during the early phase of HgCl2-induced nephropathy (Yanagisawa et al., Citation1998); however, the role of this hormone in the inflammatory events during this nephropathy has not been reported.
Therefore, the current study was designed to investigate the role of Ang II in the inflammatory events during HgCl2-nephropathy. In this regard, Losartan (antagonist of angiotensin II AT-1 receptor) and Enalapril (inhibitor of angiotensin I converting enzyme) were used to evaluate the renal expression of Ang II and molecules associated with its known pro-inflammatory activity: the intercellular adhesion molecule-1 (ICAM-1), ED-1 (monocytes/macrophages) and inducible nitric oxide synthase (iNOS).
Materials and methods
Experimental design
Male Sprague-Dawley rats weighing 200–250 g (n = 30) obtained from the Instituto de Investigaciones Cientificas (IVIC, Caracas, Venezuela) were used in these studies. All rats were housed in pathogen-free facilities maintained at 25°C and a 12-h light/dark cycle. All rats had ad libitum access to standard chow and filtered water. All protocols used in these studies were approved by and followed the ethical guidelines of the Committee of Bioethics and Biosecurity of FONACIT (Caracas, Venezuela) and the Committee of Bioethics of the Medical School (Universidad del Zulia, Maracaibo, Venezuela).
For these studies, rats to be injected subcutaneously (SC) with a single dose of HgCl2 (2.5 mg/kg BW, in 500 µl of 0.9% saline solution) were first randomly assigned into three groups (n = 10 each). Rats in the Losartan (Cozaar, Merck & Co., Whitehouse Station, NJ) group received this antagonist of AT-1 receptors (angiotensin II receptor type-1) at 30 mg Losartan/kg BW daily by gastric gavage (Abdi and Johns, Citation1997). Rats in the Enalapril (Renitec, Merck) group received this inhibitor of angiotensin I converting enzyme at 30 mg Losartan/kg BW daily by gavage (Guo et al., Citation2008). Rats in the non-drug-treated group received distilled water only daily by gavage. Each of these treatments occurred daily for 3 days prior to the HgCl2 injection and daily thereafter (up to 4 days post-HgCl2 injection). As was previously reported by our laboratories (Muñoz et al., Citation2011), anti-AngII pre-treatments 3 days before and then throughout the experimental period was a successful protocol for diminishing expression of Ang II and pro-inflammatory molecules in the kidney in a model of rat nephrosis induced by adriamycin. A separate set of control rats (n = 10) were SC injected only with saline solution instead of HgCl2.
The dose of HgCl2 (2.5 mg/kg) selected for use and the time selected to permit evolution of nephropathy (i.e. 96 h) were based on the two following criteria: (1) it has been demonstrated previously that doses ranging from 1.0–3.5 mg HgCl2/kg induce dose-dependent alterations not only in the cytoplasm but also the nucleus of proximal tubule cells in the rat kidney (Stacchiotti et al., Citation2003); and (2) simultaneous ICAM-1 and ED-1 expression is related to the evolution time of HgCl2 nephropathy (Ghielli et al., Citation2000; de Greef et al., Citation2003). Specifically, at 24 h post-HgCl2 exposure, increased expression of ED-1 is not associated to ICAM-1 (de Greef et al. Citation2003), but their co-expression is observed 2–6 days post-exposure (Ghielli et al. Citation2000). Accordingly, since simultaneous expression of both molecules is associated with pro-inflammatory activity of Ang II, the experiments here were performed at day 4 (96 h) post-HgCl2 injection.
At 72 h post-injection, each of the 30 HgCl2-treated and 10 control rats was placed individually into metabolic cages for 24 h to collect urine samples. This was essential to determine protein urinary concentration (by the sulfosalicylic acid method). All rats were then anesthetized 24 h later (i.e. at 96 h post-HgCl2 administration) with ether and blood samples were collected via abdominal aortic puncture in order to determine blood creatinine levels (by a direct colorimetric method using an Astra 4 Model [Beckman Instruments Inc., Fullerton, CA]).
Thereafter, the kidneys were perfused in situ with saline solution. Renal biopsies from the left kidney were then rapidly removed, embedded in Tissue Tek (Miles Inc., Kankakee, IL), frozen in acetone-dry ice solution, and stored at −70°C until used in immunohistochemical studies.
Renal histopathological studies
The left kidney recovered at necropsy was processed for light microscopy. Specifically, tissue samples were fixed in 10% formalin, embedded in paraffin, sections (4 μm-thick) were prepared, and the sections were stained with the periodic acid-Schiff reaction and hematoxylin. Each section was then examined using a light microscope (Axioscop, Zeiss, Germany) and pathological alterations were quantified. Analyses were performed blinded to sample identity. The extension of tubular injury was assessed as focal tubular necrosis and complete tubular necrosis. At least 100 S1–S2 and S3 proximal tubular segments were analyzed and reported as a percentage of tubular segments showing a given histological damage. For these analyses, the proximal tubule as a part of the nephron was ‘divided’ into those zones (S1–S2, S3), as each reflects different populations/types of cells and, therefore, presumably, different functions too.
Immunofluorescence studies
The expression of Ang II, macrophage, iNOS and ICAM-1 was determined in the acetone-fixed, frozen renal sections (4-µm) by indirect immunofluorescence. For these analyses, polyclonal rabbit anti-angiotensin II (Bachem, Torrance, CA), monoclonal mouse anti-rat ED-1 for monocytes/macrophages (Chemicon International, Inc., Temecula, CA), monoclonal mouse anti-rat iNOS (Chemicon), and monoclonal mouse anti-rat ICAM-1 (Pierce Biotechnology, Rockford, IL), antibodies were, respectively, used. Secondary fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG (Fab’2) antibody (Pierce) was used to localize the monoclonal antibodies in the renal tissues. The rabbit IgG was localized using TRITC (tetramethyl rhodamine isothiocyanate)-conjugated secondary donkey anti-rabbit IgG antibody (Chemicon).
Each section was then examined using an epifluorescence microscope (Axioskop, Carl Zeiss, Germany), and total positive cells for each antibody type used were quantified. Analyses were performed blinded to sample identity. All positive cells were expressed as the number of cells per glomerular cross-section (GCS) in at least 20 glomeruli and in 20 randomly selected fields of tubulointerstitial areas and expressed as number of cells per mm2. Glomerular ICAM-1 expression was determined with a semi-quantitative score according to extension and intensity of fluorescence as follows: 0 = negative; 1 = weak; 2 = mild; and 3 = intense.
Statistical analysis
Data were expressed as mean ± standard deviation (SD). Statistical comparison between groups was performed using one-way analysis of variance (ANOVA) followed by a Bonferroni post-hoc test. Correlations were determined by Pearson’s correlation test. Statistical significance was assumed when a two-tailed p-value was < 0.05.
Results
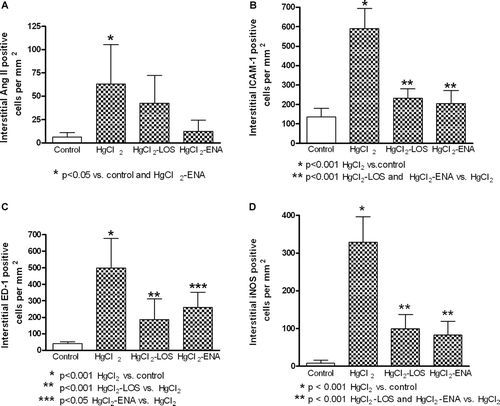
Increased interstitial expression of Ang II, ICAM-1, and iNOS, along with increased levels of monocyte-macrophage infiltration, were found in rats treated with HgCl2 (). Treatment with both Losartan and Enalapril were capable of diminishing these induced expressions of ICAM-1, iNOS, and ED-1. As expected, Enalapril decreased renal Ang II expression, but Losartan did not (). In the case of Ang II, the level of decrease from that in the rats that received only HgCl2 was 38% and 77% for the rats in the Losartan and Enalapril groups, respectively. With regard to effects on HgCl2-induced ICAM-1, iNOS, and ED-1 expression, both drugs reduced expression by 60%, 72.06%, and 62.75% (Losartan) and 63.8%, 76.47%, and 50.99% (Enalapril), respectively.
Figure 1. Renal interstitial expression of angiotensin II (AngII), intercellular adhesion molecule-1 (ICAM-1), inducible nitric oxide synthase (iNOS), and macrophages (ED-1) in HgCl2-induced nephropathy. Increased expressions of (a) Ang II, (b) ICAM-1, (c) ED-1 (macrophages), and (d) iNOS were observed in all HgCl2-treated rats. Losartan and Enalapril diminished the induced expression of those molecules. Values shown are mean (± SD) from 10 rats/group.
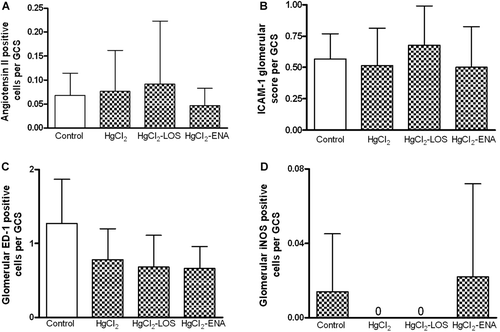
Similarly, tubular expressions of Ang II, ICAM-1, and iNOS were also increased in the HgCl2 group, expressions decreased by drug treatment (). A representative picture of immunofluorescence findings is observed in . At 96 h after HgCl2 injection, glomerular expression of Ang II and pro-inflammatory molecules remained similar in controls and in the different groups treated with the metal ().
Figure 2. Renal tubular expressions of angiotensin II (AngII), intercellular adhesion molecule-1 (ICAM-1), and inducible nitric oxide synthase (iNOS) in HgCl2-induced nephropathy. Increased expressions of (a) Ang II, (b) ICAM-1, and (c) iNOS were observed in all HgCl2-treated rats. Enalapril diminished the expression of AngII and both Enalapril and Losartan diminished the expressions of ICAM-1 and INOS.
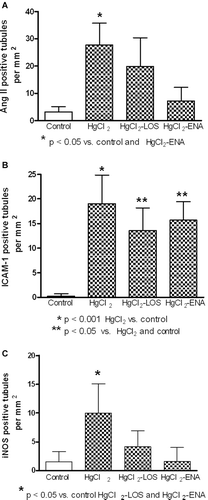
Figure 3. Immunofluorescence of renal angiotensin II (AngII), intercellular adhesion molecule-1 (ICAM-1), inducible nitric oxide synthase (iNOS), and macrophages (ED-1) in HgCl2-induced nephropathy. (a) Intense tubular expression of Ang II in tubules (arrows) and in interstitial cells (arrowheads). Magnification: ×100. (b) ICAM-1 expression in brush border of proximal tubules (arrows) and in interstitial cells (arrowheads). Magnification: ×400. (c) Increased infiltration of ED-1+ cells in tubulo-interstitial areas (arrows). Magnification: ×400. (d) iNOS+ cells in tubulo-interstitial areas (arrows). Magnification: ×400.
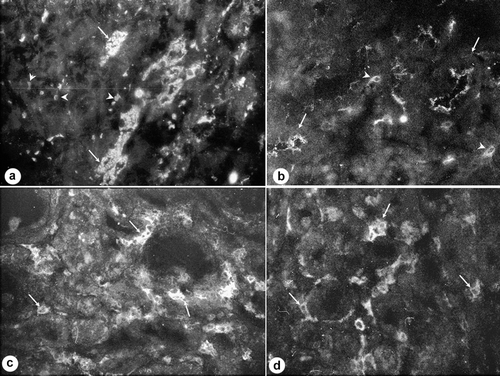
Figure 4. Glomerular expression of angiotensin II (AngII), intercellular adhesion molecule-1 (ICAM-1), inducible nitric oxide synthase (iNOS), and macrophages (ED-1) in HgCl2-induced nephropathy. In general, expression of (a) Ang II and the (b, c, d) pro-inflammatory molecules remained similar to control values. Values shown are mean (± SD) from 10 rats/group.
The histological findings are reported in . Toxic tubular damage was more intense in untreated HgCl2 animals. In contrast, the percentage of necrotic tubules was reduced in the rats treated with Losartan or Enalapril. These immunological and histological events were not accompanied by altered renal functions as observed by normal serum creatinine levels and values of proteinuria (). The expressions of Ang II and Ang II associated-pro-inflammarory molecules (i.e. ICAM-1, ED-1, and iNOS) were partially positively correlated in the interstitial area and the tubules ().
Table 1. Proximal tubule changes in HgCl2 nephropathy.
Table 2. Serum creatinine and renal protein excretion in HgCl2 nephropathy.
Table 3. Interstitial and tubular correlations of angiotensin II, ICAM-1, iNOS, and infiltration of ED-1+ cells.
Discussion
Mercury chloride (HgCl2) induces acute renal failure (ARF) by damaging proximal tubule epithelial cells without pathological findings in the glomeruli (Hostetter et al., Citation1983; Kreisberg et al., Citation1983). Previous reports have shown that inorganic mercury can induce renin release from isolated glomeruli (Kozma et al., Citation1996) and Ang II production by arterial endothelial cells (Wiggers et al., Citation2008). This study showed that HgCl2 could also induce Ang II production by interstitial and tubular cells, suggesting that this metal is capable of stimulating diverse cell types to produce Ang II.
The role of Ang II in HgCl2-induced nephropathy has been reported and focused on the vasoconstrictive activity of this hormone (Russell, Citation1975; Yanagisawa et al., Citation1998). However, besides being a potent vasoactive peptide, Ang II has pro-inflammatory effects (Marchesi et al., Citation2008; Hayashi et al., Citation2010). The present study demonstrated expression of Ang II and Ang II-associated pro-inflammatory molecules in this experimental model. Increased expression of Ang II, ICAM-1, ED-1, and iNOS in the rat kidney during the acute phase of HgCl2-induced renal damage (96 h) was demonstrated here using immunohistochemical techniques. A blockade of the biological action of Ang II by an AT-1 receptor antagonist (Losartan) or by an angiotensin I-converting enzyme inhibitor (Enalapril) mitigated the degree of expression of those molecules. This indicated to us that ICAM-1, ED-1, or iNOS expressions were, in part, up-regulated by endogenous Ang II. In this regard, the blocking of Ang II was associated with diminished expression of pro-inflammatory molecules, an outcome that has been reported in other renal and vascular pathologies (Guo et al., Citation2008; Marchesi et al., Citation2008; Hayashi et al., Citation2010).
The pro-inflammatory events here were observed mainly in the interstitium and tubules; glomerular expression remained at basal levels, suggesting that interstitial and tubular mechanisms were involved in the development of HgCl2-induced renal damage. Controversial findings regarding the role of glomerular alterations in HgCl2 renal damage have been reported (Flamenbaum et al., Citation1972; Yanagisawa et al., Citation1998). Increased glomerular Ang II, endothelin-1, and nitric oxide expression at 20 h after exposure to HgCl2 have been found, suggesting a role for these molecules in the progression of HgCl2-induced acute renal failure, mostly through an acceleration of proximal tubule epithelial cell injury and deterioration of glomerular hemodynamics (Yanagisawa et al., Citation1998). Attempts to diminish azotemia in rennin-immunized rats with HgCl2 nephropathy at 48 h were unsuccessful (Flamenbaum et al., Citation1972). Our results at 96 h were not associated to acute renal failure, since creatinine values were observed to be normal and up-regulation of Ang II and pro-inflammatory molecule levels were not observed. This indicated to us that glomerular inflammatory events induced by Ang II were absent and that any glomerular Ang II effect in this model was related to post-exposure time. It is plausible that the initial activity of Ang II could occur at early timepoints post-exposure, and subsequently return to normal condition. In this regard, it has been reported that glomerular damage in this nephropathy was reversible (Kreisberg et al., Citation1983).
The mechanisms responsible for development of cell injury caused by the pro-inflammatory effects of Ang II in the tubulo-interstitial area remain unclear. The biochemical mechanisms by which increased expression of iNOS leads to progression of proximal tubule injury may be due to a cytotoxic effect from large amounts of free radicals, like nitric oxide (NO) (Giacco and Brownlee, Citation2010). It may be worth exploring further whether a factor such as ischemia resulting from the vasoconstrictor(s) could also be involved in the mechanism(s) occurring here. Effects related to local Ang II-induced vasoconstriction in the microcirculation with ischemia could be responsible for the development of the epithelial cell injury (Vicaut et al., Citation1991; Reslan and Khalil, Citation2010). Anti-Ang II treatment associated with decreased expressions of endogenous Ang II and iNOS found in this study could induce decreased vasopressor effects. In addition, Ang II is capable of inducing endothelin-1 production (Touyz and Schiffrin, Citation2003), a more powerful vasoconstrictor (Kon and Badr, Citation1991; Marsen et al., Citation1994); in this case, decreased Ang II activity could lead to decreased ET-1, and this could represent a beneficial status during this nephropathy. Via the AT1 receptor, angiotensin II also stimulates IL-6 production by vascular smooth muscle cells (Funakoshi et al., Citation1999). This pro-inflammatory effect of Ang II could thus be related to hypertension (Harrison et al., Citation2012) and could promote mesangial cell activation, leading to increases in the mesangial extracellular matrix (Simonson and Ismail-Beigi, Citation2011).
Adhesion molecules expressed on cell surfaces mediate specific cell–cell interactions in embryogenesis, histogenesis, immune responses, and inflammatory reactions (Galkina and Ley, Citation2007). Several studies using different anti-ICAM-1 strategies have shown attenuation of functional impairment and histological changes during acute renal failure (Kelly et al., Citation1991, Citation1996; Haller et al., Citation1996). During this study, increased expression of ICAM-1 in interstitial area and in tubules was observed. ICAM-1 may be critical in the pathophysiology of renal injury following mercury intoxication, perhaps by its effects on leukocyte–endothelial interactions and further renal leukocyte infiltration and inflammatory damage (Galkina and Ley, Citation2007). In this regard, interstitial over-expression of ICAM-1 was associated (and correlated) with ED-1 expression. In addition, anti-Ang II treatment decreased ICAM-1 and ED-1 expression, suggesting that there was a sequential event, i.e., increases in Ang II activity led to ICAM-1 expression and subsequently to monocyte/macrophage (ED-1+ cell) infiltration. ED-1+ monocytes/macrophages could represent an important factor in the induction of the tissue damage (Wang and Harris, Citation2011).
ICAM-1 over-expression was also observed in proximal tubule epithelium. This finding has been previously reported in different nephropathies (Daniel et al., Citation2001; Fernandez et al., Citation2003), suggesting that tubular ICAM-1 expression may represent a common event during renal injury. Interestingly, cadmium and mercury induce endothelial ICAM-1 gene expression in humans and ICAM-1 expression on immortalized proximal tubule cells (Martinotti et al., Citation1995; Pritchard et al., Citation2000; Jiang et al., Citation2002). The over-expression of ICAM-1 in proximal tubules could be a deleterious factor, since it has been associated to apoptosis and proliferation during experimental nephropathy (Shappell et al., Citation2000). The expression of ICAM-1 on capillaries, interstitial cells, and epithelial tubular cells suggest a non-cellular restricted ICAM-1-inducer effect of HgCl2. The association between Ang II expression and both pro-inflammatory molecules and leukocyte infiltration could be related to the binding of Ang II to the AT-1 receptor; the pro-inflammatory effect could occur mainly through down-stream activation of an intracellular signaling cascade that involves nuclear factor-κB (NF-κB) activation (Capettini et al., Citation2012).
Anti-Ang II treatment diminished tubular damage as expressed by a lower percentage of tubular necrosis, suggesting a role for Ang II in tubular damage during this experimental model. Accordingly, Ang II-mediated tubular damage has been reported in diverse renal pathologies (Ruster and Wolf, Citation2006; Hayashi et al., Citation2010; Muñoz et al., Citation2011). According to previous reports using a similar HgCl2 dose and timeline to induce nephropathy (Ghielli et al., Citation2000), normal values of serum creatinine were found in animals treated with HgCl2. During this experimental model, ARF is secondary to the back leak of tubular fluid due to the necrotic lesions induced by this toxin. Tubular cell proliferation leading to tubular repair is associated with a decrease of serum creatinine at ~ 48 h after HgCl2 injection (Diamond and Zalups, Citation1998). Therefore, it was not expected to find increased values of serum creatinine in this study (96 h); in addition, increased glomerular expressions of pro-inflammatory molecules and Ang II were not observed. However, levels of serum creatinine in mercuric nephropathy do not seem to be a good biomarker for renal tissue damage activity. In this regard, a marked elevations in urinary kidney injury molecule-1 concentration and viral cellular receptor-1 (Kim-1/Havcr1) expression were observed in HgCl2 nephropathy at 72 h when serum creatinine levels were not different from controls (Zhou et al., Citation2008), thereby suggesting tissue damage even with normal kidney function.
In humans, HgCl2 is primarily a skin and mucous membrane irritant that is rapidly absorbed. However, acute poisoning has been reported following oral ingestion and dermal applications of HgCl2 solutions. HgCl2 intoxication can induce death accompanied by gastrointestinal and renal lesions (Nordlind, Citation1990; National Toxicological Program. Citation1993; Singer et al., Citation1994). The beneficial effects of Losartan and Enalapril in reducing pro-inflammatory events and tubular damage are observed when the drugs are given before the administration of HgCl2. Under this condition, the role of Ang II in the inflammation during the intoxication was experimentally demonstrated. Further experiments are needed to determine the treatment of mercuric intoxication using anti-Ang II drugs.
In conclusion, the results of the present study suggest that, in HgCl2-treated rats, Ang II, ICAM-1, iNOS, and monocyte/macrophage infiltration may participate in local inflammatory events, especially through the Ang II production and its interaction with AT-1 receptors.
Acknowledgments
This study was supported by Consejo Científico y Humanístico de la Universidad del Zulia (CONDES: CC-0513-10), Maracaibo, Venezuela.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Abdi, A., Johns, E. J. 1997. The effect of angiotensin II receptor antagonists on kidney function in two-kidney, two-clip Goldblatt hypertensive rats. Eur. J. Pharmacol. 23:185–192.
- Capettini, L. S., Montecucco, F., Mach, F., Stergiopulos, N., Santos, R. A., and da Silva, R. F. 2012. Role of renin-angiotensin system in inflammation, immunity and aging. Curr. Pharm. Des. 18:963–970.
- Daniel, L., Sichez, H., Giorgi, R., Dussol, B., Figarella-Branger, D., Pellissier, J. F., Berland, Y. 2001. Tubular lesions and tubular cell adhesion molecules for the prognosis of lupus nephritis. Kidney Int. 60:2215–2221.
- de Greef, K. E., Ysebaert, D. K., Persy, V., Vercauteren, S. V., de Broe, M. E. 2003. ICAM-1 expression and leukocyte accumulation in inner stripe of outer medulla in early phase of ischemic compared to HgCl2-induced ARF. Kidney Int. 63:1697–1707.
- Diamond, G. L., Zalups, R. K. 1998. Understanding renal toxicity of heavy metals. Toxicol. Pathol. 26:92–103.
- Fernandez, L., Romero, M., Rincón, J., Mosquera, J. 2003. Increased expression of CD54, CD18, MHC Class II molecules, and proliferating cell nuclear antigen in acute puromycin aminonucleoside nephrosis. Nephron Exp. Nephrol. 94:e55–65.
- Flamenbaum, W., Kotchen, T. A., Oken, D. E. 1972. Effect of renin immunization on mercuric chloride and glycerol-induced renal failure. Kidney Int. 1:406–412.
- Funakoshi, Y., Ichiki, T., Ito, K., Takeshita, A. 1999. Induction of IL-6 expression by angiotensin II in rat vascular smooth muscle cells. Hypertension 34:118–125.
- Galkina, E., Ley, K. 2007. Vascular adhesion molecules in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 27:2292–2301.
- Giacco, F., Brownlee, M. 2010. Oxidative stress and diabetic complications. Circ. Res. 107:1058–1070.
- Ghielli, M., Verstrepen, W. A., de Greef, K. E., Helbert, M. H., Ysebaert, D. K., Nouwen, E. J., de Broe, M. E. 2000. Antibodies to both ICAM-1 and LFA-1 do not protect the kidney against toxic (HgCl2) injury. Kidney Int. 58:1121–1134.
- Guo, S., Kowalewska, J., Wietecha, T. A., Iyoda, M., Wang, L., Yi, K., Spencer, M., Banas, M., Alexandrescu, S., Hudkins, K. L, Alpers, C. E. 2008. Renin-angiotensin system blockade is renoprotective in immune complex-mediated glomerulonephritis. J. Am. Soc. Nephrol. 19:1168–1176.
- Haller, H., Dragun, D., Miethke, A., Park, J. K., Weis, A., Lippoldt, A., Gross, V., Luft, F. C. 1996. Antisense oligonucleutides for ICAM-1 attenuate reperfusion injury and renal failure in the rat. Kidney Int. 50:473–480.
- Harrison, D. G., Marvar, P. J., and Titze, J. M. 2012. Vascular inflammatory cells in hypertension. Front. Physiol. 3:128.
- Hayashi, T., Takai, S., Yamashita, C. 2010. Impact of the renin-angiotensin-aldosterone-system on cardiovascular and renal complications in diabetes mellitus. Curr. Vasc. Pharmacol. 8:189–197.
- Hostetter, T. H., Wilkes, B. M., Brenner, B. M. 1983. Renal circulatory and nephron function in experimental acute renal failure. In: Acute Renal Failure (Brenner, B. M.Lazarus, J. M., Eds.).Philadelphia, PA:W. B. Saunders Company, pp. 99–115.
- Hu, H. 2000. Exposure to metals. Occup. Environ. Med. 27:983–996.
- Jiang, J., McCool, B. A., Parrish, A. R. 2002. Cadmium- and mercury-induced intercellular adhesion molecule-1 expression in immortalized proximal tubule cells: Evidence for a role of decreased transforming growth factor-1. Toxicol. Appl. Pharmacol. 179:13–20.
- Kelly, K. J., Williams, W. W. Jr., Colvin, R. B., Bonventre, J. V. 1991. Antibody to intercellular adhesion molecule 1 protects the kidney against ischemic injury. Proc. Natl. Acad. Sci. USA 2:812–816.
- Kelly, K. J., Williams, W. W. Jr., Colvin, R. B., Meehan, S. M., Springer, T. A. 1996. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J. Clin. Invest. 97:1056–1063.
- Kon, V., Badr, K. F. 1991. Biological actions and pathophysiologic significance of endothelin in the kidney. Kidney Int. 40:1–12.
- Kozma, L., Lenkeyb, A., Gombaa, V. S. 1996. Induction of renin release from isolated glomeruli by inorganic mercury (I1). Toxicol. Lett. 85:49–54.
- Kreisberg, J. I., Matthys, E., Venkatachalam, M. A., 1983. Morphologic factors in acute renal failure. In: Acute Renal Failure (Brenner, B. M.Lazarus, J. M., Eds.).Philadelphia, PA:W. B. Saunders Company, pp. 21–46.
- Madden, E. F., Fowler, B. A. 2000. Mechanisms of nephrotoxicity from metal combinations: A review. Drug Chem. Toxicol. 23:1–12.
- Marchesi, C., Paradis, P., Schiffrin, E. L. 2008. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol. Sci. 29:367–374.
- Marsen, T. A., Schramek, H., Dunn, M. J. 1994. Renal action of endothelin: Linking cellular signaling pathways to kidney disease. Kidney Int. 45:336–344.
- Martinotti, S., Toniato, E., Colagrande, A., Alesse, E., Alleva, C., Screpanti, I., Morrone, S., Scarp, S., Frati, L., Hayday, A. C. 1995. Heavy-metal modulation of the human intercellular adhesion molecule (ICAM-1) gene expression. Biochim. Biophys. Acta 1261:107–114.
- Muñoz, M., Rincón, J., Pedreañez, A., Viera, N., Hernández-Fonseca, J. P., Mosquera, J. 2011. Pro-inflammatory role of angiotensin II in a rat nephrosis model induced by adriamycin. J. Renin Angiotensin Aldosterone System 12:404–412.
- National Toxicological Program. 1993. Toxicology and carcinogenesis studies of mercuric chloride (CAS No. 7487-94-7) in F344 rts and B6C3F1 mice. Gavage Stud. 408:1–260.
- Nordlind, K. 1990. Biological effects of mercuric chloride, nickel sulphate and nickel chloride. Progress Med. Chem. 27:189–233.
- Pritchard, K. A. Jr., Ackerman, A., Kalyanaraman, B. 2000. Chromium(VI) increases endothelial cell expression of ICAM-1 and decreases nitric oxide activity. J. Environ. Pathol. Toxicol. Oncol. 19:251–260.
- Reslan, O. M., Khalil, R. A. 2010. Molecular and vascular targets in the pathogenesis and management of the hypertension associated with preeclampsia. Cardiovasc. Hematol. Agents Med. Chem. 8:204–226.
- Russell, S. B. 1975. The mechanism of action of mercuric chloride on the isolated perfused rat kidney. Eur. J. Clin. Invest. 5:319–325.
- Ruster, C., Wolf, G. 2006. Renin–angiotensin–aldosterone system and progression of renal disease. J. Am. Soc. Nephrol. 17:2985–2991.
- Shappell, S. B., Mendoza, L. H., Gurpinar, T., Smith, C. W., Suki, W. N., Truong, L. D. 2000. Expression of adhesion molecules in kidney with experimental chronic obstructive nephropathy: The pathogenic role of ICAM-1 and VCAM-1. Nephron 85:156–166.
- Simonson, M. S., and Ismail-Beigi, F. 2011. Endothelin-1 increases collagen accumulation in renal mesangial cells by stimulating a chemokine and cytokine autocrine signaling loop. J. Biol. Chem. 286:11003–11008.
- Singer, A. J., Mofenson, H. C., Caraccio, T. R., Ilasi, J. 1994. Mercuric chloride poisoning due to ingestion of a stool fixative. J. Toxicol. Clin. Toxicol. 32:577–582.
- Stacchiotti, A., Borsani, E., Rodella, L., Rezzani, R., Bianchi, R. 2003. Dose-dependent mercuric chloride tubular injury in rat kidney. Ultrastruc. Pathol. 27:253–259.
- Touyz, R. M., Schiffrin, E. L. 2003. Role of endothelin in human hypertension. Can. J. Physiol. Pharmacol. 81:533–541.
- Vicaut, E., Trouvé, R., Hou, X. 1991. Microvascular effects of cocaine: Interaction with nitrendipine and enalaprilat. J. Toxicol. Clin. Toxicol. 29:165–175.
- Wang, Y., Harris, D. C. 2011. Macrophages in renal disease. J. Am. Soc. Nephrol. 22:21–27.
- Wiggers, G. A., Stefanon, I., Padilha, A. S., Peçanha, F. M., Vassallo, D. V., Oliveira, E. M. 2008. Low nanomolar concentration of mercury chloride increases vascular reactivity to phenylephrine and local angiotensin production in rats. Comp. Biochem. Physiol. 147:252–260.
- Yanagisawa, H., Nodera, M., Umemori, Y., Shimoguchi, Y., Wada, O. 1998. Role of angiotensin II, endothelin-1, and nitric oxide in HgCl2-induced acute renal failure. Toxicol. Appl. Pharmacol. 152:315–326.
- Zhou, Y., Vaidya, V. S., Brown, R. P., Zhang, J., Rosenzweigm, B. A., Thompson, K. L., Miller, T. J., Bonventre, J. V., Goering, P. L. 2008. Comparison of kidney injury molecule-1 and other nephrotoxicity biomarkers in urine and kidney following acute exposure to gentamicin, mercury, and chromium. Toxicol. Sci. 101:159–170.