Abstract
The present study was designed to examine and compare the effects of three suppressors on the cytokine response in tandem with examining: the synthesis of inducible forms of heat shock proteins; HSP72 and HSP90α; activities of NF-κB and SAPK/JNK signaling pathways; and TLR4 expression. Pre-treatment with inhibitors offers promise as protective means to lower the activity of these cascades, thereby circumventing the formation of excessive amounts of pro-inflammatory molecules. Three inhibitors of TLR4, SAPK/JNK, and NF-κB signaling, namely CLI-095, SP600125, and IKK Inhibitor XII, respectively, were added to cultured RAW 264.7 macrophages before the Escherichia coli lipopolysaccharide (LPS) application. Treatments of RAW 264.7 cells with each of the inhibitors resulted in a reduced response to LPS as was visualized by a decrease of TNF-α, IL-1, and IFN-γ production. In addition, inhibitors of the NF-κB and SAPK/JNK signaling reduced IL-6 production in LPS-treated cells, whereas the IKK inhibitor XII also decreased IL-10 production. Further, the NO production in LPS-stimulated macrophages was significantly reduced following application of CLI-095 or IKK inhibitor XII. The results also showed that the inhibitors suppressed TLR4 production and decreased phosphorylation of NF-κB and SAPK/JNK proteins, thereby preventing the activation NF-κB and SAPK/JNK signaling pathways in LPS-activated cells. In addition, the production of inducible heat shock proteins, HSP72 and HSP90-α, was reduced in LPS-stimulated RAW 264.7 cells pre-treated with inhibitors. These results suggest that inhibitors CLI-095, SP600125, and IKK inhibitor XII demonstrate potential effectiveness in the reduction of the inflammatory response by mechanisms involving both the cellular defense system and cellular signaling. In conclusion, suppressor of NF-κB cascade, IKK inhibitor XII, seems to be the most effective anti-toxic agent among studied inhibitors.
| Abbreviations | ||
| LPS, | = | lipopolysaccharide; |
| NO, | = | nitric oxide; |
| TNF, | = | tumor necrosis factor; |
| IFN, | = | interferon; |
| IL, | = | interleukin; |
| TLR, | = | toll like receptor; |
| IKK, | = | IκB kinase; |
| HSP, | = | heat shock protein; |
| SAPK, | = | Stress-Activated Protein Kinase; |
| JNK, | = | Jun N-terminal kinase; |
| NF-κB, | = | nuclear factor-κB |
Introduction
The discovery of toll-like receptors (TLRs) has revealed specificity in terms of ligand recognition, expression in different cell types and a role for TLRs in the pathogenesis of diseases involving both the innate and the adaptive immune system (Akira et al., Citation2006; Kawai and Akira, Citation2009; O’Neill et al., Citation2009). The mammalian TLR family comprises more than 12 members (Akira et al., Citation2006), and TLR4 was the first TLR-identified receptor initially characterized as a pattern recognition receptor in the experiments using lipopolysaccharide-resistant mice (Medzhitov et al., Citation1997).
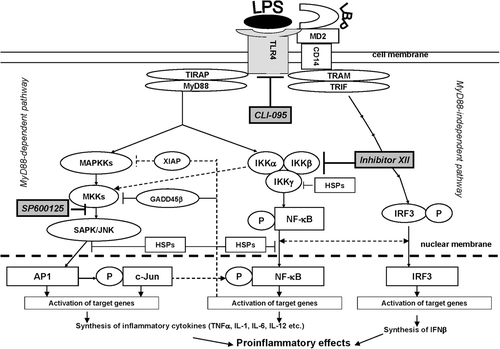
Lipopolysaccharide (LPS) from gram-negative bacteria is well known as the most potent stimulator of macrophages. LPS has high specificity for the TLR4, which seems to be unique among the TLR proteins, because it uses three additional proteins, i.e. LBP, CD14, and MD-2, as co-receptors (Schumann et al., Citation1990; Wright et al., Citation1990; Shimazu et al., Citation1999) (). The TLR4-mediated response to LPS can be divided into two categories: an early MyD88-dependent response and a delayed MyD88-independent response. The activation of the MyD88-dependent pathway by LPS leads to activation of the NF-κB and MAPK pathways (Zeytun et al., Citation2010). Substances decreasing the toxin-induced activation of TLR4-expressing cells may be used as pharmacological tools to protect against inflammation. Furthermore, as was reported earlier, the inhibitor of heat shock protein HSP90, geldanamycin, decreased murine macrophage responses to LPS via mechanisms involving TLR4 and NF-κB signaling pathways, resulting in an effect similar to TLR4-related inhibitors (Vega and De Maio, Citation2003; Glushkova et al., Citation2010).
Figure 1. TLR4 signaling pathway and inhibitors of TLR4 and TLR4-related pathways used in the study. CD14, LBP, and TLR4 are part of the LPS recognition/response pathway. The most extensively studied signaling pathways activated by TLR4 in response to LPS require adaptor proteins including myeloid differentiation factor 88 (MyD88), MyD88 adaptor-like protein (Mal or, alternatively, TIRAP), TIR-containing adapter molecule (TRIF), and TRIF-related adaptor molecule (TRAM) (Krishnan et al., Citation2007). The differences in induction of MyD88-dependent and -independent signaling by endotoxin suggests that the initial interaction of an endotoxin with the extracellular domain of the TLR4 complex results in changes that lead to the recruitment of different adaptor proteins (Zughaier et al., Citation2005). MyD88 activation involves NF-κB and AP-1 signaling cascades, resulting in inflammatory cytokine over-production. The TLR4-mediated MyD88 independent pathway leads to dimerization and activation of IRF3 and IFNβ expression. It follows that the TLR4 signaling pathway leading to LPS-mediated NF-κB, AP-1, and IRF3 activation constitutes an important therapeutic target for sepsis therapy. Recognition of LPS and subsequent signaling pathways are not determined by one single receptor but through the orchestration of multiple receptors that are concentrated at so-called lipid rafts. For instance, in monocytes and macrophages several LPS signaling molecules, namely CD14, HSP70, and HSP90, are constitutively present at lipid rafts, while others, such as TLR4, CXCR4, and MyD88, are recruited upon LPS stimulation. It is known that LPS-mediated activation of the transcription factors NF-κB and AP-1 requires phosphorylation of IKK and JNK kinase, respectively. JNK subsequently phosphorylates AP-1, inducing its transactivation, whereas activated IKK phosphorylates the family of I-κB proteins, targeting them for degradation via the ubiquitin proteasome pathway (Medvedev et al., Citation2000; Haddad and Abdel-Karim, Citation2011). The early MyD88-dependent response to LPS also induces an early activation of IRF3 and induction of IFNκ. The MyD88-independent pathway is responsible for the delayed activation of NF-κB and IRF3. Intriguingly, there is a sequence of data indicating co-operation between the NF-κB and IRF3 pathways. Down-stream NF-κB, IRF3, and MAPK activation can result in intracellular over-production of pro-inflammatory cytokines, including TNFκ, IFNγ, IL-1, and IL-6, as well as reactive oxygen species, including nitric oxide (NO) (Akira et al., Citation2006; Novoselova et al., Citation2006; O’Neill et al., Citation2009). CLI-095 was used for specific inhibition of the TLR4 receptor protein, SP600125 for that of JNK 1/2/3 (which blocks the SAPK/JNK signaling pathway), and IKK Inhibitor XII for inhibition of IKKα/β/ϵ (which leads to NF-κB-pathway blockade).
Studies that utilized a direct blockade of the enzymatic activities of IKK, JNK, or p38 MAPK have demonstrated the great potential for such treatments to elicit anti-inflammatory effects. Targeting intracellular protein kinase pathways is particularly promising. Indeed, some new antagonists of the TLR4 receptor, including CLI-095—a novel cyclohexene derivative that blocks the intracellular domain of TLR4—have become available (Loiarro et al., Citation2010; Wittebole et al., Citation2010; Zhu and Mohan, Citation2010). Over the last decade, two inhibitors of signaling pathways NF-κB and SAPK/JNK, IKK Inhibitor XII and inhibitor SP600125, respectively, have been investigated for clinical potential in inflammatory diseases (Bennett et al., Citation2001; Christopher et al., Citation2007). These inhibitors are mainly synthetic or natural small hydrophobic molecules that can penetrate into the cell.
In a few of the studies mentioned above, attempts were made to evaluate separately the efficacy of inhibitors via their effects on cytokines and NO production. The present study was designed to examine and compare the effects of three suppressors on the cytokine response in tandem with examining: the synthesis of inducible forms of heat shock proteins; HSP72 and HSP90α; activities of NF-κB and SAPK/JNK signaling pathways; and TLR4 expression. Pre-treatment with inhibitors offers promise as a protective means to lower the activity of these cascades, thereby circumventing the formation of excessive amounts of pro-inflammatory molecules. A schematic illustration of TLR4 signaling and possible mechanisms involved in the inhibitors’ activities is shown in .
Materials and methods
Cell culture and reagents
RAW 264.7 cells (murine macrophage cells, TIB-71, ATCC, Rockville, MD) were cultured in DMEM (Sigma, St. Louis, MO) supplemented with 10% fetal calf serum (FCS), 100 U penicillin G/ml, 100 μg gentamycin/ml, and 100 μg streptomycin/ml (all Sigma) at 37°C in 5% CO2. In each of the experiments, the cells from a separate passage (over the range from passages 3–8) were pooled, and the cell’s aliquots were treated separately with 1 μg/ml CLI-095 (TLR4 inhibitor, InvivoGen, San Diego, CA), or 2.2 μg/ml SP600125 (JNK-1, -2, -3 inhibitor, StressGen, Farmingdale, NY), or 1 μg/ml IKK Inhibitor XII (inhibitor of IKK-α, -β, and -ϵ-activity, Merck, Whitehouse Station, NJ). In the presence of inhibitors, the cells were cultured for 1 h (SP600125, IKK Inhibitor XII) or for 15 min (CLI-095) before lipopolysaccharide (LPS) addition (2.5 μg/ml, Escherichia coli Type J5; Merck). Optimal dosages of inhibitors were calculated using their pIC50 values indicated in certificates of quality. To state the efficacy of studied inhibitors against the LPS-induced tumor necrosis factor (TNF)-α and nitric oxide (NO) production, some approximate experiments were initially performed using different concentrations of inhibitors (data not shown). The inhibitors in optimal dosages did not show cytotoxicity by using trypan blue exclusion method (Strober, Citation1997).
ELISA
RAW 264.7 cells (1 × 106) were suspended in RPMI 1640 (Sigma) supplemented with gentamycin, HEPES, and 10% FCS, and cultured in 24-well plates with LPS and with/without the inhibitors for 24 h at 37°C in 5% CO2. Lysates of the cells were obtained by three cycles of freezing–refreezing. The concentration of cytokines in the cell samples was then determined using ELISA Development Kits for mouse TNF-α, interleukin (IL)-1, IL-6, IL-10, and interferon (IFN)-γ; all kits were purchased from Peprotech (Rocky Hill, NJ). To visualize binding in the assays, 100 µl of ABTS green dye (dissolved in 0.05 M citrate buffer [pH 4.0] containing 0.01% H2O2) (Sigma) was added to each well. The optical density in each well was then measured at 405 nm with a Multiscan EX plate-reader/spectrophotometer (Thermo Electron Corporation, Vantaa, Finland).
Nitric oxide assay
RAW 264.7 macrophages (1 × 106 cells/ml) were cultivated in 24-well plates in DMEM without pH indicator (Sigma) at 37°C in a humidified atmosphere containing 5% CO2. The medium was supplemented with 1 mM sodium pyruvate, 25 mM HEPES, 2 mM L-glutamine, and 3% fetal calf serum (Nakamura et al., Citation1994). Supernatants were collected at 21 h and assayed for NO2. Griess reagent (Sigma) containing a mixture of 1% sulphanilamide in 0.1% naphthylethylenediamine dihydrochloride, and 2.5% H3PO4 (1:1) was used to estimate NO2, an indicator of NO production. After cultivation, 100 μl of each sample was placed into a well of a 96-well plate and 100 μl Griess reagent was then added to each. After 10 min at 23°C, the optical density in each well was measured on the plate reader at 546 nm. A calibration curve was plotted using standard NaNO2 solutions.
Western blot analysis
To prepare specimens, RAW 264.7 cells (1 × 108) suspended in RPMI 1640 supplemented with 10% FCS, 2 mM glutamine, and 100 µg streptomycin/ml, were cultivated in 24-well culture plates for 6 h at 37°C in a humidified atmosphere containing 5% СO2. The cells were then lysed using an ultrasonic disintegrator with constant stirring for 2 min. Total protein concentration in each sample was then determined by the Bradford method. Thereafter, the proteins in each sample were precipitated in acetone, solubilized, boiled for 5 min, and stored at −70°C. Proteins were then resolved by electrophoresis over a 10% PAGE gel, as described previously (Towbin et al., Citation1976). The proteins were transferred from the gel onto a nitrocellulose membrane (GE Healthcare, Amersham, UK) in a transblot chamber.
After blocking, the membrane was exposed for 2 h to antibodies against the mouse proteins: HSP70 (rabbit anti-mouse HSP 72, clone SPA-812, inducible form, StressGen), HSP90 (rabbit anti-Hsp90α [Hsp86], StressGen) or phospho-NF-κB antibody (phospho-NF-κB p65 [Ser 536], #3031, Cell Signaling Technology, Danvers, MA), or rabbit phospho-SAPK/JNK antibody to synthetic phospho-peptide SAPK/JNK or rabbit TLR4 antibody (#2246, Cell Signaling). After washing, the nitrocellulose membranes were incubated for 1 h with the anti-rabbit biotinylated antibody (Jackson ImmunoResearch, West Grove, PA), and peroxidase-conjugated streptavidin (IMTEK, Moscow, Russia) was added for 1 h. The loading control was mouse monoclonal anti-human tubulin-β (US Biological, Swampscott, MA). An ECL-plus chemiluminescent cocktail (Amersham/GE) was then used to develop the blots (according to manufacturer instructions) and the blot was then exposed to film. Quantitative evaluation of protein bands was then performed using the Qapa computer program (Pushchino, Russia).
Statistical analysis
Statistical analysis was performed using Statistica/Win 6.0 software (Tulsa, OK). One-way analysis of variance (ANOVA) followed by a post-hoc Tukey test was used to determine the significance of differences among groups. p-values ≤ 0.05 were considered significant. All values were expressed as mean (± SE).
Results
Cell viability
The 24 h cultivation of RAW 264.7 cells with 2.5 µg LPS/ml and/or with each of inhibitors did not result in significant changes in cell death. Indeed, in LPS-treated macrophages, cellular viability was no less than 80 (±5)%; in RAW 264.7 cells treated separately with each of inhibitors, cellular viability was no less than 91 (±5)% (p < 0.05) (data not shown).
Inhibitors of TLR4, NF-κB, and JNK pathways decreased pro-inflammatory cytokine and NO responses in LPS-treated cells
To characterize effects of inhibitors on LPS-induced inflammatory responses, production of interleukins (IL-1, IL-6, IL-10), TNF-α, IFN-γ, and NO was measured concurrently in five sample clusters of RAW 264.7 cells (). Treatment with LPS alone resulted in an expected increase in production of all pro-inflammatory cytokines, e.g. TNF-α, IL-1, IL-6, IFN-γ, and NO in the RAW 264.7 cells. Production of anti-inflammatory IL-10 was unchanged (). We showed earlier that LPS increased IL-10 production in peritoneal mouse macrophages (CitationNovoselova et al., 2009). In contrast to native LPS-stimulated mouse macrophages, endotoxin did not affect IL-10 production in RAW 264.7 cells. One explanation for this difference is that there could be a lower sensitivity of the RAW 264.7 cells to LPS as compared to that by native macrophages.
Figure 2. The effect of inhibitors on production of cytokines and NO by RAW 264.7 cells treated with LPS. RAW 264.7 cells from one separate passage were pooled, and aliquots of 1 × 106 cells (cultivated for 24 h) were used for each sample. NO was measured in supernatants, and cytokines were estimated using cellular lysates. Lysates of the cells were obtained by three cycles of freezing–refreezing, and cytokine concentrations in each sample were determined using ELISA. Results are expressed as the mean and standard error from four independent experiments that were performed separately with different passages; the measurements in separate experiments were made for each individual cellular sample (six repetitions). The effectiveness of inhibitors in comparison with the most effective, i.e. the IKK Inhibitor XII, is shown using upper staples. Value is significantly different from control, *p < 0.05, **p < 0.01; from LPS, #p < 0.05, ##p < 0.01; or from IKK Inhibitor XII, ^p < 0.05, ^^p < 0.01.
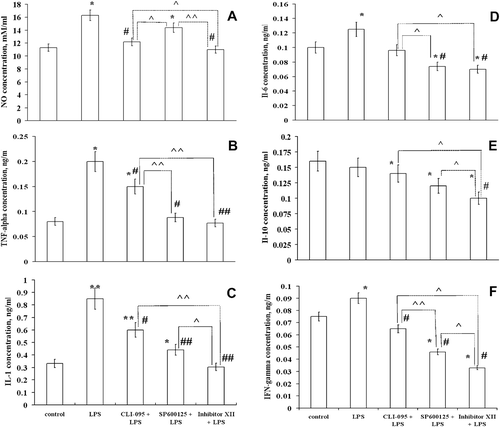
Pre-treatment of RAW 264.7 cells with each of the inhibitors differently affected their response to LPS; however, in most cases it resulted in normalization of pro-inflammatory cytokines and NO concentrations. Specifically, CLI-095 or IKK Inhibitor XII, but not SP600125, decreased NO production in these cells (). The inhibition of NF-κB or SAPK/JNK signaling, and to a lesser extent the TLR4 activity, resulted in a significant decrease in IL-6 production in LPS-stimulated RAW 264.7 cells (). In addition, in LPS-stimulated cells pre-treated with each of the inhibitors, a decrease in the pro-inflammatory response was reflected as a reduced production of TNF-α, IL-1, and IFN-γ (, , and ). Despite the fact that no changes in anti-inflammatory IL-10 production were observed in LPS-treated cells, the effects of inhibitors were quite noticeable (). Indeed, the inhibitors significantly reduced IL-10 production in LPS-stimulated RAW 264.7 cells. These results showed that the efficacy of IKK Inhibitor XII was significantly higher than the others ().
Inhibitors of TLR4, NF-κB, and JNK signaling decreased LPS-mediated TLR4 expression
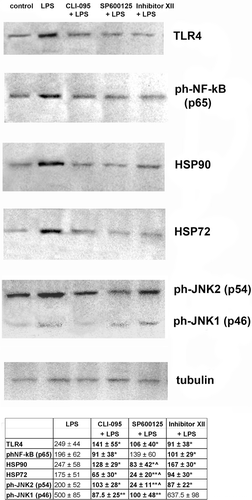
The results described above suggest that the over-production of cytokines and NO in macrophages could be corrected by exposure of the cells to inhibitors of pro-inflammatory signaling pathways. To elucidate the role of TLR4 (an essential receptor for LPS), we analyzed TLR4 levels in the cells using Western blot analysis. As shown in , LPS application to the RAW 264.7 cells induced a significant increase in TLR4 levels; however, cells pre-treated with each of the inhibitors demonstrated reduced levels of TLR4. Pre-incubation of the cells with inhibitors of NF-κB and SAPK/JNK pathways (SP600125 and IKK Inhibitor XII, respectively) resulted in a decrease of TLR4 production to the control level. In cells pre-treated with CLI-095, TLR4 expression was reduced, but did not attain the control value. The results of this experiment demonstrated that an effectiveness of the inhibitor of the extra-cellular domain of TLR4 (CLI-095) was somewhat lower than the inhibitor SP600125 or IKK Inhibitor XII, but the differences between inhibitor’s efficacies were not statistically significant.
Figure 3. Inhibitors of cellular signaling pathways decrease expression of some of the proteins involved in LPS-induced response in RAW 264.7 cells. Expression of proteins was measured in cells lysed by boiling in Laemmli sample buffer. Equal amounts of protein were analyzed by Western blot analysis using corresponding antibodies. The values shown at the bottom in the Table were calculated as relative units and are the results of tested protein blots densitometry by program QAPA from four independent experiments, values are expressed as a percentage of control. Control values of the left blots were in all cases assumed to be 100%; tubulin is shown on a parallel blotting panel. Value is significantly different from LPS (*p < 0.05, **p < 0.01) or from IKK Inhibitor XII (^p < 0.05).
Effects of inhibitors of TLR4, NF-κB, and SAPK/JNK cascades on the activation of NF-κB and SAPK/JNK signaling pathways
To reveal possible mechanisms of the inhibitor effects on LPS-mediated responses, the activation of two signal cascades, NF-κB and SAPK/JNK, was evaluated by measuring phosphorylation of some signal proteins. Indeed, the phosphorylation of NF-κB is an essential step in the pathway, ensuring NF-κB translocation to the nucleus and induction of gene expression. We have found that the LPS treatment resulted in a significant increase in NF-κB phosphorylation, while the pre-treatment of RAW 264.7 cells with inhibitor CLI-095 or IKK Inhibitor XII down-regulated phosphorylation of NF-κB (). In contrast, the effect of the JNK pathway inhibitor SP600125 on NF-κB phosphorylation was insignificant. This finding is expected based on the inter-relationship between the pathways (). JNK phosphorylation is a marker of the SAPK/JNK pathway activation, which in turn induces an activation of the transcription factor AP1 (see ). We examined CLI-095, IKK Inhibitor XII, and SP600125 as potential negative regulators of LPS-mediated activation of SAPK/JNK signaling. In the RAW 264.7 cells, LPS induced a significant increase in JNK1 and JNK2 phosphorylation. Western blot analysis showed that the pre-treatment of RAW 264.7 cells with CLI-095 or SP600125 resulted in an unequal reduction in the levels of the ph-SAPK/JNK1 and ph-SAPK/JNK2. It should be noted that the NF-κB-pathway inhibitor (IKK Inhibitor XII) significantly decreased activity of the pro-apoptotic form of SAPK/JNK (JNK2) and increased the SAPK/JNK1, which is usually associated with cell survival. Taken together, these data suggests that the phosphorylation of SAPK/JNK was significantly increased after LPS application, and all of the inhibitors prevented the rise in production of the phosphorylated p54 isoform of SAPK/JNK protein.
Inhibitors prevented over-production of HSP72 and HSP90-α in LPS-treated cells
In animal models as well as in cell cultures, an expression of heat shock proteins (HSP) plays a central role in promoting cell survival after stress induced by many damaging factors. Using inflammation-bearing mice, we have recently reported that both HSP72 and HSP90α are involved in LPS-induced pro-inflammatory cell responses (CitationNovoselova et al., 2009). Therefore, we next evaluated whether LPS could also influence HSP72 and HSP90α expression. To analyze the defense system in macrophages, production of HSP72 and HSP90 (inducible forms of heat-shock proteins) were measured in RAW 264.7 cells with or without TLR4, JNK, and IKK inhibitors. In cells treated with LPS, the expected increase in the total concentrations of both HSP72 and HSP90α was observed (). The results showed that all three inhibitors individually significantly decreased both HSP72 and HSP90α levels in these macrophages. It is of note that the inhibitory effect of SP600125 was significantly greater than that of the IKK Inhibitor XII.
Discussion
The present study aimed to compare the effects of several inhibitors of cellular signaling on the response to LPS in murine RAW 264.7 macrophage-like cells. Our findings indicate that RAW 264.7 cells showed a potent inflammatory response to LPS as visualized by the induced cytokine and NO production. In addition, the LPS application resulted in the activation of NF-κB and SAPK/JNK signaling, plus an increase of the expression of inducible HSP72 and HSP90 proteins. Therefore, it can be concluded that RAW 264.7 macrophages are a suitable cellular model to study the effects of signal transduction inhibitors.
The results of the present study demonstrate the difference in the anti-toxic efficacy of inhibitors of the TLR4, NF-κB and JNK pathways in RAW 264.7 macrophage-like cells. Indeed, treatment of the cells with CLI-095 (a TLR4 inhibitor) that binds directly to a specific amino acid, Cys747, in the intracellular domain of TLR4 (O’Neill et al., Citation2009), suppressed LPS-mediated TLR4 expression. This inhibitor also decreased NF-κB and SAPK/JNK activation, reduced expression of heat shock proteins HSP72 and HSP90, lowered production of NO, and suppressed production of cytokines, namely TNF-α, IL-1, and IFNγ.
IKK Inhibitor XII, a cell-permeable amino-diarylbenzamide compound that acts as a potential ATP site targeting inhibitor against IKK-1 and IKK-2, had more pronounced effects than inhibitor CLI-095. In fact, pre-treatment of cells by IKK Inhibitor XII led to the greatest reduction in IL-1, IL-2, IL-6, TNF-α, and IFNγ production in LPS-treated cells. Interestingly, Inhibitor XII, a selective inhibitor of NF-κB signaling, which functions by decreasing NF-κB phosphorylation, thus reducing the probability of translocation of the NF-κB into the nucleus, also prevented SAPK/JNK activation in LPS-treated cells. These results coincide with findings demonstrating that SAPK/JNK signaling is regulated by the NF-κB pathway (Papa et al., Citation2006). Indeed, it is generally suggested that NF-κB activation plays a critical role in LPS-induced responses, and suppression of the NF-κB pathway activity prevents its ability to activate the transcription of a wide variety of genes encoding immunologically relevant proteins. The appropriate mechanisms are shown in .
Interestingly, the selective JNK1, -2, and -3 inhibitor SP600125 decreased the SAPK/JNK cascade activity but did not affect the NF-κB signaling. At the same time, there is direct evidence showing that NF-κB stimulated by TNFα exerts a control on apoptosis by engaging in a negative regulation of the SAPK/JNK pathway (CitationPapa et al., 2004; Luo et al., Citation2005). On the other hand, the JNK pathway could regulate NF-κB-mediated gene transcription through its phosphorylation and activation of c-Jun (Tuyt et al., Citation1999). Subsequently, it can be assumed that interaction between the two signaling cascades, NF-κB and SAPK/JNK, is dependent on various asymmetrical mechanisms. SP600125 also suppressed LPS-mediated HSP90, HSP72, and TLR4 over-expression as well as TNFα, IL-1, IL-6, IL-10, and IFNγ production, but did not change the degree of NO production.
It is well established that intracellular events that lead to the activation of NF-κB are complex. They involve phosphorylation and proteolytic reactions and seem to be controlled by the cellular redox status. Thus, the intensive production of reactive oxygen species, including NO associated with LPS application, generally results in oxidative stress (Macdonald et al., Citation2003; CitationNovoselova et al., 2009). Some authors have assumed that the expression of the iNOS gene is regulated by the activation of both NF-κB and STAT1 signaling, but it does not touch on the JNK pathway (Gao et al., Citation1998; Ratajczak-Wrona et al., Citation2011). Indeed, in the present study, we have shown that inhibition of JNK (by SP600125) incited a weaker anti-inflammatory effect when compared to CLI-095 or IKK Inhibitor XII.
Based on these findings, it can be concluded that the greatest protective effect in LPS-treated cells can be achieved through inhibition of NF-κB signaling (IKK Inhibitor XII). So, the critical role of NF-κB activation, and the effectiveness of inhibitors in preventing the NF-κB activation in LPS-stimulated cells indicate that targeting NF-κB is a promising therapeutic strategy with respect to septic pathologies.
Stimulation of macrophages by LPS results in the formation of HSP72 and HSP90 clusters with known LPS-binding receptors: TLR4, CD14, and MD2 (Triantafilou et al., Citation2008). Moreover, HSP70 over-expression may regulate LPS-induced cytokine expression through NF-κB (Dokladny et al., Citation2010). Nevertheless, our findings indicate that the expression of HSP72 and HSP90-α was significantly reduced in the RAW 264.7 cells pre-treated separately with all of the tested inhibitors. However, the maximal inhibitory effect on the production of heat shock proteins in LPS-stimulated RAW 264.7 cells resulted from the treatment with SP600125. This finding confirms the potential efficacy of SAPK/JNK inhibitor as an anti-stress agent.
The TLR4 signaling pathway has shown itself to be an important therapeutic target for treatment of a septic-related pathology. Natural and synthetic inhibitors of the TLR4 signaling pathway are known and act at different stages of the signal transduction (Loiarro et al., Citation2010; Wittebole et al., Citation2010; Zhu and Mohan, Citation2010). However, none of these drugs have been introduced into clinical practice for inflammation and sepsis treatment, and the mechanisms underlying their action are still unclear. Additional laboratory and clinical investigations are required to confirm and to compare their molecular mechanisms and possible side-effects. Some TLR4 antagonists have already been investigated and it should be noted that they caused side-effects. Indeed, blocking TLR4 may lead to ‘inappropriate’ immune responses, such as allergic T-helper (TH)-2 responses or immunological tolerance (Broad et al., Citation2006; Ishii et al., Citation2006). Moreover, due to the ubiquitous involvement of these molecules in many biological processes, modulation of their activity could manifest in unforeseen outcomes. Given this overlap, the blockade of a single pathway may be merely inefficient, but has the potential to be highly deleterious (Thiefes et al., Citation2005; Haddad and Abdel-Karim, Citation2011). Therefore, anti-toxic protection by specific inhibitors of cellular signaling requires further study of the essential mechanisms that control toxic signals.
Conclusions
In conclusion, the findings of the present study confirmed the ability of several inhibitors of a signal transduction system to reduce the LPS-mediated response in RAW 264.7 cells. As a whole, the efficacy of IKK Inhibitor XII against the LPS-induced pro-inflammatory response was shown to be greater when compared to others.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper. This work was supported by the Russian Foundation for Support of Leading Scientific Schools (project SS-1853.2012.4), the Russian Foundation for Basic Research: projects No 10-04-00351-a, No 11-04-00023-а, and No 12-04-00113-а.
Related Research Data
References
- Akira, S., Uematsu, S., Takeuchi, O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801.
- Bennett, B. L., Sasaki, D. T., Murray, B. W., O’Leary, E. C., Sakata, S. T., Xu, W., Leisten, J. C., Motiwala, A., Pierce, S., Satoh, Y., Bhagwat, S. S., Manning, A. M., Anderson, D. W. 2001. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 98:13681–13686.
- Broad, A., Jones, D. E., Kirby, J. A. 2006. Toll-like receptor (TLR) response tolerance: A key physiological “damage limitation” effect and an important potential opportunity for therapy. Curr. Med.Chem. 13:2487–2502.
- Christopher, J. A., Avitabile, B. G., Bamborough, P., Champigny, A. C., Cutler, G. J., Dyos, S. L., Grace, K. G., Kerns, J. K., Kitson, J. D., Mellor, G. W., Morey, J. V., Morse, M. A., O’Malley, C. F., Patel, C. B., Probst, N., Rumsey, W., Smith, C. A., Wilson, M. J. 2007. The discovery of 2-amino-3,5-diarylbenzamide inhibitors of IKKα and IKKβ kinases. Bioorg. Med. Chem. Lett. 17:3972–3977.
- Dokladny, K., Lobb, R., Wharton, W., Ma, T. Y., Moseley, P. L. 2010. LPS-induced cytokine levels are repressed by elevated expression of HSP70 in rats: Possible role of NF-κB. Cell Stress Chaperon. 15:153–163.
- Gao, J. J., Filla, M. B., Fultz, M. J., Vogel, S. N., Russell, S. W., Murphy, W. J. 1998. Autocrine/paracrine IFNγ mediates the lipopolysaccharide-induced activation of transcription factor Stat1 in mouse macrophages: Pivotal role of Stat1 in induction of the inducible nitric oxide synthase gene. J. Immunol. 161:4803–4810.
- Glushkova, O. V., Novoselova, T. V., Khrenov, M. O., Parfenyuk, S. B., Lunin, S. M., Fesenko, E. E., Novoselova, E. G. 2010. Role of heat shock protein hsp90 in formation of protective reactions in acute toxic stress. Biochemistry (Moscow) 75:702–707.
- Haddad, J. J., Abdel-Karim, N. E. 2011. NF-κB Cellular and molecular regulatory mechanisms and pathways: Therapeutic pattern or pseudo-regulation. Cell. Immunol. 271:5–14.
- Ishii, K. J., Uematsu, S., Akira, S. 2006. ‘Toll’ gates for future immunotherapy. Curr. Pharm. Des. 12:4135–4142.
- Kawai, T., Akira, S. 2009. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 21:317–337.
- Krishnan, J., Selvarajoo, K., Tsuchiya, M., Lee, G., Choi, S. 2007. Toll-like receptor signal transduction. Exp. Mol. Med. 39:421–438.
- Loiarro, M., Ruggiero, V., Sette, C. 2010. Targeting TLR/IL-1R signalling in human diseases. Mediat. Inflamm. Available online at: http://www.hindawi.com/journals/mi/2010/674363/. Accessed 10 May 2012
- Luo, J. L, Kamata, H., Karin, M. 2005. IKK/NFкB signaling: Balancing life and death - a new approach to cancer therapy. J. Clin. Invest. 115:2625–2632.
- Macdonald, J., Galley, H. F., Webster, N. R. 2003. Oxidative stress and gene expression in sepsis. Brit. J. Anaesth. 90:221–232.
- Medvedev, A. E., Kopydlowski, K. M., Vogel, S. N. 2000. Expression chemokine, and toll-like receptor 2 and 4 gene macrophages: Deregulation of cytokine, transduction in endotoxin-tolerated mouse inhibition of lipopolysaccharide-induced signal. J. Immunol. 164:5564–5574.
- Medzhitov, R., Preston-Hurlburt, P. Jr., andJaneway, C. A. 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394–397.
- Nakamura, L. T., Wu-Hsieh, B. A., Howard, D. H. 1994. Recombinant murine γ-interferon stimulates macrophages of the RAW cell line to inhibit intracellular growth of Histoplasma capsulatum. Infect. Immun. 62:680–684.
- Novoselova, E. G., Glushkova, O. V., Cherenkov, D. A., Parfenyuk, S. B., Novoselova, T. V., Lunin, S. M., Khrenov, M. O., Guzhova, I. V., Margulis, B. A., Fesenko, E. E. 2006. Production of heat shock proteins, cytokines, and nitric oxide in toxic stress. Biochemistry (Moscow) 71:376–383.
- Novoselova, E. G., Lunin, S. M., Novoselova, T. V., Khrenov, M. O., Glushkova, O. V., Avkhacheva, N. V., Safronova, V. G., Fesenko, E. E. 2009. Naturally occurring anti-oxidant nutrients reduce inflammatory response in mice. Eur. J. Pharmacol. 615:234–240.
- O’Neill, L. A., Bryant, C. E., Doyle, S. L. 2009. Therapeutic targeting of Toll-like receptors for infectious and inflammatory diseases and cancer. Pharm. Rev. 61:177–197.
- Papa, S., Bubici, C., Zazzeroni, F., Pham, C. G., Kuntzen, C., Knabb, J. R., Dean, K., Franzoso, G. 2006. The NF-κB-mediated control of the JNK cascade in the antagonism of programmed cell death in health and diseases. Cell Death Differ. 13:712–729.
- Papa, S., Zazzeroni, F., Pham, C. G., Bubici, C., Franzoso, G. 2004. Linking JNK signaling to NF-кB: A key to survival. J. Cell Sci. 117:5197–5208.
- Ratajczak-Wrona, W., Jablonska, E., Garley, M., Jablonski, J., Radziwon, P. 2011. Effect of N-nitrosodimethylamine on inducible nitric oxide synthase expression and production of nitric oxide by neutrophils and mononuclear cells: The role of JNK signalling pathway. Acta Pathol. Microbiol. Immunol. Scand. 119:431–441.
- Schumann, R. R., Leong, S. R., Flaggs, G. W., Gray, P. W., Wright, S. D., Mathison, J. C., Tobias, P. S., Ulevitch, R. J. 1990. Structure and function of lipopolysaccharide binding protein. Science 249:1429–1431.
- Shimazu, R., Akashi, S., Ogata, H., Nagai, Y., Fukudome, K., Miyake, K., Kimoto, M. 1999. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 189:1777–1782.
- Strober, W. 1997. Trypan blue exclusion test of cell viability. Curr. Prot. Immunol. 21:A.3B.1–A.3B.2.
- Thiefes, A., Wolter, S., Mushinski, J. F., Hoffmann, E., Dittrich-Breiholz, O., Graue, N., Dorrie, A., Schneider, H., Wirth, D., Luckow, B., Resch, K., Kracht, M. 2005. Simultaneous blockade of NF-κB, JNK, and p38 MAPK by a kinase-inactive mutant of the protein kinase TAK1 sensitizes cells to apoptosis and affects a distinct spectrum of tumor necrosis target genes. J. Biol. Chem. 280:27728–27741.
- Towbin, H., Staehelin, T., Gordon, J. 1976. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some application. Proc. Natl. Acad. Sci. USA 76:4350–4354.
- Triantafilou, M., Sawyer, D., Nor, A., Vakakis, E., Triantafilou, K. 2008. Cell surface molecular chaperones as endogenous modulators of the innate immune response. Novartis Found. Symp. 291:74–79.
- Tuyt, L. M., Dokter, W. H., Birkenkamp, K., Koopmans, S. B., Lummen, C., Kruijer, W., Vellenga, E. 1999. Extracellular-regulated kinase 1/2, Jun N-terminal kinase, and c-Jun are involved in NF-κB-dependent IL-6 expression in human monocytes. J. Immunol. 162:4893–4902.
- Vega, V. L., de Maio, A. 2003. Geldanamycin treatment ameliorates the response to LPS in murine macrophages by decreasing CD14 surface expression. Mol. Biol. Cell. 14:764–773.
- Wittebole, X., Castanares-Zapatero, D., Laterre, P. F. 2010. Toll-like receptor 4 modulation as a strategy to treat sepsis. Mediat. Inflamm. Available online at: http://www.hindawi.com/ journals/mi/2010/568396, accessed 2 March 2010.
- Wright, S. D., Ramos, R. A., Tobias, P. S., Ulevitch, R. J., Mathison, J. C. 1990. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 249:1431–1433.
- Zeytun, A., Chaudhary, A., Pardington, P., Cary, R., Gupta, G. 2010. Induction of cytokines and chemokines by Toll-like receptor signaling: strategies for control of inflammation. Crit. Rev. Immunol. 30:53–67.
- Zhu, J., Mohan, C. 2010. Toll-like receptor signaling pathways - therapeutic opportunities. Mediat. Inflamm. Available online at: http://www.hindawi.com/journals/mi/2010/781235/, accessed 20 July 2010.
- Zughaier, S. M., Zimmer, S. M., Datta, A., Carlson, R. W., Stephens, D. S. 2005. Differential induction of the toll-like receptor 4-MyD88-dependent and -independent signaling pathways by endotoxins. Infect. Immun. 73:2940–2950.