Abstract
Contact dermatitis is the second most reported occupational injury associated with workers compensation. Inflammatory cytokines are closely involved with the development of dermatitis, and their modulation could exacerbate skin damage, thus contributing to increased irritancy. IL-6 is a pro-inflammatory cytokine paradoxically associated with both skin healing and inflammation. To determine what role this pleiotropic cytokine plays in chemically-induced irritant dermatitis, IL-6 deficient (KO), IL-6 over-expressing transgenic (TgIL6), and corresponding wild-type (WT) mice were exposed to acetone or the irritants JP-8 jet fuel or benzalkonium chloride (BKC) daily for 7 days. Histological analysis of exposed skin was performed, as was tissue mRNA and protein expression patterns of inflammatory cytokines via QPCR and multiplex ELISA. The results indicated that, following JP-8 exposure, IL-6KO mice had greatly increased skin IL-1β, TNFα, CCL2, CCL3, and CXCL1 mRNA and corresponding product protein expression when compared to that of samples from WT counterparts and acetone-exposed control mice. BKC treatment induced the expression of all cytokines examined as compared to acetone, with CCL2 significantly higher in skin from IL-6KO mice. Histological analysis showed that IL-6KO mice displayed significantly more inflammatory cell infiltration as compared to WT and TgIL6 mice in response to jet fuel. Analysis of mRNA for the M2 macrophage marker CD206 indicated a 4-fold decrease in skin of IL-6KO mice treated with either irritant as compared to WT. Taken together, these observations suggest that IL-6 acts in an anti-inflammatory manner during irritant dermatitis, and these effects are dependent on the chemical nature of the irritant.
Keywords::
Introduction
Of the reported occupational injuries associated with workers compensation, contact dermatitis ranks second most prevalent of all (Beltrani, Citation2003) and can affect workers in all industries. Contact dermatitis is divided into two main manifestations, those of allergic and irritant dermatitis. The major difference between the two pathologies is often described as whether the disease is of immunological origin (allergic), where T-cells are involved, or non-immunological origin (irritant), where physical damage is thought to be the major initiating event. Dermatitis is generally characterized at the histological level by neutrophil and macrophage infiltration and at the molecular level by inflammatory cytokine production (Corsini and Galli, Citation2000). The major difference between the two types of contact dermatitis appears to be the source of inflammatory cytokines. Whereas allergic dermatitis depends on T-cells, irritant type depends initially on dermal or epidermal cell sources. Keratinocytes themselves are a reservoir of the primary inflammatory cytokine interleukin (IL)-1, and mere disruption of these cells causes release of this cytokine that, in turn, induces other inflammatory cytokines, such as chemokines and IL-6 (Sugawara et al., Citation2001). Thus, the skin itself acts as an immune organ and can initiate inflammation without the assistance of lymphocytic cells. Acute skin inflammation is actually protective as it serves as a defense against infection, clears cell debris, and is closely associated with healing (Martin, Citation1997; Gallucci et al., Citation2000). It is only when inflammation is chronic that it becomes pathologic.
Numerous skin irritants have been identified, including detergents, silica, dusts, food, solvents, lubricants, machine oils, and even water (Mathias, Citation1988). Irritants can be sub-classified based on the type of pathology initiated by exposure (Effendy et al., Citation2000; Beltrani, Citation2003). Acute irritant contact dermatitis is caused by overt and rapid damage to the skin, usually by corrosive substances such as acids or bases. Acute delayed irritant contact dermatitis is caused by substances that elicit an inflammatory response well after exposure, e.g., the widely-used disinfectant benzalkonium chloride (BKC). Machining or cutting oils (Morris and Maloof, Citation1952) and jet fuel (Koschier, Citation1999) may also fall into this classification, as usually repeated contact is required for pathology to ensue. However, these petroleum-based mixtures seem to have a far more complex mechanism than merely barrier disruption (McDougal and Robinson, Citation2002; Monteiro-Riviere et al., Citation2006).
While inflammation is a common link between irritant and allergic dermatitis, the mechanism of the latter has been more closely investigated. Chemicals that promote allergic dermatitis tend to induce a specific cytokine profile wherein T-cell chemotactic (CXCR3) chemokines such as CXCL10 are induced (Flier et al., Citation1999; Meller et al., Citation2007). However, classifying and predicting the inflammatory response elicited by irritants is less cut and dry. As often referenced, Patrick et al. (Citation1987) stated, ‘… chemicals do not produce skin irritation by a common inflammatory pathway’ (P. 24).
One inflammatory cytokine that has been alternately associated with both allergic and irritant dermatitis is IL-6. Similar to chemokines, this cytokine is also induced by immediate early inflammatory cytokines such as IL-1 and tumor necrosis factor (TNF)-α. In addition to its immunomodulatory activities, IL-6 is involved in the growth and differentiation of numerous cell types, including those of dermal and epidermal origin (Sehgal, Citation1990), and is closely linked to skin wound healing (Gallucci et al., Citation2000; Lin et al., Citation2003). IL-6 treatment also appears to modulate stratum corneum regeneration and skin barrier function (Wang et al., Citation2004) to maintain skin homeostasis.
Previous work has shown that JP-8 differentially modulates IL-6 mRNA and protein expression in the skin of rats, following 5 and 7 days of JP-8 exposure (Gallucci and Mickle, Citation2006). Since IL-6 is closely linked to wound healing (Gallucci et al., Citation2000) and inflammation—where it is known to modulate the expression of various chemokines that influence inflammatory cell trafficking (Hurst et al., Citation2001; Fielding et al., Citation2008)—modulation of its gene could profoundly affect the pathology of dermatitis. Indeed, the expressions of specific chemokines that influence the recruitment of monocytes and dendritic cells (i.e., CCL2 and CCL3), PMN (i.e., CXCL1), as well as T-cells (i.e., CXCL10) were altered in skin following JP-8 exposure (Gallucci et al., Citation2004; Gallucci and Mickle, Citation2006). Thus, the purpose of this study was to further investigate the observation that IL-6 protein was decreased in dermatitis lesions caused by JP-8 jet fuel (Gallucci and Mickle, Citation2006), an irritant associated with moderate-to-severe skin pathology.
Materials and methods
Animals
IL-6KO and WT C57BL/6, as well as CD-1 mice of 8–12-weeks-old, were acquired from the Jackson Laboratory (Bar Harbor, ME). Transgenic IL-6 over-expressing mice (TgIL6) on the CD-1 background (graciously provided by Elaine Fuchs, Rockefeller University) (Turksen et al., Citation1992) were bred in-house. Mice were group-housed in polycarbonate cages containing hardwood chip bedding at room temperature (21 ± 3°C) on a 12-h light/dark cycle. Animals were allowed to acclimate to the animal facility for at least 1 week prior to JP-8 exposure. Throughout the studies, animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health (Citation2011).
Dermal JP-8 exposure
One day prior to exposure, mice were sedated with Isoflurane and a ~ 9-cm2 section of fur was clipped. Treatments were initiated 24 h post-hair removal to ensure that minimal irritation occurred from the hair removal process. Treatment groups (n = 8/chemical) of each mouse genotype and the respective WT were placed in separate fume hoods to minimize exposure to JP-8 vapor. Filtered (0.45 µm) JP-8 (lot #UN1863) was provided by the Air Force Research Laboratory (AFRL/HEPB) at Wright-Patterson Air Force Base and the Air Force Office for Scientific Research (AFOSR). Jet fuel (neat), 1% aqueous benzalkonium chloride (Sigma, St. Louis, MO) (BKC, positive control), or acetone (negative control) was applied (100 µl) to the denuded skin of the animals daily for 7 consecutive days.
Twenty-four hours following the final exposure, skin samples were collected via a 4-mm full thickness punch biopsy. Harvested skin was immediately homogenized in TriReagent (Molecular Resource Center, Cincinnati, OH) for RNA or RIPA buffer plus PMSF and protease inhibitor (Sigma) for protein isolation or fixed in 10% buffered formalin for histology.
Real-time quantitative RT-PCR
Total RNA from mouse skin was prepared and cDNA was synthesized using random decamer primers essentially as previously described (Simeonova and Luster, Citation1995). Primers were produced utilizing Genbank sequences and synthesized by Invitrogen (Grand Island, NY). Real-time quantitative RT-PCR was performed on an ABI PRISM 7000 SDS (Life Technologies, Carlsbad, CA). Results are reported as percent (%) acetone-treated C57 control, as it was previously noted that these animals had minimal, if any, skin inflammation (Gallucci et al., Citation2004).
Pro-inflammatory cytokine protein expression
Total protein from frozen skin samples was prepared by homogenizing skin samples in RIPA buffer containing protease inhibitor cocktail (#P8340 Sigma, St. Louis, MO) as described (Luckett-Chastain and Gallucci, Citation2009). Skin expression of pro-inflammatory cytokines was determined by MILLIPLEX MAP multiplex assay (Millipore, Billerica, MA) and presented relative to total protein as determined by Bradford assay (BioRad, Hercules, CA).
Histopathology
Formalin-fixed skin biopsies were embedded in paraffin and 5-µm skin cross-sections were hematoxylin and eosin (H&E) stained. Digital images of the skin histopathology (under 20× objective) were acquired utilizing a Leica 4000b microscope (Leica Microsystems, Buffalo Grove, IL). Five random fields from each H&E-stained slide were analyzed for dark-stained inflammatory cells utilizing Image J (NIH, http://rsbweb.nih.gov/ij/) and data was presented as mean events (inflammatory cells) per field.
Statistical analysis
All experiments were replicated and representative findings are shown. Statistical significance was determined by two-way analysis of variance (ANOVA) and Bonferoni post-hoc analysis. In all statistical comparisons, a p-value of < 0.05 indicated a significant difference.
Results
Modulation of pro-inflammatory cytokine expression in JP-8-treated mice
No outward signs of systemic toxicity, such as weight loss or overt behavioral changes, were noted with any skin irritant, regardless of strain (data not shown). While there were apparent strain differences between C57 and CD-1 WT mice, it was most applicable to compare each transgenic strain to respective WT strains. JP-8 exposure did not alter skin TNFα mRNA expression regardless of strain (, dark bars). However, protein expression was significantly higher in IL-6KO skin as compared to WT (, dark bars). BKC elicited a significantly difference response in skin. TNFα mRNA (, gray bars) and protein (, gray bars) levels were profoundly increased following BKC treatment of the IL-6KO animals (~25- and 5-fold, respectively); no difference was observed between TgIL6 and CD-1 controls.
Figure 1. BKC and JP-8 exposure induces TNF-α, but not IL-1β expression in mouse skin. Mice were treated daily for 7 days with acetone, benzalkonium chloride, or JP-8, and 4-mm skin biopsies were collected and processed for mRNA and protein analysis. Expression of (a) TNFα and (b) IL-1β mRNA was analyzed via real-time RT-PCR; expression differences were normalized to 28s rRNA expression and presented as a percentage of level in samples from acetone-treated C57BL/6 control animals. Skin (c) TNFα and (d) IL-1β protein expression was determined by Milliplex MAP multiplex ELISA as per manufacturer instructions. Data presented as means ± SE (n = 8). *Value significantly different from corresponding WT control (p ≤ 0.05).
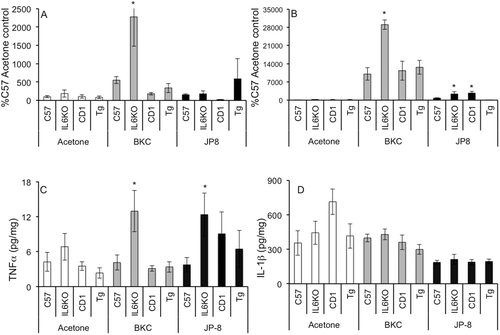
BKC treatment greatly induced IL-1β mRNA expression in all strains, with the greatest increase (~ 2.5-fold over WT levels) observed in IL-6KO animals (). Interestingly, despite this profound induction of mRNA, no significant differences were observed concerning IL-1β protein expression, regardless of irritant or mouse strain ().
Modulation of chemokine expression in skin of JP-8-treated mice
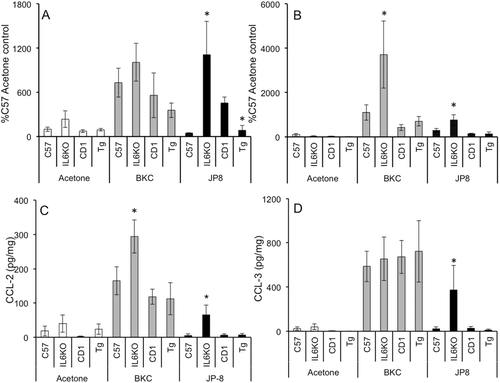
BKC treatment did not result in a significant increase in CCL2 mRNA expression in any strain (, gray bars), while CCL3 mRNA was significantly induced in IL-6KO mouse skin (, gray bars). Conversely, CCL2 protein was significantly induced following BKC exposure in IL-6KO mice (, gray bars), and CCL3 protein was induced nearly uniformly regardless of strain (). The effects of jet fuel exposure appeared to be markedly affected by IL-6 expression, where both CCL2 and CCL3 mRNA levels were significantly higher in IL-6KO mice ( and , dark bars). As well, CCL2 mRNA was significantly lower in Tg over-expressing mice as compared to that in respective WT hosts (, dark bars). Jet fuel-induced CCL2 and CCL3 protein levels ( and , dark bars) were both significantly increased during IL-6 deficiency, but not changed with over-expression.
Figure 2. BKC and JP-8 differentially modulate CCL2 and CCL3 expression in mouse skin. Mice were treated daily for 7 days with acetone, benzalkonium chloride, or JP-8, and 4 mm skin biopsies were collected and processed for mRNA and protein analysis. Expression of (a) CCL2 and (b) CCL3 mRNA was analyzed via real-time RT-PCR; expression differences were normalized to 28s rRNA expression and presented as a percentage of level in samples from acetone-treated C57BL/6 controls. Skin (c) CCL2 and (d) CCL3 protein expression was determined by Milliplex MAP multiplex ELISA as per manufacturer instructions. Data presented as means ± SE (n = 8). *Value significantly different from corresponding WT control (p ≤ 0.05).
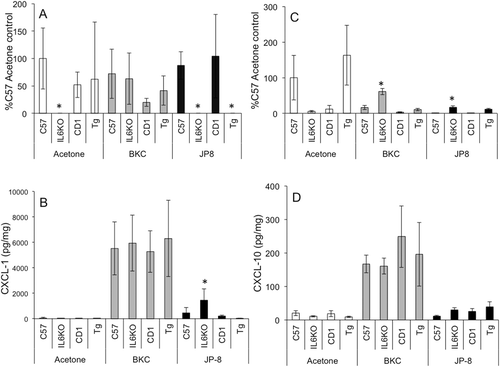
Interestingly, CXCL1 mRNA expression was decreased in acetone treated IL-6KO mice as compared to WT (, white bars). BKC exposure did not significantly change CXCL1 mRNA expression (, gray bars), while JP-8 treatment significantly reduced expression in both IL-6KO and TgIL6 mice compared to levels in respective WT controls. CXCL10 mRNA expression was only increased in IL-6KO skin in response to either irritant (). Similar to CCL3, the expression of CXCL1 and CXCL10 proteins were almost uniformly induced by BKC exposure animals, regardless of strain (, gray bars). JP-8 exposure induced CXCL1 protein exclusively in IL-6KO mice (, dark bars); no differences were noted in CXCL10 expression in any mouse strain ().
Figure 3. BKC and JP-8 differentially induced CXCL1 and CXCL10 expression in mouse skin. Mice were treated daily for 7 days with acetone, benzalkonium chloride, or JP-8, and 4 mm skin biopsies were collected and processed for mRNA and protein analysis. Expression of (a) CXCL1 and (b) CXCL10 mRNA was analyzed via real-time RT-PCR; expression differences were normalized to 28s rRNA expression and presented as a percentage of level in samples from acetone-treated C57BL/6 controls. Skin (c) CXCL1 and (d) CXCL10 protein expression was determined by Milliplex MAP multiplex ELISA as per manufacturer instructions. Data presented as means ± SE (n = 8). * Value significantly different from corresponding WT control (p ≤ 0.05).
IL-6 expression alters inflammatory cell infiltration into skin
Treatment with either BKC or JP-8 resulted in obvious erythema in all strains, but clinical scores based on visualization did not significantly differ between the irritants (data not shown). To examine whether changes in cytokine expression were associated with inflammatory cell migratory changes, skin samples from BKC- and JP-8-treated WT, IL-6KO, and TgIL6 mice were collected for histological examination. Image analysis of hematoxylin and eosin (H&E)-stained skin sections showed that, while both irritants induced similar inflammatory cell infiltration, JP-8 treatment revealed significant differences with respect to genotype. Specifically, following JP-8 treatment, WT controls displayed similar leukocyte migration (), IL-6KO skin ( and ) showed significantly higher inflammatory infiltrate, whereas TgIL6 animals appeared to display the lowest ( and ). Acetone exposure induced the migration of few, if any, inflammatory cells into the skin, regardless of strain (data not shown).
Figure 4. IL-6 deficiency results in greater skin inflammatory cell infiltration following irritant exposure. Mice were treated daily for 7 days with acetone, benzalkonium chloride, or JP-8, and 4-mm skin biopsies were collected and processed for mRNA and protein analysis, or embedded in paraffin for histological analysis. (a) Digital images of skin histopathology from JP-8-treated mice were acquired (under 20× objective) and analyzed for dark-stained inflammatory cells using Image J (NIH); data are represented by mean events (inflammatory cells) per field. Shown are representative images of H&E-stained JP-8-treated skin: (b) IL-6KO and (c) Tg(IL6). Expression of (d) CD86 and (e) CD206 mRNA was analyzed via real-time RT-PCR; expression differences were normalized to 28s rRNA expression and presented as a percentage of level in samples from acetone-treated C57BL/6 controls. Data presented as means ± SE (n = 8). * Value significantly different from corresponding WT control (p ≤ 0.05).
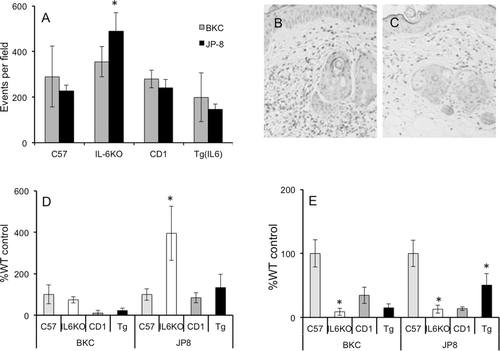
While the vast majority of inflammatory cells appeared to be neutrophils (PMN; and ), it was of interest to assess whether chemical or mouse strain affected discrete macrophage population infiltration into skin following chemical exposure. Since macrophage populations were quite low, histological analysis was difficult to interpret. Thus, to examine this, CD86 (M1, or classical) and CD206 (M2, healing or anti-inflammatory) mRNA expression was assessed in skin samples. Corresponding with inflammatory cytokine expression, JP-8-treated IL-6KO animals had the highest levels of CD86 mRNA expression compared to levels in WT counterparts (). Likewise, CD206 mRNA expression was significantly lower in IL-6KO skin regardless of chemical exposure (). IL-6 over-expression only seemed to affect CD206 mRNA expression in JP-8 treated skin, where it was significantly increased ().
Discussion
It is believed that the overall mechanism of JP-8-induced dermatitis involves increased oxidative stress or the physical disruption of membranes, which initiates irritant responses such as inflammation, growth, proliferation, and apoptosis (McDougal and Garrett, Citation2007). In previous studies, this laboratory has shown that specific cytokine profiles in skin were observed following JP-8 exposure and that the inflammatory cytokine IL-6 was modulated disparately at the mRNA and protein levels in rats (Gallucci et al., Citation2004; Gallucci, and Mickle, Citation2006). However, it was not known what role IL-6 played in the pathology of JP-8 irritant dermatitis, or if these observed effects were specifically associated with jet fuel exposure. To further investigate these findings, IL-6-deficient and transgenic IL-6-over-expressing mouse models were utilized to examine the effects of IL-6 on the severity of dermatitis caused by JP-8 and another well-characterized irritant, benzalkonium chloride. Indeed, IL-6 deficiency resulted overall in increased inflammation that appears to be associated with modulation of specific cytokines and chemokines. The type of irritant also played a significant role in alteration of cytokine and chemokine expression, whereas jet fuel appeared to induce a much more intense and complex response.
Inflammation and the resultant recruitment of inflammatory cells into irritant exposed skin have been associated with increased primary inflammatory cytokine expression. Indeed, here it was shown that TNFα and IL-1β were variably modulated based on irritant and mouse strain. Notably, IL-6 deficiency greatly increased TNFα mRNA and protein expression following either BKC or JP-8 exposure as compared to outcomes seen in C57 WT control mice (see and ). Previous studies utilizing rats have reported increases in both IL-1α and IL-1β in response to jet fuel exposure (Kabbur et al., Citation2001; Gallucci et al., Citation2004; Chatterjee et al., Citation2006; Gallucci and Mickle, Citation2006). In the present study, IL-1β mRNA expression was induced by either irritant, with the highest expression associated with IL-6 deficiency (see ). However, increased protein expression did not accompany this increase in mRNA, regardless of treatment or strain. This may indicate a post-transcriptional modulation or increased degradation of this cytokine in exposed skin (compare vs 1d) or, more likely, it may be associated with the ‘7 day’ timepoint at which samples were taken. Other studies have noted IL-1 protein levels early as 1 h post-exposure, indicating that the cytokine may originate from storage sites rather than via de novo synthesis (Kabbur et al., Citation2001). Indeed, McDougal and Garrett (Citation2007) postulated a mechanism by which JP-8 induces dermatitis, and the JP-8 causes release of pre-formed IL-1α from epidermal keratinocytes, thereby inducing a rapid activation of IL-6 signaling. However, the present study seems to indicate that of the two primary inflammatory cytokines, TNFα may dominate the later inflammatory response associated with these chemicals in mice rather than IL-1.
Since IL-6 is associated with wound healing (Gallucci et al., Citation2001) and skin barrier maintenance (Wang et al., Citation2004), it is tempting to speculate that increased skin damage caused by certain irritant chemicals may be associated with the modulation of this important cytokine. Based on the pro-inflammatory nature of IL-6, one might expect that Tg-IL-6 would have the most severe dermatitis, while mice lacking IL-6 (i.e., IL-6KO) would be expected to have the least amounts of these signs of skin inflammation. This in fact was not the case, as IL-6KO animals had the greatest and JP-8 treated Tg-IL-6 animals the least amount of inflammatory cell infiltration into skin (see ). Interestingly, the characterization of the Tg-IL-6 animals by Turksen et al. (Citation1992) produced similar findings. While these authors suggested that the IL-6 may need to work synergistically with other inflammatory factors, the observations reported here would indicate that, rather than promoting inflammation, IL-6 appears to suppress it in skin.
Chemokines, or chemotactic cytokines, have a range of activities that vary from exclusive neutrophil chemoattraction to T-cell infiltration. These cytokines can be up-regulated by numerous inflammatory stimuli, including primary inflammatory cytokines such as TNFα or IL-6. Previous studies utilizing rats (Gallucci et al., Citation2004; Gallucci and Mickle, Citation2006) showed significantly greater CCL2, CCL3, and CXCL1 mRNA expression following JP-8 exposure. In the present study, BKC exposure induced profound induction of these chemokines as well as CXCL10 at the protein level (see and , gray bars), with IL-6KO mice having significantly higher protein expression of CCL2. Conversely, JP-8 induction of CCL2, CCL3, and CXCL1 seemed much more associated with IL-6 deficiency (see and , dark bars), while CXCL10 was not induced in any strain by this chemical.
While novel, the link between IL-6 deficiency and irritant induced chemokine expression in this model is perplexing and becomes further complicated when considering jet fuel exposure. However, based on the fact that irritant dermatitis lesions are predominantly populated by PMN and monocytes, the overall observations made concerning the pattern of chemokine expression may not be surprising. CC chemokines attract a variety of cells depending on their receptor specificity, including monocytes, eosinophils, basophils, immature dendritic cells, and T-cells (Alam, Citation1997). Aside from monocytes, the latter cell types tend to be involved in chronic or hypersensitivity reactions and are not generally observed in simple irritant dermatitis such as that induced by JP-8 (Gallucci et al., Citation2004). Furthermore, it is well known that JP-8 inhibits acquired immune responses (Ullrich and Lyons, Citation2000; Ramos et al., Citation2002, Citation2007). CXC chemokines can be classified based on receptor specificity as either relatively strict neutrophil chemoattractants (CXCR1 or 2) or lymphocyte chemoattractants (CXCR3), wherein CXCR1/2 is associated with acute inflammation and PMN chemotaxis, and CXCR3 is associated with chronic inflammation (for review, see Baggiolini et al., Citation1998).
Indeed, it is well known that jet fuel is a very weak sensitizer and that the resulting inflammatory dermatitis is acute, lacking significant lymphocyte infiltration. Interestingly, allergic contact dermatitis is decreased in IL-6-deficient mice (Hope et al., Citation2000), and the lack of IL-6 signaling has been recently shown to increase neutrophil accumulation and decrease macrophage chemotaxis into a site of inflammation (Fielding et al., Citation2008). Neutrophil clearance is thought to be essential in resolution of acute inflammation, and the persistence of these cells may greatly exacerbate damage in the tissue from reactive oxygen species and protease production (Moraes et al., Citation2006). Indeed, this laboratory reported that JP-8 dermatitis was characterized almost exclusively by neutrophil infiltration (Gallucci et al., Citation2004). As shown previously (Gallucci and Mickle, Citation2006), and herein, the production of PMN-associated chemokines CXCL1 (see ), CCL2, and CCL3 dominate as compared to the lymphocyte chemoattractant CXCL10 and . These data seem to indicate IL-6 is an important discriminatory cytokine when assessing allergic vs irritant dermatitis.
The nature of chemokines is to attract cells to a site of inflammation. Based on the chemokine patterns shown in the present study relative to IL-6 deficiency, it was of interest to determine the populations of macrophages that traffic into the site relative to irritant and dermatitis severity. While historically considered inflammatory only, two different macrophage cell types have been described. The M1 macrophage is pro-inflammatory and produces inflammatory cytokines such as TNFα, while the M2 type is anti-inflammatory, producing cytokines such as IL-10 and transforming growth factor (TGF)-β, and thought to be associated with tissue repair (for review, see Laskin, Citation2009) and tumor progression (Mantovani et al., Citation2006). While the M2 macrophage appears to be protective, little is known concerning the effects of IL-6 on the trafficking or function of these cell types during irritant dermatitis. It was readily apparent that increased inflammatory cell infiltration was associated with IL-6 deficiency (see and ). Furthermore, assessment of the mRNA for the M1 marker CD86 showed the greatest expression in JP-8 treated skin of IL-6KO mice, suggesting greater inflammatory macrophage infiltration (see ). Not surprisingly, CD206 expression was significantly lower in IL-6KO animals as a result of exposure to either irritant (see ). More telling perhaps is that the ratio of CD86 to CD206 was ~ 10- and ~ 30-fold higher in the skin of IL-6KO mice exposed to BKC and JP-8 (respectively) as compared to in samples from WT controls (compare vs 4e). Indeed, while IL-6 over-expression did not significantly affect most parameters measured herein, TgIL6 mouse skin showed an ~ 2-fold higher CD206:CD86 ratio compared to that in skin from WT mice after JP-8 exposure (compare vs ). These data suggest that IL-6 may function in skin inflammation to shift the relative degree of macrophages recruited to skin to reflect a greater percentage of M2 macrophages, thus minimizing the influence of inflammatory M1 macrophages.
Summary and conclusions
In the studies reported here, it was shown that IL-6 deficiency resulted (overall) in increased inflammatory cytokine (i.e., IL1β and TNFα) and chemokine, i.e., CCL2, CCL3, and CXCL1 []) expression. However, these results differed markedly, depending on irritant. Furthermore, infiltration of inflammatory macrophages was higher, but M2 macrophage infiltration was lower, in the skin of irritant-exposed IL-6KO mice (). The cumulative observations of decreased inflammatory cell recruitment, decreased chemokine expression, decreased inflammatory cytokine expression, and altered macrophage recruitment suggest an anti-inflammatory rather than a pro-inflammatory role for IL-6 in JP-8-induced dermatitis. While IL-6 is traditionally viewed as a pro-inflammatory cytokine, it is also known to be extremely pleiotropic, and other researchers have reported anti-inflammatory activities of IL-6 (Xing et al., Citation1998). The observations presented herein strongly suggest an anti-inflammatory role for IL-6 in dermatitis (see ), and it is possible that variations in the expression or function of this cytokine may lead to modulation of irritant sensitivity in humans.
Table 1. Summary of chemical-induced skin cytokine expression relative to strain.
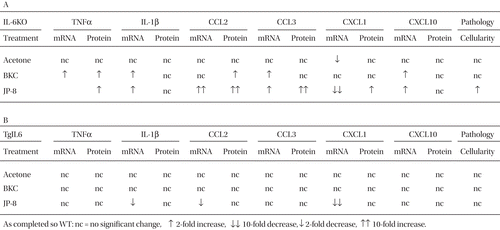
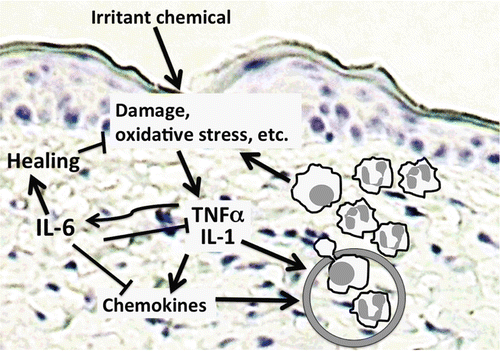
Figure 5. Proposed model of the role of IL-6 in irritant dermatitis. Following chemical exposure, damage or irritation occurs in the epidermis, causing release or synthesis of primary inflammatory cytokines like TNFα and IL-1. These, in turn, induce expression of chemokines as well as IL-6. Chemokines and primary inflammatory cytokines then act on the capillary endothelium to mediate inflammatory cell extravasation and accumulation in the tissue, resulting in further damage. IL-6 counters this by inhibiting the expression of primary inflammatory cytokines, as well as some chemokines. IL-6 itself mediates skin healing, and may promote the influx or differentiation of anti-inflammatory macrophage populations that further promote repair.
Acknowledgment
This work was funded by CDC/NIOSH grant 5R03OH009662.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- 2011. Guide for the Care and Use of Laboratory Animals. 8th ed. Washington DC.
- Alam, R. 1997. Chemokines in allergic inflammation. J. Allergy Clin. Immunol. 99:273–277.
- Baggiolini, M., Dewald, B., Moser, B. 1998. Chemokines and leukocyte traffic. Human chemokines: An update. Nature 392:565–568.
- Beltrani, V. S. 2003. Occupational dermatoses. Curr. Opin. Allergy Clin. Immunol. 3:115–123.
- Chatterjee, A., Babu, R. J., Klausner, M., Singh, M. 2006. In vitro and in vivo comparison of dermal irritancy of jet fuel exposure using EpiDerm (EPI-200) cultured human skin and hairless rats. Toxicol. Lett. 167:85–94.
- Corsini, E., Galli, C. L. 2000. Epidermal cytokines in experimental contact dermatitis. Toxicology 142:203–211.
- Effendy, I., Loffler, H., Maibach, H. I. 2000. Epidermal cytokines in murine cutaneous irritant responses. J. Appl. Toxicol. 20:335–341.
- Fielding, C. A., McLoughlin, R. M., McLeod, L., Colmont, C. S., Najdovska, M., Grail, D., Ernst, M., Jones, S. A., Topley, N., Jenkins, B. J. 2008. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J. Immunol. 181:2189–2195.
- Flier, J., Boorsma, D. M., Bruynzeel, D. P., van Beek, P. J., Stoof, T. J., Scheper, R. J., Willemze, R., Tensen, C. P. 1999. The CXCR3 activating chemokines IP-10, Mig, and IP-9 are expressed in allergic but not in irritant patch test reactions. J. Invest. Dermatol. 113:574–578.
- Gallucci, R. M., Mickle, B. 2006. Inflammatory cytokine expression patterns in rat skin exposed to JP-8 jet fuel. Am. J. Pharm. Toxicol. 1:48–53.
- Gallucci, R. M., O’Dell, S. K., Rabe, D., Fechter, L. D. 2004. JP-8 jet fuel exposure induces inflammatory cytokines in rat skin. Int. Immunopharmacol. 4:1159–1169.
- Gallucci, R. M., Simeonova, P. P., Matheson, J. M., Kommineni, C., Guriel, J. L., Sugawara, T., Luster, M. I. 2000. Impaired cutaneous wound healing in IL-6-deficient and immunosuppressed mice. FASEB J. 14:2525–2531.
- Gallucci, R. M., Sugawara, T., Yucesoy, B., Berryann, K., Simeonova, P. P., Matheson, J. M., Luster, M. I. 2001. Interleukin-6 treatment augments cutaneous wound healing in immunosuppressed mice. J. Interferon Cytokine Res. 21:603–609.
- Hope, J. C., Campbell, F., Hopkins, S. J. 2000. Deficiency of IL-2 or IL-6 reduces lymphocyte proliferation, but only IL-6 deficiency decreases the contact hypersensitivity response. Eur. J. Immunol. 30:197–203.
- Hurst, S. M., Wilkinson, T. S., McLoughlin, R. M., Jones, S., Horiuchi, S., Yamamoto, N., Rose-John, S., Fuller, G. M., Topely, N., Jones, S. A. 2001. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 14:705–714.
- Kabbur, M. B., Rogers, J. V., Gunasekar, P. G., Garrett, C. M., Geiss, K. T., Brinkley, W. W., McDougal, J. N. 2001. Effect of JP-8 jet fuel on molecular and histological parameters related to acute skin irritation. Toxicol. Appl. Pharmacol. 175:83–88.
- Koschier, F. J. 1999. Toxicity of middle distillates from dermal exposure. Drug Chem. Toxicol. 22:155–164.
- Laskin, D. L. 2009. Macrophages and inflammatory mediators in chemical toxicity: A battle of forces. Chem. Res. Toxicol. 22:1376–1385.
- Lin, Z. Q., Kondo, T., Ishida, Y., Takayasu, T., Mukaida, N. 2003. Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J. Leukocyte Biol. 73:713–721.
- Luckett-Chastain, L. R., Gallucci, R. M. 2009. Interleukin (IL)-6 modulates transforming growth factor-β expression in skin and dermal fibroblasts from IL-6-deficient mice. Br. J. Dermatol. 161:237–248.
- Mantovani, A., Schioppa, T., Porta, C., Allavena, P., Sica, A. 2006. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metast. Rev. 25:315–322.
- Martin, P. 1997. Wound healing - aiming for perfect skin regeneration. Science 276:75–81.
- Mathias, C. G. 1988. Occupational dermatoses. J. Am. Acad. Dermatol. 19:1107–1114.
- McDougal, J. N., Garrett, C. M. 2007. Gene expression and target tissue dose in the rat epidermis after brief JP-8 and JP-8 aromatic and aliphatic component exposures. Toxicol. Sci. 97:569–581.
- McDougal, J. N., Robinson, P. J. 2002. Assessment of dermal absorption and penetration of components of a fuel mixture (JP-8). Sci. Total Environ. 288:23–30.
- Meller, S., Laumerma, A. I., Kopp, F. M., Winterberg, F., Anthoni, M., Muller, A., Gombert, M., Haahtela, A., Alenius, H., Rieker, J., Dieu-Nosjean, M. C., Kubitza, R. C., Gleichmann, E., Ruzicka, T., Zlotnick, A., Homey, B. 2007. Chemokine responses distinguish chemical-induced allergic from irritant skin inflammation: Memory T-cells make the difference. J. Allergy Clin. Immunol. 119:1470–1480.
- Monteiro-Riviere, N. A., Inman, A. O., Barlow, B. M., Baynes, R. E. 2006. Dermatotoxicity of cutting fluid mixtures: In vitro and in vivo studies. Cutan. Ocul. Toxicol. 25:235–247.
- Moraes, T. J., Zurawska, J. H., Downey, G. P. 2006. Neutrophil granule contents in the pathogenesis of lung injury. Curr. Opin. Hematol. 13:21–27.
- Morris, G. E., Maloof, C. C. 1952. Cutting-oil dermatitis. Ind. Med. Surg. 21:573–574.
- Patrick, E., Burkhalter, A., Maibach, H. I. 1987. Recent investigations of mechanisms of chemically induced skin irritation in laboratory mice. J. Invest. Dermatol. 88:24s–31s.
- Ramos, G., Limon-Flores, A. Y., Ullrich, S. E. 2007. Dermal exposure to jet fuel suppresses delayed-type hypersensitivity: A critical role for aromatic hydrocarbons. Toxicol. Sci. 100:415–422.
- Ramos, G., Nghiem, D. X., Walterscheid, J. P., Ullrich, S. E. 2002. Dermal application of jet fuel suppresses secondary immune reactions. Toxicol. Appl. Pharmacol. 180:136–144.
- Sehgal, P. B. 1990. Interleukin-6: Molecular pathophysiology. J. Invest. Dermatol. 94:2S–6S.
- Simeonova, P. P., Luster, M. I. 1995. Iron and reactive oxygen species in the asbestos-induced TNFα response from alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 12:676–783.
- Sugawara, T., Gallucci, R. M., Simeonova, P. P., Luster, M. I. 2001. Regulation and role of IL-6 in wounded human epithelial keratinocytes. Cytokine 15:328–336.
- Turksen, K., Kupper, T., Degenstein, L., Williams, I., Fuchs, E. 1992. Interleukin-6: Insights to its function in skin by over-expression in transgenic mice. Proc. Natl. Acad. Sci. USA 89:5068–5072.
- Ullrich, S. E., Lyons, H. J. 2000. Mechanisms involved in the immunotoxicity induced by dermal application of JP-8 jet fuel. Toxicol. Sci. 58:290–298.
- Wang, X. P., Schunck, M., Kallen, K. J., Neumann, C., Trautwein, C., Rose-John, S., Proksch, E. 2004. The IL-6 cytokine system regulates epidermal permeability barrier homeostasis. J. Invest. Dermatol. 123:124–131.
- Xing, Z., Gauldie, J., Cox, G., Baumann, H., Jordana, M., Lei, X. F., Achong, M. K. 1998. IL-6 is an anti-inflammatory cytokine required for controlling local or systemic acute inflammatory responses. J. Clin. Invest. 101:311–320.