Abstract
The failure of regulatory science to keep pace with and support the development of new biological medicines was very publically highlighted in March 2006 when the first-in-man Phase I clinical trial of the immunomodulatory CD28-specific monoclonal antibody (mAb) TGN1412 ended in disaster when all six volunteers suffered a life-threatening adverse reaction termed a ‘Cytokine Storm’. The poor predictive value of standard pre-clinical safety tests and animal models applied to TGN1412 demonstrated the need for a new generation of immunotoxicity assays and animal models that are both sensitive and predictive of clinical outcome in man. The non-predictive result obtained from pre-clinical safety testing in cynomolgus macaques has now been attributed to a lack of CD28 expression on CD4+ effector memory T-cells that therefore cannot be stimulated by TGN1412. In contrast, high levels of CD28 are expressed on human CD4+ effector memory T-cells, the source of most TGN1412-stimulated pro-inflammatory cytokines. Standard in vitro safety tests with human cells were also non-predictive as they did not replicate in vivo presentation of TGN1412. It was subsequently shown that, if an immobilized therapeutic mAb-based assay or endothelial cell co-culture assay was used to evaluate TGN1412, then these would have predicted a pro-inflammatory response in man. New in vitro assays based on these approaches are now being applied to emerging therapeutics to hopefully prevent a repeat of the TGN1412 incident. It has emerged that the mechanism of pro-inflammatory cytokine release stimulated by TGN1412 is different to that of other therapeutic mAbs, such that standard pro-inflammatory markers such as TNFα and IL-8 are not discriminatory. Rather, IL-2 release and lymphoproliferation are optimal readouts of a TGN1412-like pro-inflammatory response.
Introduction
In March 2006, the Phase I clinical trial of TGN1412, a therapeutic CD28-specific monoclonal antibody (mAb), ended in a disaster that brought into question the safety of new biological medicines. TGN1412 was a new class of immunomodulatory therapeutic intended for the treatment of rheumatoid arthritis, leukemia and multiple sclerosis, but instead it caused near fatal reactions in all six healthy volunteers. Within 2 h of receiving a single intravenous dose of 0.1 mg/kg, all six volunteers had a systemic inflammatory response characterized by a rapid induction of pro-inflammatory cytokines, later termed a ‘cytokine storm’ (Suntharalingham et al., 2006). Within 16 h, all six volunteers became critically ill with acute lung injury, multiple organ failure, and disseminated intravascular coagulation that would result in amputation of fingers and toes for the worst affected. All six patients were transferred to an intensive care unit where they received intensive organ support and high-dose immunosuppressive therapy, but two developed prolonged cardiovascular shock and acute respiratory distress. Fortunately, all six patients survived but have been warned that they have a greatly increased chance of developing cancers and immune system disorders in later life. The company that developed TGN1412, TeGenero AG, filed for insolvency 4 months later.
The adverse drug response to TGN1412 had been unanticipated since it had passed a standard battery of pre-clinical safety tests without raising any alarms and been approved by both UK and German regulatory agencies for first-in-man Phase I clinical trials. Initially a contaminant was suspected of causing the adverse drug responses, but subsequent analysis showed that the trial material was not contaminated and had been correctly formulated (Duff, Citation2006; Stebbings et al., Citation2007). When standard pre-clinical safety tests were independently repeated, using recovered clinical trial material, no pro-inflammatory cytokine release could be detected either in vitro with human white blood cells or when infused into cynomolgus macaques (Duff, Citation2006; Stebbings et al., Citation2007). The conclusion was that an unexpected biological effect of TGN1412 in man had caused an adverse drug response that was not predicted by pre-clinical safety testing (Duff, Citation2006; Stebbings et al., Citation2009). This posed a serious dilemma for regulators asked to grant licences for trials of new biological medicines where the potential to cause a ‘cytokine storm’ in man could not be confidently assessed. Concomitantly, fears of another TGN1412 incident made it difficult for research-based pharmaceutical companies to attract new investment in therapeutic mAbs, postponing the implementation of new and potentially life-saving drugs.
Why did pre-clinical safety testing in cynomolgus macaques not predict human responses?
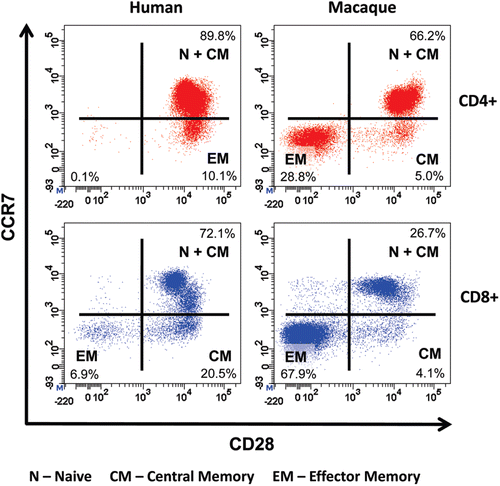
Cynomolgus macaques were chosen for pre-clinical safety testing of TGN1412 because they share 100% sequence homology for the extracellular loop of CD28 with man, demonstrated equivalent binding affinity for TGN1412, and did not reveal differences in patterns of tissue staining (Hanke et al., Citation2006). However, cynomolgus macaques were the wrong species for pre-clinical safety testing of TGN1412 because the CD4+ effector memory sub-set of this species does not express CD28 and, therefore, cannot be stimulated by anti-CD28 agonists (Eastwood et al., Citation2010; ). In contrast the CD4+ effector memory T-cells of man express high levels of CD28 so can, therefore, be stimulated by TGN1412 (Sallusto et al., Citation1999; Eastwood et al., Citation2010). The evolutionary reason for this subtle difference in CD28 expression on human CD4+ effector memory T-cells is not understood, but likely enhances antigen responsiveness by providing co-stimulatory signals. Human and cynomolgus macaque CD8+ effector memory T-cells do not express CD28, thus explaining the CD4+ T-cell bias of the TGN1412 response in man (Pitcher et al., Citation2002; Moniuszko et al., Citation2004; Sallusto et al., Citation2004; ). Only a comparative analysis of target cell subsets would have identified the species difference in CD4+ effector memory T-cells prior to pre-clinical safety testing.
Figure 1. Comparative immunophenotyping of human and cynomolgus macaque CD4+ T-cells. Analysis gated on CD4+ T-cells shown in top panels and on CD8+ T cells shown in bottom panels. Human and cynomolgus macaque naive (N) and central memory (CM) CD4+ and CD8+ T-cells are CCR7+CD28+. Human CD4+ effector memory (EM) T-cells are CCR7−CD28+ in contrast to cynomolgus macaque CD4+ EM T-cells, which are CCR7−CD28−. Cynomolgus macaque CD4+CCR7−CD28+ T-cells are a transitional sub-set with CM properties. Both human and cynomolgus macaque CD8+ N and CM T-cells express CD28 compared to CD8+ EM T-cells that lack CD28 expression.
Although cynomolgus macaque naive and central memory T-cells express CD28, these subsets were not stimulated in vitro by TGN1412 unless exogenous IL-2 was added to cultures, producing a weak proliferative response compared to human lymphocytes (Stebbings et al., Citation2007). It is possible that initial release of cytokines, including IL-2, by activated CD4+ effector memory T-cells in concert with TGN1412 similarly drove proliferation and then differentiation of naive and central memory CD4+ T-cells in man, resulting in a second reactive phase. This may correspond with the time line of events after infusion of TGN1412 where a transient recovery after the first corticosteroid dose was followed by a rapid deterioration and more severe adverse response including multi-organ failure (Suntharalingham et al., 2006). The driving force for the second reactive phase was most likely IL-2, highlighting the importance of measuring release of this cytokine and associated lymphoproliferation during pre-clinical safety testing of new therapeutics.
Why were pre-clinical in vitro safety tests with human cells non-predictive?
TGN1412 is an immunomodulatory drug that targets the surface marker CD28 in order to exploit co-stimulatory signals required for T-cell activation (Jenkins et al., Citation1991; Tacke et al., Citation1997; Lühder et al., Citation2003). In rat models of experimental autoimmune encephalomyelitis and adjuvant arthritis, treatment with anti-CD28 mAbs orthologous to TGN1412 stimulated the release of anti-inflammatory cytokines and the expansion of regulatory T-cells believed to counter autoimmune responses (Rodríguez-Palmero et al., Citation1999; Lin and Hünig, Citation2003; Beyersdorf et al., Citation2005). Since TGN1412 stimulated massive pro-inflammatory cytokine release in man it was postulated that pre-clinical in vitro safety tests with human lymphocytes did not replicate in vivo presentation due to the biology of CD28 (Stebbings et al., Citation2007). The natural ligands for CD28, i.e., CD80 and CD86, are present on antigen-presenting cells that interact with T-cells through localized engagement at the point of cell-to-cell contact (Dustin et al., Citation1998). This promotes formation of an immunological synapse that consists of a central cluster of T-cell receptors surrounded by a ring of adhesion molecules that sequesters kinases required for T-cell activation from across the inner cell membrane (Dustin et al., Citation1998; Monks et al., Citation1998; Wang et al., Citation2004). Conversely, engagement of CD28 outside of the immunological synapse or disruption of its formation impedes T-cell activation (Sanchez-Lockhart and Miller, Citation2006; Thoulouze et al., Citation2006). Standard in vitro safety tests employ aqueous phase presentation of mAb to human lymphocytes where binding occurs over the whole cell surface that does not replicate localized cell receptor engagement.
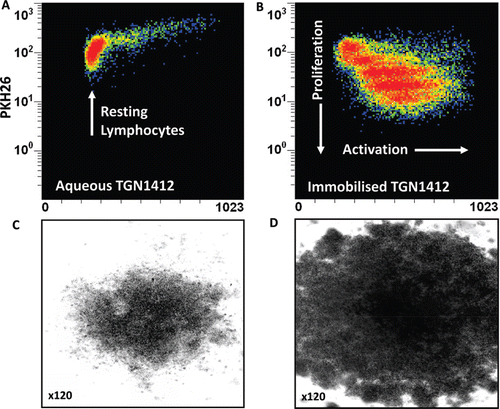
In order to mimic delivery of a localized signal TGN1412 was immobilized onto the surface of tissue culture plates so that human T-cells could only interact with the mAb at one interface (Stebbings et al., Citation2007). The result of this simple change in presentation was dramatic; massive lymphoproliferation and pro-inflammatory cytokine release that mirrored trial volunteer responses to TGN1412 (Stebbings et al., Citation2007; ). The possibility that this response was an artefact was ruled out by the observation that lymphocytes from cynomolgus macaques were not stimulated by immobilized TGN1412 at any dose (Stebbings et al., Citation2007). The species-specific response of human lymphocytes to TGN1412 in vitro mirrored the in vivo adverse drug response in man but not cynomolgus macaques. Moreover, the calculation of a safe starting dose in man based upon pre-clinical safety testing of TGN1412 in cynomolgus macaques was flawed. A no-observable adverse effect level (NOAEL) of 50 mg/kg in cynomolgus macaques was used to justify a low safe starting dose of 0.1 mg/kg in man (TeGenero, Citation2005). In reality, this dose correlated with 45–80% receptor occupancy in man and a near maximum immunostimulatory dose in vitro (Stebbings et al., Citation2007; Waibler et al., Citation2008). A NOAEL approach is no longer preferred; a minimum anticipated biological effect level (MABEL) is now recommended for calculation of safe starting doses of therapeutics in man (Duff, Citation2006; CHMP, Citation2007).
Figure 2. Proliferation of PKH26-stained human PBMC incubated 3 days with (a) 1 μg/well aqueous TGN1412 vs (b) 1 μg/well immobilized TGN1412. Lymphocyte proliferation appears as bands of cells with decreasing PKH26 fluorescence as dye is divided equally between daughter cells and increased forward scatter (FSC) corresponding to increased size due to blasting. No proliferation is observed with aqueous TGN1412 (a) compared to a strong proliferative response with immobilized TGN1412 (b). Low power light microscope images of 2 × 105 human PBMC in tissue culture after 3 days culture with (c) 1 μg/well aqueous TGN1412 vs (d) 1 μg/well immobilized TGN1412.
Predictivity of newly-proposed cytokine release assays
Initial in vitro studies of pro-inflammatory cytokine release from human lymphocytes stimulated with immobilized TGN1412 measured high levels of tumor necrosis factor (TNF)-α and interleukin (IL)-8 (Stebbings et al., Citation2007). Yet, when later compared with immobilized Campath-1H (an anti-CD52 mAb used for the treatment of B-cell chronic lymphocytic leukemia) and Rituximab, (an anti-CD20 mAb used to treat hematological neoplasm such as leukemia and lymphomas), two therapeutics known to induce cytokine release syndrome in some patients (Moreau et al., Citation1996; Winkler et al., Citation1999), levels of TNFα (3000–5000 pg/ml) and IL-8 (50,000–100,000 pg/ml) release after 24 h stimulation were not significantly different to TGN1412 (Eastwood et al., Citation2010; ). After 72 h, TGN1412 does stimulate significantly (p < 0.05) more TNFα than Rituximab, but not Campath-1H (). This result does not correlate well with the severity of the response to TGN1412 compared to Campath-1H and Rituximab, so TNFα and IL-8 release are poorly predictive of the severity of response (Eastwood et al., Citation2010).
Figure 3. Cytokine release from human PBMCs stimulated in vitro for 24, 48, and 72 h with 1 µg/well immobilized isotype-matched control (IgG4κ), Rituximab (IgG1), Campath-1H (IgG1), and TGN1412 (IgG4κ). The lectin PHA at 10 µg/ml was used as mitogen control. Cytokine release into culture supernatants was measured by ELISA. (a) TNFα, (b) IL-8, (c) IFNγ, and (d) IL-2 release are expressed in pg/ml. Donors responses shown (n = 9). * Denotes p < 0.05 (one-way analysis of variance with bonferroni correction).
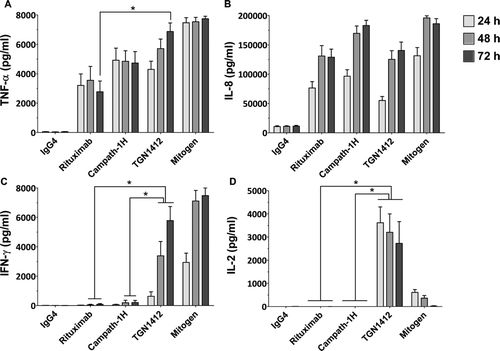
Subsequently, both interferon (IFN)-γ and IL-2 were found to be better predictive markers of a TGN1412-like cytokine release using immobilized mAbs (Eastwood et al., Citation2010). All three of these therapeutic mAbs stimulate IFNγ release in vitro when immobilized, but levels are significantly (p < 0.05) higher with TGN1412 at 48 and 72 h compared to Rituximab and Campath-1H, correlating with the severity of the response (). However, the best predictive marker for TGN1412 is IL-2, with high levels released after 24, 48, and 72 h (4000–5000 pg/ml) compared to no response with Campath-1H and Rituximab (). The only other therapeutic mAb found to stimulate IL-2 release is Muromonab-CD3, an anti-CD3 mAb used to treat allograft rejection, but whose administration is often complicated by serious adverse reactions related to cytokine release (Suthanthiran et al., Citation1989). In renal allograft patients, not pre-treated with immunosuppressant, serum levels of IL-2 peak around 400 pg/ml 4 h after Muromonab-CD3 administration (Chatenoud et al., Citation1990). In contrast, TGN1412 stimulated release of > 5000 pg/ml IL-2 in trial volunteer sera after 4 h (Suntharalingam et al., Citation2006), consistent with the greater severity of response compared to Muromonab-CD3. Thus, a level of T-cell activation beyond that seen with Muromonab-CD3 is increasingly likely to cause a TGN1412-like response.
Whilst it is tempting to suggest that levels of IL-2 release up to those caused by Muromonab-CD3 may be acceptable it should be remembered that this response is self-limiting, cytokine producing cells are depleted by treatment. For immunomodulatory mAbs, lower levels of IL-2 release will have the potential to cause a severe response. The ability of IL-2 release to amplify pro-inflammatory T-cell cytokine responses by promoting lymphoproliferation suggests that this was the driving force with respect to the adverse effects caused by TGN1412. It was established that TGN1412 stimulates CD4+ T-cells to undergo lymphoproliferation and release pro-inflammatory cytokines (Stebbings et al., Citation2007). In contrast, Campath-1H and Rituximab stimulate cytokine release from natural killer (NK) and CD8+, but not CD4+, T-cells (Wing et al., Citation1996; Eastwood et al., Citation2010: ). Muromonab-CD3 was found to represent a third pattern of cytokine release stimulating subsets of CD4+ and CD8+ T-cells and NK cells (). Since IL-2 is produced by mainly CD4+ T-cells, not NK cells; this observation explained why immobilized Campath-1H and Rituximab stimulation did not induce release of this cytokine. Stimulation of more CD4+ T-cells by TGN1412 than Muromonab may also explain quantitative differences in IFNγ and IL-2 release ().
Figure 4. Intracellular IFN-γ staining of human PBMC stimulated for 24 h with 1 μg/well of immobilized TGN1412, Rituximab, Muromonab, and Campath-1H. CD4+ T cells, CD8+ TT cells and NK cells are identified using the key above. TGN1412 stimulates IFN-γ secretion from CD4+ T-cells, Rituximab, and Campath-1H stimulate IFNγ secretion from NK cells and CD8+ T-cells and Muromonab stimulate IFN-γ secretion from CD4+ and CD8+ T-cells and NK cells.
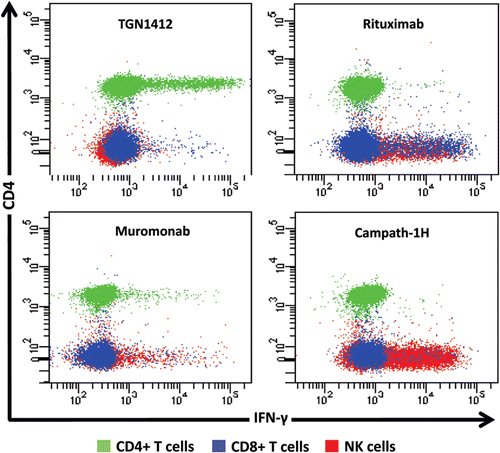
TGN1412 stimulates IFNγ release from CD4+ T-cells, whereas Campath-1H and Rituximab stimulate release of this cytokine from NK and CD8+ T-cells, showing that there are at least two different mechanisms for inducing IFNγ release (). However, a cytokine ELISA will not discriminate between cellular sources giving the impression that these pro-inflammatory responses are the same if equivalent amounts of cytokines are released. Differences between Campath-1H and Rituximab cytokine release are also evident in the proportions of CD8+ T-cells and CD8+/− NK cells stimulated implying activation of different subsets. Understanding the cellular sources of these cytokines is important for assessing safety of new therapeutics, since each mediates a different pro-inflammatory mechanism with different clinical outcomes. The mechanism of TGN1412 cytokine release (a CD4+ T-cell-restricted response) is different to that reported for other therapeutic mAbs and is mediated by binding to antigen via the complementary determining regions (Findlay et al., Citation2011a). In contrast, the Fc component of therapeutic mAbs mediates NK cell cytokine release. A therapeutic mAb of the IgG1 isotype, such as Campath-1H and Rituximab, with high affinity for Fc receptors, will likely stimulate NK cell cytokine release, whereas an IgG4 mAb such as TGN1412 with low Fc receptor affinity will likely not (Bruhns et al., Citation2009). Whilst IL-2 release is an important marker of a TGN1412-like cytokine response, development of a robust assay for future trials of therapeutic mAbs would have to include cytokines that are expressed by all target cell types, such as TNFα or IFNγ.
Reconciling differences between in vivo and in vitro cytokine release
High concentrations (> 5000 pg/ml) of the cytokines TNFα, IL-2, and IFNγ were measured in the plasma of trial volunteers within 4 h of TGN1412 infusion (Suntharalingam et al., Citation2006). In comparison, the kinetics of cytokine release in vitro using immobilized TGN1412 was much slower, particularly in the case of IFNγ where an incubation time of 48–72 h was required to achieve parity (Eastwood et al., Citation2010; ). The search for an explanation of this led to the identification of CD4+ effector memory T-cells being the source of TGN1412-stimulated IFNγ release (Eastwood et al., Citation2010; ).
Figure 5. Intracellular TNFα, IL-2, and IFNγ staining of human PBMCs and effector memory T-cell (EM) enriched PBMC following stimulation with either 1 µg/well immobilized IgG4 isotype matched control mAb or TGN1412. Analysis shown is gated on CD3+CD4+ T-cells. Cells in the right hand quadrants are positive for TNFα, cells in the upper quadrants are positive for IL-2, cells positive and negative for IFN-γ are identified using the key above.Cells in the upper right corner in red are positive for TNFα, IL-2, and IFNγ. The frequency of cytokine positive cells is expressed as a percentage of CD3+CD4+ T-cells. Quadrant statistics for cytokine positive CD3+CD4+ T-cells are shown in the table below.
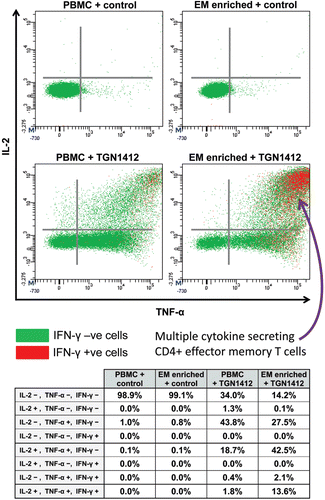
CD4+ effector memory T-cells are known to have the capacity for rapid release of considerable levels of cytokines, with TNFα, IL-2, and IFNγ being major components that are consistent with the pro-inflammatory response to TGN1412 (Sallusto et al., Citation1999; Chung, Citation2008). In the body, high concentrations of CD4+ effector memory T-cells are found in the lungs and gastrointestinal mucosa (Campbell et al., Citation2001; Picker, Citation2006). Moreover, up to 90% of all the lymphocytes in the body are contained within the lungs and gastrointestinal mucosa, compared to only 2–5% in the peripheral blood (Cerf-Bensussan and Guy-Grand, Citation1991; Smit-McBride et al., Citation1998). It is, therefore, likely that infusion of TGN1412 into trial volunteers caused high concentrations of cytokines to be rapidly released into the blood by large numbers of CD4+ effector memory T-cells within the mucosal tissues. In contrast, peripheral blood lymphocytes contain low numbers of CD4+ effector memory T-cells, so their use for in vitro assays is likely to under-estimate TGN1412-like cytokine responses such as IFNγ release, with the caveat that the method of presentation in vivo is different.
Delay in release of high levels of IFNγ in vitro by peripheral blood mononuclear cells (PBMC) was most likely due to the time required for naive and central memory CD4+ T-cells to undergo proliferation and differentiation into effector memory CD4+ T-cells with the capacity to secrete this cytokine (Geginat et al., Citation2001). For ethical and practical reasons, it is not feasible to use lymphocytes harvested from healthy human tissues in order to perform in vitro safety tests that replicate mucosal and lymphoid responses. Therefore, it is important to understand the differences between peripheral blood, lymphoid, and mucosal tissue lymphocytes when interpreting results of in vitro safety tests. Alternatively, enrichment of CD4+ effector memory T-cells from peripheral blood lymphocytes for in vitro assays will better replicate in vivo cytokine responses to TGN1412 in terms of both kinetics and cytokine secretion (Eastwood et al., Citation2010; ), but is limited by low cell yields.
Assay refinements and co-culture alternatives
Levels of cytokine release with immobilized mAbs treated are hugely dependent upon assay conditions such as antibody coating method, serum concentration, cell concentration, and whether dilute whole blood or isolated lymphocytes are used. For example, low serum concentrations (e.g., 2%) are quickly exhausted by stimulated lymphocytes, blunting cytokine release. Originally air-drying, but not wet-coating, of TGN1412 onto polystyrene tissue culture plates was necessary to elicit a response (Stebbings et al., Citation2007). It is now known that superior responses can be obtained with polypropylene tissue culture plates and that both wet coating and air drying methods can be employed (Eastwood et al., Citation2010; Findlay et al., Citation2010). This was originally thought to be a property of the plastic used, but non-tissue culture treated polystyrene plates are equally effective and can be wet-coated with mAb. The process of modifying tissue culture plates to make it easier for adherent cells to grow appears to reduce the amount of mAb that can be bound (Findlay et al., Citation2010). The density of immobilized mAb appears critical for cytokine release, suggesting a threshold below which no response is seen. This is reminiscent of ‘trigger pharmacology’. Conversely, a prozone effect is observed with high concentrations of mAb and air drying, probably due to leaching of mAb into aqueous phase where it can inhibit responses to immobilized TGN1412 (Findlay et al., Citation2010).
Another approach to immobilize therapeutic mAbs for presentation to lymphocytes in cytokine release assays is to employ polystyrene beads coated with protein A or anti-human IgG to act as artificial antigen-presenting cells (Walker et al., Citation2011). This method greatly increases the surface area available for interaction with lymphocytes, allowing responses to lower concentrations of therapeutic mAbs. Regardless of the approach employed, immobilization of TGN1412 onto plastic is an effective in vitro method for replicating TGN1412 cytokine release in man, but does not mimic in vivo presentation.
Attempts to model in vivo presentation of TGN1412 employed a co-culture of human endothelial cell monolayers with donor lymphocytes have been shown to result in pro-inflammatory cytokine release, particularly IL-6, IL-8, and TNFα (Stebbings et al., Citation2007; Findlay et al., Citation2011b; Dhir et al., Citation2012). It has subsequently been reported that the cytokine profile of this model does not fully match that of the trial volunteers or assays with immobilized TGN1412, as only low levels of the cytokines IL-2 and IFNγ are produced (Findlay et al., Citation2011b). If endothelial cell interactions are only part of the story then other mechanisms are likely to account for in vivo presentation, for example Fc-capture, but since TGN1412 is of the IgG4 sub-class, this would then be restricted to the FcγRI receptor (Bruhns et al., Citation2009), possibly involving a monocyte component (Sandilands et al., Citation2010). Alternatively, pre-culture of human lymphocytes at high density, to promote tissue-like conditions, has recently been reported to increase the sensitivity of circulating T-cell responses to TGN1412 in aqueous phase (Romer et al., 2011). Other methods being investigated include 3D cell and skin biopsy cultures to attempt to replicate tissue responses.
Conclusions
The mechanism of cytokine release caused by TGN1412 is different to that caused by other therapeutic mAbs because it involved a different arm of the immune system, the CD4+ effector memory T-cells. Unlike other therapeutic mAbs, aqueous presentation of TGN1412 in vitro is not immunostimulatory and gives a false negative result that, combined with use of the wrong species for pre-clinical safety testing, led to the erroneous prediction that it was safe to test in man. Although immobilization has since been demonstrated to render TGN1412 immuno-stimulatory in vitro it should be noted that this is an artificial means of stimulation unlikely to replicate in vivo presentation, but nevertheless it is the only method that induces substantial cytokine release similar to that experienced by trial volunteers. Stimulation of different pro-inflammatory pathways produces different cytokine profiles and clinical outcomes so it is important to measure the relevant responses in order to discriminate between them. In the case of TGN1412, measurement of IL-2 and lymphoproliferation provide the most unambiguous biomarkers. The likelihood that another therapeutic mAb or immunomodulatory agent is capable of stimulating a TGN1412 like ‘cytokine storm’ can now be linked to the ability to activate CD4+ effector memory T-cells. However, activation of other arms of the immune system also has the potential to cause a ‘cytokine storm’.
Acknowledgments
This work was supported by National Institute for Health Research Funding.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Beyersdorf, N., Gaupp, S., Balbach, K., Schmidt, J., Toyka, K. V., Lin, C. H., Hanke, T., Hünig, T., Kerkau, T., Gold, R. 2005. Selective targeting of regulatory T-cells with CD28 super-agonists allows effective therapy of experimental autoimmune encephalomyelitis. J. Exp. Med. 202:445–455.
- Bruhns, P., Iannascoli, B., England, P., Mancardi, D. A., Fernandez, N., Jorieux, S., Daëron, M. 2009. Specificity and affinity of human Fcγ receptors and their polymorphic variants for human IgG subclasses. Blood 113:3716–3725.
- Campbell, J. J., Murphy, K. E., Kunkel, E. J., Brightling, C. E., Soler, D., Shen, Z., Boisvert, J., Greenberg, H. B, Vierra, M. A, Goodman, S. B, Genovese, M. C, Wardlaw, A. J, Butcher, E. C, Wu, L 2001. CCR7 expression and memory T cell diversity in humans. J. Immunol. 166:877–884.
- Cerf-Bensussan, N., Guy-Grand, D. 1991. Intestinal intra-epithelial lymphocytes. Gastroenterol. Clin. North Am. 20:549–576.
- Chatenoud, L., Ferran, C., Legendre, C., Thouard, I., Merite, S., Reuter, A., Gevaert, Y., Kreis, H., Franchimont, P., Bach, J. F. 1990. In vivo cell activation following OKT3 administration. Systemic cytokine release and modulation by corticosteroids. Transplantation 49:697–702.
- CHMP (Committee for Medicinal Products for Human Use). 2007. Guideline on Strategies to Identify and Mitigate Risks for First-In-Human Clinical Trials with Investigational Medicinal Products. Available online at: http://www.ema.europa.eu/pdfs/human/swp/2836707enfin.pdf.
- Chung, C. H. 2008. Managing premedications and the risk for reactions to infusional monoclonal antibody therapy. Oncologist 13:725–732.
- Dhir, V., Fort, M., Mahmood, A., Higbee, R., Warren, W., Narayanan, P., Wittman, V. 2012. A predictive biomimetic model of cytokine release induced by TGN1412 and other therapeutic monoclonal antibodies. J. Immunotoxicol. 9:34–42.
- Duff, G. W. 2006. Expert Group on Phase One Clinical Trials: Final report. Norwich, UK: TSO Publications Centre.
- Dustin, M. L., Olszowy, M. W., Holdorf, A. D., Li, J., Bromley, S., Desai, N., Widder, P., Rosenberger, F., van der Merwe, P. A., Allen, P. M., Shaw, A. S. 1998. A novel adaptor protein orchestrates receptor patterning and cytoskeletal polarity in T-cell contacts. Cell 94:667–677.
- Eastwood, D., Findlay, L., Poole, S., Bird, C., Wadhwa, M., Moore, M., Burns, C., Thorpe, R., Stebbings, R. 2010. Monoclonal antibody TGN1412 trial failure explained by species differences in CD28 expression on CD4+ effector memory T-cells. Br. J. Pharmacol. 161:512–526.
- Findlay, L., Eastwood, D., Ball, C., Robinson, C. J., Bird, C., Wadhwa, M., Thorpe, S. J., Thorpe, R., Stebbings, R., Poole, S. 2011a. Comparison of novel methods for predicting the risk of pro-inflammatory clinical infusion reactions during monoclonal antibody therapy. J. Immunol. Meth. 371:134–142.
- Findlay, L., Eastwood, D., Stebbings, R., Sharp, G., Mistry, Y., Ball, C., Hood, J., Thorpe, R., Poole, S. 2010. Improved in vitro methods to predict the in vivo toxicity in man of therapeutic monoclonal antibodies including TGN1412. J. Immunol. Meth. 352:1–12.
- Findlay, L., Sharp, G., Fox, B., Ball, C., Robinson, C. J., Bird, C., Stebbings, R., Eastwood, D., Wadhwa, M., Poole, S., Thorpe, R., Thorpe, S. J. 2011b. Endothelial cells co-stimulate peripheral blood mononuclear cell responses to monoclonal antibody TGN1412 in culture. Cytokine 55:141–151.
- Geginat, J., Sallusto, F., Lanzavecchia, A. 2001. Cytokine-driven proliferation and differentiation of human naive, central memory, and effector memory CD4+ T-cells. J. Exp. Med. 194:1711–1719.
- Hanke, T. 2006. Lessons from TGN1412. Lancet 368:1569–1570.
- Jenkins, M. K., Taylor, P. S., Norton, S. D., Urdahl, K. B. 1991. CD28 delivers a co-stimulatory signal involved in antigen-specific IL-2 production by human T-cells. J. Immunol. 147:2461–2466.
- Lin, C. H., Hünig, T. 2003. Efficient expansion of regulatory T-cells in vitro and in vivo with a CD28 super-agonist. Eur. J. Immunol. 33:626–638.
- Lühder, F., Huang, Y., Dennehy, K. M., Guntermann, C., Müller, I., Winkler, E., Kerkau, T., Ikemizu, S., Davis, S. J., Hanke, T., Hünig, T. 2003. Topological requirements and signaling properties of T-cell-activating, anti-CD28 antibody super-agonists. J. Exp. Med. 197:955–966.
- Moniuszko, M., Fry, T., Tsai, W. P., Morre, M., Assouline, B., Cortez, P., Lewis, M. G., Cairns, S., Mackall, C., Franchini, G. 2004. Recombinant IL-7 induces proliferation of naive macaque CD4+ and CD8+ T-cells in vivo. J. Virol. 78:9740–9749.
- Monks, C. R., Freiberg, B. A., Kupfer, H., Sciaky, N., Kupfer, A. 1998. Three-dimensional segregation of supramolecular activation clusters in T-cells. Nature 395:82–86.
- Moreau, T., Coles, A., Wing, M., Isaacs, J., Hale, G., Waldmann, H., Compston, A. 1996. Transient increase in symptoms associated with cytokine release in patients with multiple sclerosis. Brain 119:225–237.
- Picker, L. J. 2006. Immunopathogenesis of acute AIDS virus infection. Curr. Opin. Immunol. 18:399–405.
- Pitcher, C. J., Hagen, S. I., Walker, J. M., Lum, R., Mitchell, B. L., Maino, V. C., Axthelm, M. K., Picker, L. J. 2002. Development and homeostasis of T-cell memory in rhesus macaque. J. Immunol. 168:29–43.
- Rodríguez-Palmero, M., Hara, T., Thumbs, A., Hünig, T. 1999. Triggering of T-cell proliferation through CD28 induces GATA-3 and promotes T-helper type 2 differentiation in vitro and in vivo. Eur. J. Immunol. 29:3914–3924.
- Römer, P. S., Berr, S., Avota, E., Na, S. Y., Battaglia, M., ten Berge, I., Einsele, H., Hünig, T. 2011. Pre-culture of PBMC at high cell density increases sensitivity of T-cell responses, revealing cytokine release by CD28 super-agonist TGN1412. Blood 118:6772–6782.
- Sallusto, F., Geginat, J., Lanzavecchia A. 2004. Central memory and effector memory T-cell subsets: Function, generation, and maintenance. Annu. Rev. Immunol. 22:745–763.
- Sallusto, F., Lenig, D., Förster, R., Lipp, M., Lanzavecchia, A. 1999. Two subsets of memory T-lymphocytes with distinct homing potentials and effector functions. Nature 401:708–712.
- Sanchez-Lockhart, M., Miller, J. 2006. Engagement of CD28 outside of the immunological synapse results in up-regulation of IL-2 mRNA stability but not IL-2 transcription. J. Immunol. 176:4778–4784.
- Sandilands, G. P., Wilson, M., Huser, C., Jolly, L., Sands, W. A., McSharry, C. 2010. Were monocytes responsible for initiating the cytokine storm in the TGN1412 clinical trial tragedy? Clin. Exp. Immunol. 162:516–527.
- Smit-McBride, Z., Mattapallil, J. J., McChesney, M., Ferrick, D., Dandekar, S. 1998. Gastrointestinal T-lymphocytes retain high potential for cytokine responses but have severe CD4+ T-cell depletion at all stages of simian immunodeficiency virus infection compared to peripheral lymphocytes. J. Virol. 72:6646–6656.
- Stebbings, R., Findlay, L., Edwards, C., Eastwood, D., Bird, C., North, D., Mistry, Y., Dilger, P., Liefooghe, E., Cludts, I., Fox, B., Tarrant, G., Robinson, J., Meager, T., Dolman, C., Thorpe, S. J., Bristow, A., Wadhwa, M., Thorpe, R., Poole, S. 2007. “Cytokine storm” in the Phase I trial of monoclonal antibody TGN1412: Better understanding the causes to improve pre-clinical testing of immunotherapeutics. J. Immunol. 179:3325–3331.
- Stebbings, R., Poole, S., Thorpe, R. 2009. Safety of biologics, lessons learnt from TGN1412. Curr. Opin. Biotechnol. 20:673–677.
- Suntharalingam, G., Perry, M. R., Ward, S., Brett, S. J., Castello-Cortes, A., Brunner, M. D., Panoskaltsis, N. 2006. “Cytokine storm” in a Phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. New Engl. J. Med. 355:1018–1028.
- Suthanthiran, M., Fotino, M., Riggio, R. R., Cheigh, J. S., Stenzel, K. H. 1989. OKT3-associated adverse reactions: Mechanistic basis and therapeutic options. Am. J. Kidney Dis. 14:39–44.
- Tacke, M., Hanke, G., Hanke, T., Hünig, T. 1997. CD28-mediated induction of proliferation in resting T-cells in vitro and in vivo without engagement of the T-cell receptor: Evidence for functionally distinct forms of CD28. Eur. J. Immunol. 27:239–247.
- TeGenero, A. G. 2005. TGN1412 investigator’s brochure. Available online at: http://www.circare.org/foia5/tgn1412investigatorbrochure.pdf. Accessed 5 March 2012.
- Thoulouze, M. I., Sol-Foulon, N., Blanchet, F., Dautry-Varsat, A., Schwartz, O., Alcover, A. 2006. Human immunodeficiency virus type-1 infection impairs the formation of the immune-logical synapse. Immunity 24:547–561.
- Waibler, Z., Sender, L. Y., Kamp, C., Müller-Berghaus, J., Liedert, B., Schneider, C. K., Löwer, J., Kalinke, U. 2008. Toward experimental assessment of receptor occupancy: TGN1412 revisited. J. Allergy Clin. Immunol. 122:890–892.
- Walker, M. R., Makropoulos, D. A., Achuthanandam, R., Van Arsdell, S., Bugelski, P. J. 2011. Development of a human whole blood assay for prediction of cytokine release similar to anti-CD28 super-agonists using multiplex cytokine and hierarchical cluster analysis. Int. Immunopharmacol. 11:1697–1705.
- Wang, D., Matsumoto, R., You, Y., Che, T., Lin, X. Y., Gaffen, S. L., Lin, X. 2004. CD3/CD28 costimulation-induced NF-κB activation is mediated by recruitment of protein kinase C-θ, Bcl10, and IκB kinase β to the immunological synapse through CARMA1. Mol. Cell. Biol. 24:164–171.
- Wing, M. G., Moreau, T., Greenwood, J., Smith, R. M., Hale, G., Isaacs, J., Waldmann, H., Lachmann, P. J., Compston, A. 1996. Mechanism of first-dose cytokine-release syndrome by CAMPATH 1-H: Involvement of CD16 (FcγRIII) and CD11a/CD18 (LFA-1) on NK cells. J. Clin. Invest. 98:2819–2826.
- Winkler, U., Jensen, M., Manzke, O., Schulz, H., Diehl, V., Engert, A. 1999. Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8). Blood 94:2217–2224.