Abstract
Bisphenol A (BPA) is a high production volume endocrine disrupting chemical that is widely used in many consumer products and prevalent in human biological fluids. Recent studies suggest that BPA is active even at low levels, raising concern about its potential harm to human health. Given that the main route of exposure to BPA is oral, via the consumption of BPA-tainted foods and beverages, intestinal tissues could be particularly vulnerable to BPA-induced changes. A novel examination is reported here of whether oral exposure to BPA affects inflammatory bowel disease (IBD), an immune-mediated disease of the colon, using a mouse model of inflammatory colitis. In addition to direct exposure, the possible contribution of maternal BPA exposure to disease later in life is explored. It was found that daily oral exposure to BPA at the US Environmental Protection Agency described oral reference dose (50 µg/kg/day), either via direct oral route or through maternal sources (i.e. developmental exposure), did not significantly alter disease outcomes of body weight, survival, or colonic pathology. These observations suggest that oral BPA exposure, at this dose and for this exposure duration, has minimal influence on aspects of the inflammatory response that regulate immune mediated diseases of the gastrointestinal tract.
Introduction
Inflammation is an intrinsic part of many immune mediated processes, and controlled inflammation is a beneficial element of host responses to infection and injury. However, uncontrolled inflammation causes tissue damage and contributes to many complex chronic diseases, such as inflammatory bowel diseases (IBD), autoimmune diseases, and cancer (Franks & Slansky, Citation2012; Hartnett & Egan, Citation2012; Kaser et al., Citation2010; Kim & Moudgil, Citation2008). The degree and duration of inflammation is maintained at its beneficial level by a number of checkpoints; disruptions of which often give rise to or exacerbate chronic inflammatory disease. A number of physiologic molecules including cytokines, hormones, and proteolytic enzymes constitute a pool of regulatory molecules that control inflammation, while intrinsic variables such as age, sex, and genetics influence the role of these molecules during an inflammatory response. Yet, these intrinsic factors alone cannot account for the increasing incidence of chronic inflammatory diseases.
Therefore, there is growing speculation that exposure to extrinsic, environmentally derived factors modulates this intricate homeostatic balance by acting on one or more of these checkpoints of inflammation, thereby contributing to disease (Kaser et al., Citation2010; Soon et al., Citation2012). For some extrinsic factors, there is mounting experimental data that support a causal relationship; however, for many extrinsic factors, data to support or refute a potential cause-and-effect relationship between exposure and disease remain limited. Further complicating this assessment of possible causality is the realization that chronic diseases take many years to develop, and exposure to extrinsic factors that contribute to their development may have occurred well before disease symptoms present clinically.
One environmental factor that has received significant notice recently is bisphenol A (BPA), which is a component of polycarbonate plastics and epoxy resins that is used in a variety of products including food and beverage containers, electronic appliances, dental sealants, paper currency, and receipts (NTP, Citation2008). BPA is of interest for several reasons. First, there is widespread and continuous human exposure, as evidenced by its presence in a large proportion of the population (Calafat et al., Citation2005). Furthermore, fetuses and neonates are likely exposed to BPA from maternal sources (e.g., in utero and via breast milk) as well as from environmental sources after birth (Balakrishnan et al., Citation2010; Schonfelder et al., Citation2002). Maternal–fetal transfer of BPA has also been extensively demonstrated using animal models (Domoradzki et al., Citation2003; Nishikawa et al., Citation2010). Second, BPA is a selective estrogen receptor (ER) modulator; therefore, it has the potential to affect physiological processes via modulation of ER-regulated pathways. Whether BPA acts through other receptors and pathways is less clear, but there is evidence that it may (Bonefeld-Jorgensen et al., Citation2007; Moriyama et al., Citation2002; Richter et al., Citation2007; Wetherill et al., Citation2007). Indeed, BPA is generally classified as an endocrine-disrupting chemical (EDC), and in recent years there has been much discussion both in the scientific community and in general public regarding the possible adverse health effects of exposure to EDC agents. Third, estrogens and other hormones control many aspects of immune and inflammatory responses (Bebo et al., Citation2001; Gallo et al., Citation2008; Guo et al., Citation2010).
While current data do not provide a consensus on the role of ER ligands, many reports indicate that regulation of inflammatory diseases, and of the immune system in general, are influenced by agents that, at least in some capacity, bind the ER or alter ER-regulated signaling pathways. Studies with experimental animals have provided mixed information regarding the possible effects of BPA on immune cell function (Midoro-Horiuti et al., Citation2010; Ohshima et al., Citation2007; Roy et al., Citation2012; Yan et al., Citation2008; Yoshino et al., Citation2004). Most of this work has focused on T-cell responses, with less attention given to innate immune responses and inflammation. Moreover, there is significant controversy regarding whether these results are cause for concern about the potential for BPA to harm human health, because often the effects of BPA were observed only when used in high doses and/or via routes of exposure that are considered less germane to human exposure (Hengstler et al., Citation2011; Vandenberg et al., Citation2009). Therefore, it is a matter of high public interest to understand the possible effects of lower level oral doses of BPA, similar to exposure in the general population, on the regulation of inflammation and severity of inflammatory diseases.
In the work reported here, we examined whether exposure to BPA modulates tissue inflammation in a mouse model of inflammatory bowel disease (IBD). IBD is a collective term for several immune-mediated gastrointestinal tract diseases; with Crohn’s Disease (CD) and ulcerative colitis (UC) being the more prevalent ones. IBDs are chronic, relapsing diseases affecting over a million people in the US alone and millions of people worldwide, with a preponderance in industrialized nations (Molodecky et al., Citation2012). While the precise causes of IBD remain to be elucidated, it results from a complex interplay of environmental, genetic, and microbial factors that trigger an inappropriate activation of the innate and adaptive immune system. Human and mouse genetic studies have clearly identified deregulation of the innate intestinal immune system (monocyte/macrophages, dendritic cells, paneth cells, and intestinal epithelial cells) as a major susceptibility for developing IBD (Bouma & Strober, Citation2003; Uhlig & Powrie, Citation2009; Westbrook et al., Citation2010). Although microbial intestinal immune responses are being extensively investigated in IBD, few studies have examined how exposure to contaminants in our food and beverage supply, such as BPA, could be affecting the normal developing intestinal immune system to alter susceptibility towards chronic inflammatory disease. Furthermore, while acute exposure to environmental agents can have direct effects in developmentally mature adults, developmental exposure to such chemicals can also contribute to deregulated inflammation in offspring because the immune system and its interconnected networks with other systems are established during fetal development. Therefore, in the current study we utilized an established mouse model of inflammatory colitis to examine whether direct oral exposure or developmental (maternal) exposure to the current US Environmental Protection Agency (USEPA) oral reference dose of BPA affects the severity of colonic inflammation (IRIS-USEPA).
Materials and methods
Animals and BPA dosing
Adult C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed in pre-washed polysulfone microisolator cages under pathogen-free conditions. To further minimize potential exposure to background BPA or other exogenous ER ligands, mice received phytoestrogen-free irradiated rodent chow (AIN-76 A, Test Diet, Richmond, IN), and drinking water that was purified by reverse osmosis and provided in glass bottles. The specifications for the animal housing environment were based on the guidelines outlined by an NIEHS expert panel for BPA toxicity studies (vom Saal et al., Citation2007). The USEPA reference dose for daily oral exposure to BPA, i.e. 50 µg/kg [body weight]/day, was used as the oral treatment dose in this investigation (IRIS-USEPA).
For direct exposure to BPA, 6–8-week-old male mice were given BPA or vehicle control (peanut oil), by oral gavage every day for 7 days. On the 7th day of BPA treatment, DNBS was rectally instilled to induce colitis (details below), and daily oral BPA administration was continued for 21 more days. For developmental exposure to BPA, adult mice were paired for breeding, and females were checked daily for vaginal plugs as an indication of pregnancy. Singly housed pregnant females began receiving 50 µg BPA/kg/day, or vehicle (peanut oil) orally starting on gestational day (GD) 6 and continuing daily through post-natal day 21. Dosing of pregnant dams was started on GD 6 to avoid any potential for ER-signaling to diminish the rate of implantation, which occurs between GD 4–5 in mice. Each pregnant or lactating mouse received BPA dissolved in 20 µl of peanut oil absorbed to an additive-free puffed wheat cereal. A mouse consumes this spiked food treat in ≈1 min under observation, ensuring that the accurate dose is administered to each dam everyday in a stress-free manner. On post-natal Day 21, offspring were weaned and housed in same sex groups until ready for experimental procedures.
Both gavage and ingestion of spiked food yielded mean circulating BPA concentration of 5.73 (±1.55) ng/ml (Roy et al., Citation2012), which is similar to levels found in humans (Calafat et al., Citation2005; Lang et al., Citation2008; Mielke & Gundert-Remy, Citation2009). Administration of this dose of BPA directly did not affect animal body weight or health in any discernable manner (data not shown). This daily dose of BPA also did not affect pregnancy duration, litter size, the offspring sex ratio, or offspring body weight, which were monitored until termination of the experiment (Roy et al., Citation2012). All procedures involving laboratory animals were performed in accordance with protocols approved by the University Institutional Animal Care and Use Committee.
Induction of experimental colitis
Several animal models have been developed to mimic the inflammatory response in the colon in order to understand the underlying mechanisms causing disease pathology in IBD (Blumberg et al., Citation1999; Westbrook et al., Citation2010). Chemical-induced inflammatory colitis in mice is one such model, in which acute inflammation is induced by administering 2,4,6-trinitrobenzene sulfonic acid (TNBS) or 2,4-dinitrobenzene sulfonic acid (DNBS) dissolved in ethanol (Morris et al., Citation1989; Qiu et al., Citation1999; Westbrook et al., Citation2010). While ethanol helps remove the mucosal layer, DNBS or TNBS act as a hapten and induce an immune response, leading to events that mimic the clinical signs of IBD. In the current study, colitis was induced in adult (8-week-old) mice by instilling 4 mg of DNBS dissolved in 50% ethanol using catheters placed in the colon, 3.5 cm from the anus of the anesthetized animals. The distribution of animals from different exposure groups into DNBS treatment groups for induction of colitis is summarized in . Control mice in the vehicle and BPA treatment groups received ethanol alone in the same manner. Mice were kept in Trendelenburg position for 1 min to avoid reflux, and then monitored daily for body weight changes and survival. For experiments using developmentally-exposed mice, it is the dams not the offspring that are directly exposed to BPA. Therefore, treatment groups of developmentally-exposed mice were generated using adult offspring obtained from separate sets of treated dams.
Table 1. BPA exposure and DNBS treatment in various groups of animals.
Measurement of colon inflammation
Mice were sacrificed by anesthetic overdose at the indicated points in time. The abdomen was opened by mid-line incision and the colon was removed, freed from surrounding tissues, and dissected out by cutting at the small intestine end, slightly above the cecum and the rectal end as close to the anus as possible. The entire colon was evaluated for macroscopic lesions as well as physical integrity and length. Three regions (proximal, distal, and rectal) from each colon were excised and fixed with 10% neutral buffered formalin. Fixed tissues were embedded in paraffin blocks and 5 µm thick tissue sections were prepared and stained with hematoxylin and eosin. Microscope slides were coded, and evaluated by a gastrointestinal pathologist with no knowledge of treatment group, using conventional light microscopy. Colonic inflammation was graded and scored as follows: “0”: normal crypt architecture, no neutrophilic infiltrate; “1”: mild activity with lamina propria and/or crypt neutrophilic infiltrate involving < 50% of glands; “2”: moderate activity with lamina propria and/or crypt neutrophilic infiltrate involving >50% of glands; “3”: severe activity with neutrophilic infiltrate and surface erosion; and “4”: severe activity with neutrophilic infiltrate and ulceration/necrosis.
Statistical analyses
For developmental exposure, since the dams not the offspring were exposed to BPA, for statistical purposes the experimental “n” refers to the number of treated dams, not the number of offspring. Therefore, all adult offspring in each group of mice used at each timepoint of every experiment were from a different treated dam. All statistical analyses were performed using Prism 4.0 (GraphPad software, La Jolla, CA). Statistical significance of the difference in mean values between endpoints measured in vehicle versus BPA-exposed groups was calculated using the following tests, with 95% confidence interval. The specific statistical tests used were Mantel--Haenszel log-rank test for survival curves, and two-way ANOVA with a Bonferroni post-hoc test for body weight changes following DNBS treatment, and to compare differences between independent variables over time and between treatment groups. When applicable, two-tailed unpaired t-tests were used to examine differences in mean values between vehicle and BPA treatment groups at a single point in time. Independent variables included treatment with BPA or peanut oil vehicle, treatment with DNBS or ethanol (control), and time. Although male and female offspring from treated dams were used in our studies, they were not treated with DNBS at the same time; therefore, no statistical comparisons across sex were made. Error bars in all graphs represent standard error of the mean (SEM).
Results
Exposure to BPA does not alter survival or body weight loss in mice with inflammatory colitis
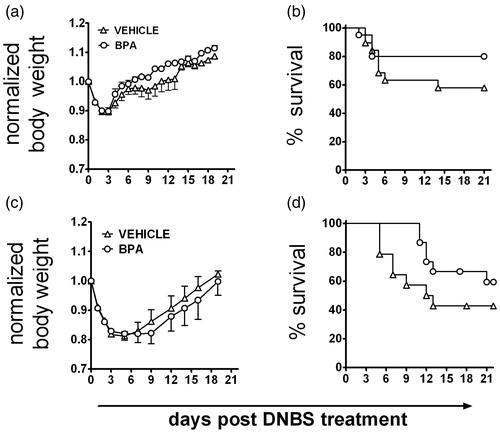
Inflammatory colitis was induced in adult mice that were either directly or developmentally exposed to BPA. Administration of DNBS consistently generated an inflammatory response in the colons of all mice regardless of BPA exposure history, and this was reflected in a transient loss in body weight (). In contrast, mice that received rectal administration of 50% ethanol without DNBS did not show any signs of disease or colon inflammation, and BPA treatment did not affect body weight of mice in this group (data not shown). Interestingly, while not statistically significant, DNBS-treated mice that received BPA either directly or via maternal exposure consistently exhibited improved survival ().
Figure 1. Neither direct nor developmental exposure to BPA alters morbidity and mortality from inflammatory colitis. Colitis was induced to adult male mice by rectal administration of DNBS, and (a and c) body weight and (b and d) mortality were recorded at regular intervals for 21 days. (a and b) Adult male mice were orally exposed to BPA starting 7 days prior to DNBS administration (Day 0), and BPA exposure continued throughout the observation period (n = 40/group). (c and d) Colitis was induced in adult male mice (8–10-weeks-of-age) that were developmentally exposed to peanut oil vehicle or BPA during gestation and via lactation (n = 15/group). Similar findings were observed in developmentally-exposed female offspring (not shown). Data are representative of two independent experiments that yielded similar results. All data are plotted as the mean ( ± SEM).
Exposure to BPA does not alter the pathological changes in the colon of mice with inflammatory colitis
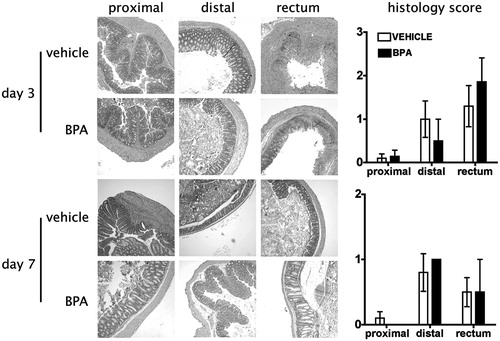
To further evaluate the degree and extent of inflammation and disease activity, sections of large intestine were collected from adult mice exposed to BPA either directly () or via maternal exposure (). Colons were obtained 3, 7, and 21 days after DNBS administration, and histopathology of the proximal colon, distal colon, and rectum were examined in BPA- and vehicle-exposed mice. Among all regions of the colon, a time-dependent difference in tissue inflammation was observed, with Day 3 presenting the most severe disease, and Day 7 appearing less severe, with inflammation more confined to the distal colon and rectum (compare the upper and lower panels, and ). Regardless of whether BPA was given directly or developmentally, by 21 days after DNBS administration, resolution of inflammation was observed in all groups, BPA and vehicle-treated (data not shown). In fact, compared to vehicle control-treated mice, the only significant difference observed was 3 days after DNBS instillation in mice that received direct oral BPA dosing (). However, this difference in overall inflammation of the rectum resolved by Day 7. No significant differences in colon inflammation were observed in adult offspring of BPA-treated dams at any point in time examined. There were no significant differences in histological parameters between mice from the two exposure paradigms.
Figure 2. Acute exposure to BPA does not alter tissue pathology during inflammatory colitis. Adult male mice were orally exposed to BPA once daily. After 7 days of oral BPA treatment, colitis was induced by rectal administration of DNBS as described in the Materials and methods section (BPA dosing was continued daily after DNBS administration). Sections of colon, from different regions, were collected for histological evaluation on 3 (two top rows), 7 (two bottom rows), and 21 (not shown) days after DNBS administration (n = 8 mice/group/time-point). The photomicrographs of colon sections are from one representative animal from each group at each time-point, and the line graphs on the right depict the mean ( ± SEM) pathology score for all animals in the group in that point in time (Day 3 and 7 post-DNBS). *, p ≤ 0.05. No detectable pathology was observed in the colon sections collected 21 days after DNBS administration in either group (not shown). Criteria for histopathologic evaluation are described in the Materials and methods section.
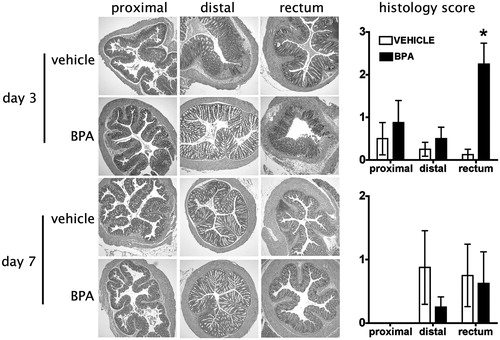
Figure 3. Developmental exposure to BPA does not alter tissue pathology during inflammatory colitis. Mice were developmentally exposed to BPA during gestation and lactation as described in the Materials and methods section. Colitis was induced to developmentally-exposed adult male mice by rectal administration of DNBS, and sections of colon were collected for histological evaluation on Days 3 (two top rows), 7 (two bottom rows), and 21 (not shown) after DNBS administration (n = 7–8 mice/group/time-point). The photomicrographs of colon sections are from one representative animal from each group at each time-point and the line graphs on the right show the mean ( ± SEM) pathology score for all animals in the group on Days 3 and 7 after DNBS treatment. No detectable pathology was observed in the colon sections collected 21 days after DNBS administration in either group (not shown). Criteria for histopathologic evaluation are described in the Materials and methods. Similar findings were observed in developmentally-exposed female offspring (not shown).
Although the difference in inflammation in the rectum of BPA-treated mice is statistically significant on Day 3, this effect did not persist, nor was it observed in other regions of the colon. Moreover, we did not find any difference in the expression of genes associated with inflammation, such as inducible nitric oxide synthase (iNOS), interleukin (IL)-6, IL-10, IL-17, IL-23, tumor necrosis factor (TNF)-α, monocyte chemotactic protein (MCP)-1, and interferon (IFN)-γ, in the colons of mice from any treatment group (data not shown). In a separate group of mice, leukocyte infiltration into the colon was also examined; the analyses found that, although the frequency of inflammatory leukocytes (macrophages and neutrophils in particular) correlated with the degree of tissue inflammation observed, there was no difference in the percentage or number of different types of leukocytes based on whether the mice had received BPA or peanut oil vehicle (data not shown).
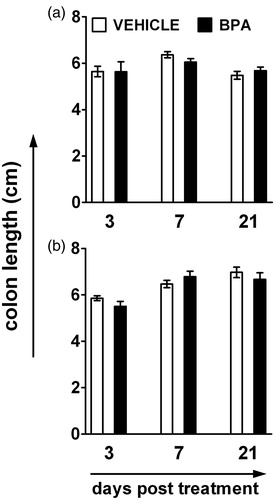
In addition to affecting tissue inflammation, severe colitis causes alterations beyond the mucosal layer that affect the overall architecture of the colon and shorten colon length during the acute phase of the disease. Consistent with our other findings, we did not observe any difference in colon length 3, 7, or 21 days after DNBS treatment () in mice developmentally or directly exposed to BPA compared to the vehicle exposed mice. Thus, when considering the overall data collected, with little to no consistent effect on several different metrics of disease, it seems unlikely that exposure to this dose of BPA, either directly or during early life development, modulates inflammatory processes in the colon.
Figure 4. Exposure to BPA does not influence the contraction in colon length following DNBS administration. (a) Mice were directly exposed to BPA via oral route starting 7 days prior to DNBS administration and daily BPA treatment continued until the day of sacrifice (n = 8 mice/group/time-point). (b) Mice were developmentally exposed to BPA during gestation and lactation (n = 7–8 mice/group/time-point). Colitis was induced to adult male mice by rectal administration of DNBS, and defined sections from different regions of the colon were collected for the evaluation of the disease 3, 7, and 21 days after DNBS administration. The bar graphs depict the mean (±SEM) length of the entire large intestine, from base of the cecum to the rectum. Similar findings were observed in developmentally-exposed female offspring (not shown).
Discussion
Our studies were designed to determine whether exposure to the endocrine disruptor BPA, either directly or during development, would alter the onset, severity, or resolution of inflammatory colitis. The major finding from our study demonstrated that neither developmental nor direct exposure of mice to BPA to the current tolerable daily intake limit for human exposure (50 µg/kg/day) had any marked effect on disease in a commonly used mouse model of inflammatory colitis. However, two modest changes were observed: in directly exposed mice, on the third day after DNBS instillation there was more severe rectal inflammation, and in both exposure paradigms, more mice in the BPA treatment group consistently survived after the induction of disease with DNBS treatment.
When considered at a population level, a transient increase in rectal inflammation in BPA-exposed animals may be significant when considering a sub-set of the human population living in a less stringently controlled environment, or that have other (e.g. genetic) factors that may predispose them to IBD (Soon et al., Citation2012). However, given that this effect was transient and did not correlate with a detectable change in colon length, expression of genes that encode immunoregulatory proteins, or leukocyte influx, speculation about human health should be considered with caution in the absence of additional data. We are unaware of any epidemiological study that has specifically examined whether BPA levels currently found in the human population correlate with the rise in inflammatory bowel diseases. Indeed, in a recent review article, Molodecky et al. (Citation2011) point out that environmental determinants of IBD are largely overlooked; this presents a critical information gap in efforts to better understand the etiology of these diseases.
Although not statistically significant, the improved survival of the DNBS-treated BPA-exposed mice compared to DNBS-treated vehicle-exposed mice is viewed with interest because we observed this phenomenon in every experiment. It suggests that BPA is affecting some aspect of the induced disease process, even if it may not directly alter the function of the immune system per se. Indeed, when these results from inflammatory colitis are combined with other recent studies (Bauer et al., Citation2012; Roy et al., Citation2012), it may be that BPA exerts its effects on non-hematopoietic cells rather than on bone marrow derived cells. For example, in mice that were developmentally exposed to BPA at the same dose used in this report, no impairment in T-cell or antibody responses to primary infection with several different influenza A viruses, no impairment in host resistance to primary or recall viral challenge, and no change in viral clearance from the lung were observed (Roy et al., Citation2012). However, tissue inflammation and the expression of several key anti-viral genes were transiently reduced in the lungs of infected adult offspring of the BPA-treated dams. This could be a result of BPA affecting lung-resident non-hematopoietic cells (e.g. lung epithelial cells), rather than changing the response of leukocytes during infection. Furthermore, using two different mouse models that recapitulate aspects of allergic asthma, only limited and moderate effects of BPA exposure were noted, with little to no effect on disease severity (Bauer et al., Citation2012). Interestingly, in both lung challenge models, slight changes in tissue inflammation were observed when comparing mice developmentally exposed to BPA versus offspring of the control-treated dams, causing speculation as to whether BPA affects epithelial cells to a greater extent than hematopoietic cells (Bauer et al., Citation2012; Roy et al., Citation2012). Moreover, consistent with the limited effect of BPA exposure on immune function in models of allergic asthma, viral infection, or inflammatory colitis, we did not observe any differences in the onset, progression, or severity of experimental autoimmune encephalomyelitis (EAE) when we compared mice that were developmentally-exposed to BPA (50 µg BPA/kg/day) and their vehicle-exposed counterparts (unpublished data).
Consistent with this idea, our study does not rule out a possible effect of oral BPA exposure on intestinal epithelial cells. These cells express ER and, although the dose administered was considerably higher than what we used (i.e. 20 mg/kg/day; 400-times higher than the dose used herein), BPA exposure during pregnancy affected the expression of genes that modulate calcium absorption and tight junctions in the GI tract (Otsuka et al., Citation2012). In another study, rats that were directly treated with 5 mg/kg/day (100-times higher than the dose used here) for 15 days showed exacerbated colorectal distension, elevated expression of the tight junction proteins occludin and JAM-A, and decreased myeloperoxidase activity in colons after TNBS treatment (Braniste et al., Citation2010). When considering these data, it is essential to bear in mind that this latter research group also administered the same dose that we used (50 µg/kg/day), and observed little to no effect of BPA exposure on the same endpoints. When viewed collectively, these data suggest that higher levels of exposure to BPA may affect intestinal permeability or other aspects of GI tract homeostasis and inflammation, whereas lower levels of exposure to BPA may not.
The effects of BPA exposure in the context of other disease models present a rather mixed picture of the potential for BPA to modulate the function of the immune system (Midoro-Horiuti et al., Citation2010; Ohshima et al., Citation2007; Yan et al., Citation2008; Yoshino et al., Citation2004). Some of the discrepancies are likely due to variation in animal strain, immunological endpoints examined, the dose of BPA administered, as well as the timings and routes of exposure. Integrating the totality of the data is challenging, but, when one attempts to do so, the picture that forms is one in which exposure to BPA at or below 50 µg/kg/day generally has little effect on adaptive immune responses, but consistently modulates some aspects of inflammation. Altered responses of cells from BPA treated animals, such as elevated T-helper (TH)-2 cell cytokine production, enhanced ex vivo lymphocyte proliferation have been reported (Gallo et al., Citation2008; Guo et al., Citation2010); however, when these endpoints were examined in studies that used multiple doses of BPA, exposures in the mg/kg/day range generally altered these endpoints, whereas doses in the µg/kg/day range were rarely shown to affect metrics of lymphocyte function or alter disease progression (Goto et al., Citation2007; Lee et al., Citation2003; Sawai et al., Citation2003).
Our findings address the concern about whether direct oral or developmental exposure to BPA contributes to human disease. While it is difficult to prove whether or not a particular environmental agent has the capacity to affect a particular disease process or physiological system, our findings suggest that BPA does not discernably influence the onset, severity, or resolution of inflammatory colitis. Furthermore, when integrated with published data from our research team and by several other groups, it is difficult to find evidence that at exposures to BPA in the µg/kg/day range strongly affect the development or function of the immune system. In contrast, when given at higher doses (e.g. mg/kg/day range), exposure to BPA may affect the immune system. Yet, even at the lower (µg/kg/day) doses, subtle but consistent changes in tissue inflammation were observed, suggesting BPA may alter aspects epithelial or endothelial cell homeostasis or responses to injury. An interesting meta-analysis of the broader question of whether environmental exposures may affect IBD onset or severity suggests this is a worthy question (Soon et al., Citation2012). While BPA exposure alone seems not to be a direct contributor, it could well pose some risk to colon inflammation and human health in general in everyday situations where exposure occurs in conjunction with other environmental agents.
Declaration of interest
The authors report no conflicts of interests. The authors alone are responsible for the content and writing of the article.
Acknowledgments
We would like to thank Ms Jill M. Gresens, Dr Katherine Schaefer, and Dr Stephen Bauer for their technical help with aspects of this study.
This work was supported by the following research and training grants from the US National Institutes of Health: RC2-ES018750, R01-ES017250, R01-HL097141, P30ES01247, and T32-ES07026.
References
- Balakrishnan, B., Henare, K., Thorstensen, E. B., et al. 2010. Transfer of bisphenol A across the human placenta. Am. J. Obstet. Gynecol. 202:393. e1–7
- Bauer, S., Roy, A., Emo, J., et al. 2012. The effects of maternal exposure to bisphenol A on allergic lung inflammation into adulthood. Toxicol. Sci. 130: 82--93
- Bebo, B. F. Jr., Fyfe-Johnson, A., Adlard, K., et al. 2001. Low-dose estrogen therapy ameliorates experimental autoimmune encephalomyelitis in two different inbred mouse strains. J. Immunol. 166:2080–2089
- Blumberg, R. S., Saubermann, L. J., and Strober, W. 1999. Animal models of mucosal inflammation and their relation to human inflammatory bowel disease. Curr. Opin. Immunol. 11:648–656
- Bonefeld-Jorgensen, E. C., Long, M., Hofmeister, M. V., and Vinggaard, M. 2007. Endocrine disrupting potential of bisphenol A, bisphenol A dimethacrylate, 4-n-nonylphenol, and 4-n-octylphenol in vitro: New data and a brief review. Environ. Health Perspect. 115:69–76
- Bouma, G., and Strober, W. 2003. The immunological and genetic basis of inflammatory bowel disease. Nat. Rev. Immunol. 3:521–533
- Braniste, V., Jouault, A., Gaultier, E., et al. 2010. Impact of oral bisphenol A at reference doses on intestinal barrier function and sex differences after perinatal exposure in rats. Proc. Natl. Acad. Sci. USA. 107:448–453
- Calafat, A. M., Kuklenyik, Z., Reidy, J. A., et al. 2005. Urinary concentrations of bisphenol A and 4-nonylphenol in a human reference population. Environ. Health Perspect. 113:391–395
- Domoradzki, J. Y., Pottenger, L. H., Thornton, C. M., et al. 2003. Metabolism and pharmacokinetics of bisphenol A (BPA) and the embryo-fetal distribution of BPA and BPA-monoglucuronide in CD Sprague--Dawley rats at three gestational stages. Toxicol. Sci. 76:21–34
- Franks, A. L., and Slansky, J. E. 2012. Multiple associations between a broad spectrum of auto-immune diseases, chronic inflammatory diseases and cancer. Anticancer Res. 32:1119–1136
- Gallo, D., Battaglia, A., Mantuano, E., et al. 2008. 17β-Estradiol and soy phytochemicals selectively induce a Type 2 polarization in mesenteric lymph nodes of ovariectomized rats. Menopause 15:718–725
- Goto, M., Takano-Ishikawa, Y., Ono, H., et al. 2007. Orally administered bisphenol A-disturbed antigen specific immunoresponses in the naïve condition. Biosci. Biotechnol. Biochem. 71:2136–2143
- Guo, H., Liu, T., Uemura, Y., et al. 2010. Bisphenol A in combination with TNFα selectively induces TH2 cell-promoting dendritic cells in vitro with an estrogen-like activity. Cell. Mol. Immunol. 7:227–234
- Hartnett, L., and Egan, L. J. 2012. Inflammation, DNA methylation and colitis-associated cancer. Carcinogenesis 33:723–831
- Hengstler, J. G., Foth, H., Gebel, T., et al. 2011. Critical evaluation of key evidence on the human health hazards of exposure to bisphenol A. Crit. Rev. Toxicol. 41:263–291
- IRIS-EPA. 2012. Integrated Risk Information System, United States Environmental Protection Agency. Bisphenol A: CASRN 80-05-7. Available at: http://www.epa.gov/iris/subst/0356.htm [last accessed 17 October 2012]
- Kaser, A., Zeissig, S., and Blumberg, R. S. 2010. Inflammatory bowel disease. Annu. Rev. Immunol. 28:573–621
- Kim, E. Y., and Moudgil, K. D. 2008. Regulation of autoimmune inflammation by pro-inflammatory cytokines. Immunol. Lett. 120:1–5
- Lang, I. A., Galloway, T. S., Scarlett, A., et al. 2008. Association of urinary bisphenol A concentration with medical disorders and laboratory abnormalities in adults. JAMA 300:1303–1310
- Lee, M. H., Chung, S. W., Kang, B. Y., et al. 2003. Enhanced IL-4 production in CD4+ T-cells and elevated IgE levels in antigen-primed mice by bisphenol A and nonylphenol, endocrine disruptors: involvement of nuclear factor-AT and Ca2+. Immunology 109:76–86
- Midoro-Horiuti, T., Tiwari, R., Watson, C. S., and Goldblum, R. M. 2010. Maternal bisphenol A exposure promotes the development of experimental asthma in mouse pups. Environ. Health Perspect. 118:273–277
- Mielke, H., and Gundert-Remy, U. 2009. Bisphenol A levels in blood depend on age and exposure. Toxicol. Lett. 190:32–40
- Molodecky, N. A., Panaccione, R., Ghosh, S., et al. 2011. Challenges associated with identifying the environmental determinants of the inflammatory bowel diseases. Inflam. Bowel Dis. 17:1792–1799
- Molodecky, N. A., Soon, I. S., Rabi, D. M., et al. 2012. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 142:46–54
- Moriyama, K., Tagami, T., Akamizu, T., et al. 2002. Thyroid hormone action is disrupted by bisphenol A as an antagonist. J. Clin. Endocrinol. Metab. 87:5185–5190
- Morris, G. P., Beck, P. L., Herridge, M. S., et al. 1989. Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology 96:795–803
- Nishikawa, M., Iwano, H., Yanagisawa, R., et al. 2010. Placental transfer of conjugated bisphenol A and subsequent reactivation in the rat fetus. Environ. Health Perspect. 118:1196–1203
- NTP (National Toxicology Program). 2008. NTP-CERHR. Monograph on the Potential Human Reproductive and Developmental Effects of Bisphenol A. NIH publication No. 08-5994. Available online at: http://ntp.niehs.nih.gov/?objectid=49C35E83-C0BC-4D5F-4AF7273D67E03195 [last accessed 10 September 2012]
- Ohshima, Y., Yamada, A., Tokuriki, S., et al. 2007. Transmaternal exposure to bisphenol A modulates the development of oral tolerance. Pediatr. Res. 62:60–64
- Otsuka, H., Sugimoto, M., Ikeda, S., et al. 2012. Effects of bisphenol A administration to pregnant mice on serum Ca and intestinal Ca absorption. Anim. Sci. J. 83:232–237
- Qiu, B. S., Vallance, B. A., Blennerhassett, P. A., and Collins, S. M. 1999. The role of CD4+ lymphocytes in the susceptibility of mice to stress-induced reactivation of experimental colitis. Nat. Med. 5:1178–1182
- Richter, C. A., Birnbaum, L. S., Farabollini, F., et al. 2007. In vivo effects of bisphenol A in laboratory rodent studies. Repro. Toxicol. 24:199–224
- Roy, A., Bauer, S. M., and Lawrence, B. P. 2012. Developmental exposure to bisphenol A modulates innate but not adaptive immune responses to influenza A virus infection. PLoS ONE 7:e38448
- Sawai, C., Anderson, K., and Walser-Kuntz, D. 2003. Effect of bisphenol A on murine immune function: modulation of IFNγ, IgG2a, and disease symptoms in NZB × NZW F1 mice. Environ. Health Perspect. 111:1883–1887
- Schonfelder, G., Wittfoht, W., Hopp, T., et al. 2002. Parent bisphenol A accumulation in the human maternal-fetal-placental unit. Environ. Health Perspect. 110:A703–707
- Soon, I. S., Molodecky, N. A., Rabi, D. M., et al. 2012. The relationship between urban environment and the inflammatory bowel diseases: A systematic review and meta-analysis. BMC Gastroenterol. 12:51
- Uhlig, H. H., and Powrie, F. 2009. Autoimmune disease: inflammatory bowel disease. Eur. J. Immunol. 39:2021–2026
- Vandenberg, L. N., Maffini, M. V., Sonnenschein, C., et al. 2009. Bisphenol-A and the great divide: A review of controversies in the field of endocrine disruption. Endocr. Rev. 30:75–95
- Vom Saal, F. S., Akingbemi, B. T., Belcher, S. M., et al. 2007. Chapel Hill Bisphenol A Expert Panel Consensus Statement: integration of mechanisms, effects in animals and potential to impact human health at current levels of exposure. Repro. Toxicol. 24:131–138
- Westbrook, A. M., Szakmary, A., and Schiestl, R. H. 2010. Mechanisms of intestinal inflammation and development of associated cancers: Lessons learned from mouse models. Mutat. Res. 705:40–59
- Wetherill, Y. B., Akingbemi, B. T., Kanno, J., et al. 2007. In vitro molecular mechanisms of bisphenol A action. Repro. Toxicol. 24:178–198
- Yan, H., Takamoto, M., and Sugane, K. 2008. Exposure to bisphenol A prenatally or in adulthood promotes TH2 cytokine production associated with reduction of CD4+CD25+ regulatory T-cells. Environ. Health Perspect. 116:514–519
- Yoshino, S., Yamaki, K., Li, X., et al. 2004. Prenatal exposure to bisphenol A up-regulates immune responses, including T-helper 1 and T-helper 2 responses, in mice. Immunology 112:489–495