
Abstract
The elicitation of anti-drug antibodies (ADA) against biotherapeutics can have detrimental effects on drug safety, efficacy, and pharmacokinetics. The immunogenicity of biotherapeutics is, therefore, an important issue. There is evidence that protein aggregation can result in enhanced immunogenicity; however, the precise immunological and biochemical mechanisms responsible are poorly defined. In the context of biotherapeutic drug development and safety assessment, understanding the mechanisms underlying aggregate immunogenicity is of considerable interest. This review provides an overview of the phenomenon of protein aggregation, the production of unwanted aggregates during bioprocessing, and how the immune response to aggregated protein differs from that provoked by non-aggregated protein. Of particular interest is the nature of the interaction of aggregates with the immune system and how subsequent ADA responses are induced. Pathways considered here include ‘classical’ activation of the immune system involving antigen presenting cells and, alternatively, the breakdown of B-cell tolerance. Additionally, methods available to screen for aggregation and immunogenicity will be described. With an increased understanding of aggregation-enhanced immune responses, it may be possible to develop improved manufacturing and screening processes to avoid, or at least reduce, the problems associated with ADA.
Introduction
Since the breakthrough of recombinant DNA technology in the 1970s, and the subsequent introduction of recombinant human insulin as a drug in the early 1980s (Johnson, Citation1983), clinical use of protein therapeutics (also known as biotherapeutics) has increased dramatically. An availability of different classes of biotherapeutics, e.g. antibodies, hormones, and enzymes, provides useful tools in treatment of a wide range of diseases, including diabetes, rheumatoid arthritis, and hemophilia. Since their launch in the mid-1980s, therapeutic monoclonal antibodies (mAb) have evolved from mice, to chimeric and humanized derivatives, to fully human molecules (Wang et al., Citation2009). Early, non-human biotherapeutics were expected to provoke an immune response, as they would be recognized as foreign. Recombinant human biotherapeutics, however, are not expected to evoke an immune response in humans given their similarity to endogenous proteins. Indeed, recombinant human proteins do display reduced immunogenicity compared with non-human sequences (Wadhwa & Thorpe, Citation2007), yet formation of anti-drug antibodies (ADA) was noted after patient treatment with such therapeutics (Sauerborn et al., Citation2010; Schernthaner, Citation1993).
ADA pose a challenge in the biotherapeutics industry and clinical medicine as they can cause adverse events (like neutralization of endogenous protein) or reduce efficacy of a biotherapeutic. Immunogenicity is therefore a key limitation for the clinical use of biotherapeutics. Protein aggregation can increase the immunogenicity of biotherapeutics, and can be a key factor in causing adverse events associated with immunogenicity in the clinic (Rosenberg, Citation2006). A reduction in yield due to protein aggregation can also have a significant impact on development and manufacturing costs. Thus, despite the success of biotherapeutics and their expansion in the market, a tendency of proteins to aggregate during production and post-production stages creates challenges for industry and in the clinic, and acts to bottleneck development (Wang et al., Citation2012).
Protein aggregation
Definition and classification of aggregates
Aggregation is a broad term, encompassing the interactions which result in the self-association of protein molecules into assemblies other than the native quaternary structure (Narhi et al., Citation2012). Protein aggregates include a diverse range of protein assemblies that can differ in their biochemical and biophysical characteristics. They can range considerably in size, from dimers up to subvisible and visible particles (see ), they can involve covalent or non-covalent linkages, be ordered or disordered in structure, soluble or insoluble, and their formation can be reversible or irreversible. To study protein aggregation and understand the pathways that result in aggregate formation, it is important to classify protein aggregates according to their characteristics; however, the size range and diversity of protein aggregates make such characterization difficult. Additionally, use of imprecise and overlapping terms in the description of some aggregates makes interpretation of reports in the literature difficult. For example, the terms ‘subvisible particles’ and ‘oligomers’ may both be used to describe the same aggregate species. Narhi et al. (Citation2012) have attempted to remove these inconsistencies by suggesting standardized nomenclature to describe protein aggregates; this classification system accounts for aggregate size, reversibility of formation, conformation, chemical modification, and morphology. In this system, size is referred to using quantitative categories rather than imprecise terms, e.g. ‘1–100 µm’ is used rather than the term ‘subvisible particles’.
Figure 1. Schematic model of protein aggregation. Bioprocessing-associated stresses can trigger partial protein unfolding and initiate the aggregation process, beginning with the association of two or more protein molecules. Oligomers made from three or more monomers can form, leading to larger aggregates or ‘subvisible particles’. Linear aggregates form where proteins associate in a uniform manner (e.g. amyloid-type), whereas amorphous aggregates form by the association of proteins in a disordered manner. Visible particulates can then form, as pre-existing aggregates act as nuclei for formation of larger aggregates. Aggregate components are not drawn to scale.
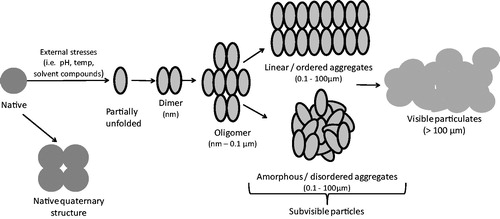
For the purpose of this review, ‘protein aggregation’ refers to the association of partially unfolded forms, which is relevant for the aggregation of therapeutic proteins during bioprocessing. It is important to note the distinction between these partially unfolded aggregate forms and amyloid fibrils, which are insoluble fibrous protein structures composed of β-sheet aggregates. Aggregation promoted by ‘amyloidogenic’ regions of a protein can be more reliably predicted on the basis of protein sequence. For example, regions with an increased capacity to form hydrogen bonds between peptide backbones and regions with a high packing density confer a higher probability of amyloid fibril formation (Garbuzynskiy et al., Citation2010). Prediction of the aggregation of partially unfolded forms is more challenging, however.
Aggregation and immunogenicity
Immunogenicity is defined as the ability of a substance (protein or chemical) to provoke an immune response. It is widely accepted in the scientific literature that protein aggregation can augment a protein-specific immune response, and lead to formation of ADA in the case of protein therapeutics (Rosenberg, Citation2006; Sauerborn et al., Citation2010). Given the ability of aggregation to enhance immune responses, aggregation can be used as a predictor for immunogenicity of biotherapeutics, and minimizing the risk of aggregation may reduce the risk of immunogenicity in the clinic. However, the majority of protein formulations contain at least a low level of aggregates, although the extent and type of aggregation that could pose a risk is not known (Weiss et al., Citation2009). Understanding the immunological mechanisms through which aggregation can influence the immunogenicity of proteins is, therefore, of considerable importance. The manufacture of biotherapeutics involves methods that can cause aggregation; these must be taken into account in bioprocess design in order to reduce aggregation-associated immunogenicity.
Aggregate formation in bioprocessing
Non-native aggregation describes the process of non-native protein structures assembling from initially native proteins (Chi et al., Citation2003). The loss of native structure is a common response of proteins to external stresses such as temperature, pH, co-solutes and adsorption to air–liquid and solid–liquid interfaces. Structurally altered proteins have a strong tendency to aggregate and additives that maintain native protein structure have been shown to reduce protein aggregation (Manning et al., Citation2010). Conformational stability, therefore, appears to be an important factor in controlling aggregation rates. Non-native aggregation is frequently observed at many stages of bioprocessing, including protein expression, purification, and storage. This process is generally irreversible, so reducing aggregation during manufacture requires controlled conditions (Weiss et al., Citation2009). Aggregation can also occur under conditions where the native state is favored thermodynamically and in the absence of stresses (Krishnan et al., Citation2002). For example, large aggregates with native conformation can form through adsorption to micro-particles, or at high concentration through ‘salting out’. This phenomenon is caused by protein exceeding the solubility limit above that required to cause precipitation as the salt concentration increases (Fink, Citation1998). More commonly in the production of protein therapeutics, aggregation is preceded by the loss of native protein structure; the resulting protein is then more susceptible to aggregation (see ). The mechanisms of protein aggregation are still not fully understood, but current evidence supports the role of partially folded intermediates in aggregation (Acosta-Sampson & King, Citation2010; Kim & Yu, Citation1996), and it is thought that exposure of hydrophobic surfaces on non-native protein structures allows inter-molecular interactions, leading to aggregate formation.
Native protein stability can be compromised by physical and chemical stressors; those associated with aggregation can be encountered at various stages of product development (Frokjaer & Otzen, Citation2005) from protein expression, to storage of the final product. Characterization of the protein is therefore required at each stage to ensure batch-to-batch uniformity and overall quality; this is particularly important for regulatory submissions. Proteins can aggregate early in product development at the protein expression stage. Therapeutic mAb are typically derived from mammalian cell culture and this process involves steps that can result in aggregation. For example, over-expression of recombinant proteins in cultures can lead to aggregation and intra-cellular inclusion body formation (Schrodel & de Marco, Citation2005). Protein purification techniques can also expose a protein to conditions like high ionic strength, pH values far from neutrality, and high protein concentrations, all which contribute to aggregation (Cromwell et al., Citation2006).
Proteins are surface-active agents, and are attracted to hydrophobic interfaces where they are prone to unfolding and subsequent aggregation (Gidalevitz et al., Citation1999; Horbett, Citation1988). Such interfaces are encountered at various points in the life of a biotherapeutic product (Dasnoy et al., Citation2011). Different biotherapeutic products have also been reported to aggregate at the air–liquid interface; here proteins can act as a detergent, migrating to the air–liquid interface, and this stress can cause partial unfolding. Susceptible proteins include insulin (Sluzky et al., Citation1991), factor VIII (Joshi et al., Citation2009), human growth hormone (hGH; Maa & Hsu, Citation1997), and IgG variants (Kiese et al., 2008; Mahler et al., Citation2005). Proteins are also subject to mechanical stress during the manufacturing process, i.e. as solutions of high protein concentration pass through piston pumps during fill-and-finish operations, molecules encounter high shear and mechanical stresses which can cause partial denaturation and, hence, aggregation (DePalma, Citation2006). Differences in product formulation can further impact on aggregation. Some solvent additives are used to stabilize protein preparations and prevent aggregation but, in contrast, some solvent compounds and contaminants promote aggregation (Arakawa et al., Citation1991). The stability of pH within formulations is important, as it is well known that acidic conditions can affect antibody structure, stability, and folding. It has been recognized that ionic strength and pH can play a key role in aggregation of mAb (Hari et al., Citation2010). Additionally, high concentration liquid formulations are often required to achieve the desired therapeutic dose: this can lead to aggregation as high concentrations promote protein association (Treuheit et al., Citation2002).
In order to optimize capacity, it is common practice to conduct large scale freezing of bulk protein preparations for storage. During this process there will be freeze/thaw stress that contributes to aggregation. The molecular mechanism of this stress-induced aggregation is not fully understood, but it is thought that proteins partially unfold at the ice–liquid interface and hydrophobic bonds weaken (Kreilgaard et al., Citation1998b). The formation of ice crystals can also be particularly damaging, as this can enhance protein unfolding (Hamada et al., Citation2009). The process of lyophilization involves dissolving a drug formulation in an aqueous matrix, which is then frozen, and the aqueous phase is removed by sublimation. This method is widely used in the biopharmaceutical industry to improve shelf life and increase stability (Tang & Pikal, Citation2004). Lyophilization can result in protein unfolding and, hence, aggregation, as the protein can be exposed to low temperature stresses (Townsend & Deluca, Citation1990). Conversely, lyophilization can also be used to avoid the aggregation seen in liquid formulations that is caused by partial unfolding due to agitation stress and exposure to liquid–solid interfaces within containers.
Some studies have suggested that the mechanism of aggregation is dependent on the nature of the stress applied (Joubert et al., Citation2011; Krishnan & Raibekas, Citation2009). As a result, aggregates can be heterogeneous with qualities attributable to a particular type of stress. Protein characterization following exposure to different stress conditions may, therefore, help to identify the stresses that cause aggregation and influence the bioprocess pipeline. Small changes to manufacturing processes, such as increases in production scale or changes to the formulation, could alter the immunogenic potential of a protein. As manufacturing processes evolve, different factors are introduced that may contribute to immunogenicity. Regulatory guidelines that are in place to ensure the safety of biotherapeutics must, therefore, accommodate these frequent changes in the manufacturing process.
Methods to reduce aggregation
Excipients
There have been recent developments in the use of excipients such as surfactants, amino acids, and pH buffers to reduce protein aggregation in formulated products (DePalma, Citation2006). Certain excipients are used specifically to inhibit aggregation at the air–liquid interface; generally these work by competing with the therapeutic protein for adsorption to the interface, thereby protecting the protein from exposure (Chou et al., Citation2005; Katakam et al., Citation1995). For example, non-ionic surfactants polysorbate 20 and 80 are typically used to prevent adsorption at the interface and subsequent aggregation (Kerwin, Citation2008). Silicone oil on the surface of syringes and stoppers can come into contact with biotherapeutics prior to delivery; this interaction has been implicated in aggregation. Silicone oil-induced aggregation can be reduced by polysorbate 20 (Thirumangalathu et al., Citation2009). Unfortunately, the use of polysorbate 80 in formulation can lead to formation of micelles that may cause aggregation and enhance immunogenicity (Villalobos et al., Citation2005). This observation highlights the need for careful selection of excipients.
Other excipients used to prevent adsorption at the air–liquid interface include cyclo-dextrin derivatives (Serno et al., Citation2010) and amino acids such as arginine, lysine, and glutamic acid (Dasnoy et al., Citation2011). Carbohydrates such as sucrose, dextrose, and trehalose have also been used as stabilizing excipients (Katakam & Banga, Citation1995; Kreilgaard et al., 1998a). These function by covering the surface of hydrophobic bonding sites to preserve the native structure (Andya et al., Citation2003). This is useful for the storage of freeze-dried proteins, as hydrophobic interactions play a key role in protein folding and support the native state; loss of these interactions under dehydration can destabilize the protein.
High throughput screening (HTS) technology may be used to assess the ability of excipients to protect against aggregation. Dasnoy et al. (Citation2011) developed a HTS stress test for studying aggregates at the air–liquid interface, allowing the evaluation of a large number of excipients for prevention of aggregation. Considerable variation (for example, in size and structure) is observed in aggregates formed during bioprocessing. Given the diversity of therapeutic proteins, the type of excipient used must be selected based on properties of each particular protein. Thus, optimal formulation for each protein of interest requires screening and optimization in order to achieve a formulation that provides minimal risk of aggregation. Research is required to understand whether formulation strategies used for typical aggregates can function effectively in the reduction of subvisible protein particles, which are believed to play an important role in aggregation of biotherapeutics (Carpenter et al., Citation2009).
Protein engineering
A protein engineering approach also can be used to minimize aggregation. For example, using a Pichia pastoris expression system, it has been shown that IgG molecules with lower levels of glycosylation were less prone to aggregation, and the presence of an N-terminal tetra-peptide extension increased the temperature at which aggregation was first induced (Schaefer & Plueckthun, Citation2012). Mutagenesis has also been used to prepare antibody variants with enhanced protein stability and reduced aggregation potential (Chennamsetty et al., Citation2009). In addition, in silico methods have been used to study protein aggregation in relation to bioprocessing; computational studies to examine the stabilizing effect of excipients look particularly promising (Cellmer et al., Citation2007). In silico methods for predicting protein aggregation and modeling optimized proteins are in their infancy, but they provide a potentially valuable tool in the design of biotherapeutics with reduced aggregation potential (Bratko et al., Citation2006).
Immunogenicity of biotherapeutics
Mechanisms of immunogenicity
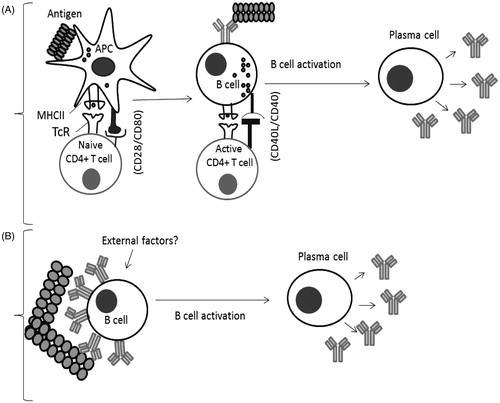
In order for a protein therapeutic to be immunogenic, it must interact with immune cells. There are essentially two ways in which a protein therapeutic may induce immune responses in the patient. The first is if the therapeutic agent is sufficiently ‘foreign’ to be recognized as such and induce an adaptive immune response. Thus, the therapeutic agent is internalized, processed, and presented by antigen-presenting cells (APC) resulting in CD4 T-cell responses and the elaboration of antibody (see ). If, however, the therapeutic agent shows high, or complete, homology with an endogenous protein to which the patient is immunologically tolerant, then that B-cell tolerance will need to be broken for an antibody response to be induced. Antibody production is the primary effector mechanism in immune responses to biotherapeutics and, while ADA can be without clinical consequences, side-effects can occur, varying from a loss of drug efficacy to anaphylaxis.
Figure 2. B-Cell activation mechanisms. (A) Classical response: Antigen is internalized by APC, and processed to peptide fragments that bind to major histocompatibility complex II (MHCII). Recognition by CD4+ T-cells stimulates cytokine secretion and B-cell activation followed by differentiation to plasma cells. (B) Breakdown of B-cell tolerance: B-cells can be activated to plasma cells by antigens possessing repetitive epitopes which cross-link antigen-specific BcR, triggering activation signals. External factors/signals could play a role in this process (Sauerborn et al., Citation2010; Ragheb & Lisak, Citation2011).
Immune tolerance
T- and B-cells express T-cell receptors (TcR) and B-cell receptors (BcR), respectively, for antigen recognition. These are generated by random gene rearrangements (somatic mutation) to ensure a very wide repertoire and the ability to recognize a vast array of foreign antigens. By chance, some receptors are often able to recognize self-antigen. Central tolerance mechanisms exist to prevent the survival and proliferation of these self-reactive immune cells, thereby preventing autoimmunity. The process of negative selection results in the deletion of developing B- and T-cells that recognize self-antigens in the environment of the bone marrow and thymus, respectively. Any remaining self-reactive lymphocytes are controlled by anergy or peripheral tolerance. B-cell peripheral tolerance is caused by exposure to circulating soluble antigen and low levels of BcR cross linkage (Andrews & Wilson, Citation2010). In CD4+ T-cells, peripheral tolerance is maintained by the following mechanisms; functional anergy rendering the T-cell unresponsive, deletion of the cell by apoptosis following cell activation, and suppression of T-cell activation by regulatory T (Treg) cells (Abbas et al., Citation2004).
Classical immune response to foreign antigen
Foreign antigens trigger a ‘classical’ immune reaction that is dependent upon T-cell activation. This mechanism requires interaction of antigen with APC that, in turn, prime naïve T-cells. Primed T-cells may then interact with B-cells displaying the antigen within a major histo-compatability complex (MHC) molecule. Interaction with co-stimulatory molecules (such as CD28/CD80) further activates T-cells and stimulates cytokine secretion, leading to the proliferation of B-cells and antibody production. Isotype switching from IgM to IgG is a hallmark of this T-cell mediated immune response (Avery et al., Citation2008). The presence of IgG ADA is often indicative of T-cell help, although some isotype switching can take place in the absence of T-cells (Sauerborn et al., Citation2010). This classical type of immune response is found in patients lacking immune tolerance to a human therapeutic protein, or in response to modified proteins containing non-self epitopes. Aggregates of recombinant human proteins may also induce this type of immune response. It is known that aggregates have the ability to activate APC, and are more easily phagocytozed (Scott & de Groot, Citation2010). Furthermore, Joubert et al. (Citation2012) recently demonstrated the ability of protein aggregates to induce an adaptive T-cell response in vitro using cultured human peripheral blood mononuclear cells stimulated with aggregated mAb. Here, aggregates induced innate signals through cell surface receptors, including Toll-like receptors that could develop into an adaptive T-cell response, characterized by CD4+ T-cell proliferation and cytokine profiling. A cytokine signature, including interleukin (IL)-1β IL-6 and tumor necrosis factor-α, was identified as a potential biomarker for aggregate immunogenicity, and this may find application in the in vitro prediction of biotherapeutic immunogenicity. However, results recorded in vitro may not be truly representative of the in vivo situation; in particular, the relationship between this biomarker and adverse effects associated with ADA formation is unknown.
Breakdown of B-cell tolerance
Unresponsive or anergic B-cells are unable under normal circumstances to respond to endogenous ‘self’ proteins that bear antigens for which they have specific receptors. However, these cells can be activated if they receive an appropriate signal from T-cells. The way in which antigen is displayed can also influence the responsiveness of tolerant B-cells, as organized structures have been found to be optimal for T-cell-independent immune responses to self-antigen in mice (Bachmann et al., Citation1993; Ohashi & DeFranco, Citation2002). Antigen organization in an ordered protein aggregate differs from the monomeric protein (see ). It has been hypothesized that repetitive epitope structures formed by aggregation may be capable of activating B-cells directly, as proposed by Bachmann and Zinkernagel (Citation1997). Aggregates may crosslink BcR in a manner that stimulates a downstream signaling cascade followed by differentiation of B-cells into plasma cells and secretion of IgG ADA. The ability of highly ordered structures to elicit more potent immune responses compared with the monomeric protein has been shown using viral coat protein from vesicular stomatitis virus (Bachmann & Zinkernagel, Citation1997). Additionally, the highly ordered structure of papaya mosaic virus, used as a vaccine platform, has been shown to be critical for generation of an efficient humoral response (Babin et al., Citation2013).
It is not unexpected that highly ordered oligomerized antigens can be more immunogenic, as they resemble the structure of foreign microorganisms such as viruses, and may be recognized as such by the immune system. Epitopes spaced 5–10 nm apart are characteristic of microbial antigens and it is hypothesized that the immune system has evolved to recognize and respond to this type of antigen (Hermeling et al., Citation2004; Kessler et al., Citation2006). Thus, a potential mechanism for the activation of the immune system by protein aggregates is the organization of protein structures into viral-like arrays and subsequent breaking of B-cell tolerance. The repetitive structure of pathogenic microbes is able to induce T-cell-independent responses (Szomolanyi-Tsuda & Welsh, Citation1998). This may occur through BcR engagement, which has been shown to lead to the breakdown of tolerance (Kouskoff et al., Citation2000), possibly through receptor cross-linking and activation of downstream proliferative signals. The mechanism responsible may not be strictly T-cell-independent as T-helper (TH) cells may also help B-cells to fully respond to antigen, possibly by the release of cytokines from activated T-cells or non-specific T-cell interaction (Hunziker et al., Citation2003; Soulas et al., Citation2005). Unlike the classical immune response that leads to the development of immunological memory, direct activation of B-cells leads predominantly to the formation of low affinity IgM in the absence of memory. Although there will only be effective class switching if there is involvement of TH cells, some isotype switching to IgG can occur via an unknown mechanism (Lange et al., Citation2008; Sauerborn et al., Citation2010).
Factors involved in immunogenicity of aggregates
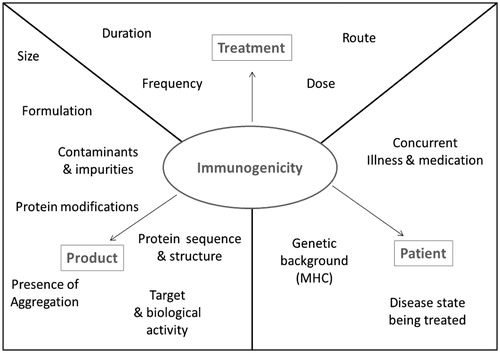
Various intrinsic and extrinsic factors can affect the propensity of a protein to stimulate an immune response. Many extrinsic factors contribute towards determining the immunogenicity of biotherapeutic proteins; these can be treatment-related factors such as the dosing regimen, patient related factors such as MHC variants, or product related factors such as product design (Singh, Citation2011). These contributing factors (illustrated in ) can all influence the immunogenicity of a protein aggregate and make it difficult to attribute immunogenicity to one source. Intrinsic factors relate to the protein itself, these include the presence of epitopes that are recognized specifically by receptors of the immune system.
Figure 3. Factors that may influence biotherapeutic immunogenicity. Treatment, product, and patient related factors that can impact upon the immunogenic potential of a biotherapeutic.
Clinical examples of aggregation associated immunogenicity
There is clinical evidence, from early studies in the 1960s through to more recent examples, that aggregation of therapeutic proteins can affect immunogenicity (Rosenberg, Citation2006; Villalobos et al., Citation2005). ADA induced by the breakdown of B-cell tolerance generally first appear months after treatment and disappear after treatment is terminated; few proteins induce a classical vaccine-type immune response where antibodies may persist for years (Schellekens, Citation2010). Biotherapeutic immunogenicity can result in no observable effect to a range of clinical manifestations; these include neutralization of therapeutic effectiveness, which may result in a worsening of the existing disease (Farrell et al., Citation2012), reactivity with host protein homologues (Casadevall et al., Citation2002), and adverse reactions such as haematotoxicity (Everds & Tarrant, Citation2013). There is also the potential for biotherapeutics to induce anaphylaxis. Although chimeric/humanized antibodies result in much reduced immunogenicity compared with original mouse antibodies, even these antibodies can result in anaphylaxis (Harding et al., Citation2010; Radstake et al., Citation2009). For example, anaphylactic shock has been recorded upon second exposure to chimeric anti IL-2 receptor (Baudouin et al., Citation2003). A patient treated with this therapy developed an IgE response that triggered anaphylactic shock on further exposure. It is possible that aggregation could exacerbate this effect. Examples of some specific biotherapeutics are discussed below to illustrate the clinical consequences of immunogenicity.
Eprex
A well-documented example of biotherapeutic immunogenicity is the antibody responses that developed in patients receiving treatment with human erythropoietin (epoetin-α) Eprex®. This was associated with the development of pure red cell aplasia (PRCA) among patients with chronic renal failure (Casadevall et al., Citation2002). Erythropoietin is a hormone that is required for red blood cell (RBC) development, and PRCA manifests as severe sudden-onset anemia that is characterized by the absence of red cell precursors in the bone marrow (Boven et al., Citation2005b). In Eprex®-treated individuals, PRCA was caused by the formation of neutralising antibodies against the administered protein, these antibodies also bound to the endogenous protein. PRCA incidence was rare in treated individuals; between 2001–2003, 50 patients out of 100,000 were affected. The mean time between initial exposure to Eprex® and diagnosis of PRCA was 9.1 months (Bennett et al., Citation2004). There is evidence to suggest that a number of factors such as micelle formation, route of exposure, and contaminants from rubber stoppers may have contributed to the immunogenicity of Eprex®; these factors will be discussed in more detail.
PRCA was observed in patients treated with the epoetin-α formulation that contained polysorbate 80 (Villalobos et al., Citation2005). An earlier formulation of Eprex® contained human serum albumin; this was replaced with polysorbate 80 as a stabilizer (Haselbeck, Citation2003) and this new formulation was shown to encourage formation of micelle-associated epoetin. Multimeric epitopes are formed when epoetin-α molecules associate with micelles, and it has been suggested these multimeric epitopes formed by the protein in the micelles may be responsible for induction of ADA following BcR recognition and cross-linking (Hermeling et al., Citation2003). This is supported by evidence that protein multimerization can lead to more efficient immune responses compared with dimers and trimers of the same protein (Rosenberg, Citation2006). It is also relevant that, in the case of Eprex®, the route of administration was an important contributory factor. Subcutaneous (SC) injection resulted in ADA formation, whereas this response was not reported following intra-venous (IV) administration. Hermeling et al. (Citation2003) proposed that micelle-associated epoetin may remain intact and interact with immune cells following SC administration, whereas following IV injection micelles are immediately dispersed in the bloodstream upon the dilution of polysorbate 80. Another factor thought to have contributed significantly to the immunogenicity of epoetin-α formulated with polysorbate 80 is the leaching of organic compounds from uncoated rubber stoppers in pre-filled syringes (Boven et al., Citation2005b); evidence indicates that these compounds could act as adjuvants (Boven et al., Citation2005a; Locatelli et al., Citation2007). Additionally, the presence of silicone oil in pre-filled syringes has been implicated in the induction of protein aggregation (Kossovsky et al. Citation1987; Thirumangalathu et al., Citation2009). Although it is clear that one or more aspects of the formulation were responsible for the immunogenicity of Eprex®, the proliferation of hypotheses that seek to explain the mechanism behind these adverse events suggests that the precise reason is still unknown (Thirumangalathu et al., Citation2009).
IFNβ
Interferon (IFN)-β is the main treatment for relapsing-remitting multiple sclerosis (MS). The induction of ADA poses a challenge for the treatment of MS with recombinant human IFNβ. Antibodies produced in patients can be a mixture of neutralizing and non-neutralizing antibodies. Neutralizing antibody prevents IFNβ binding to its receptor; this can have serious effects in MS patients by reducing drug efficacy and accelerating disease progression, with an increase in relapse rates (Farrell et al., Citation2012). Approximately 25% of patients develop a neutralizing antibody to IFNβ products; these generally appear 6–18 months after first exposure to IFNβ (Ross et al., Citation2000). In a clinical trial in which patients either received IFNβ1a by subcutaneous (Rebif®) or by intramuscular (Avonex®) injection, the subcutaneously-administered protein was more antigenic with respect to both the incidence of neutralizing antibody (25% and 2% for Rebif and Avonex, respectively) and the antibody titer. Similar numbers of patients remained relapse-free (75% and 63%, respectively), indicating that the presence of antibodies may not always be associated with loss of clinical efficacy (Panitch et al., Citation2002). However, those authors did acknowledge that the relatively short timeframe for the clinical trial (48 weeks) might have impacted on the lack of a relationship between the presence of neutralizing antibody and efficacy.
Two types of IFNβ are used therapeutically; one is IFNβ-1a produced in Chinese hamster ovary cells, with an identical amino acid sequence and similar glycosylation patterns to the endogenous human protein. The second is IFNβ-1b produced in Escherichia coli that is, therefore, unglycosylated; this protein is less potent than IFNβ-1a, which is thought to be a consequence of the lack of glycosylation, as sugar residues can stabilize some proteins. Without the glycan chain, the protein can aggregate through disulfide-linked complexes, and it is this feature that is associated with increased immunogenicity and decreased biological activity (Farrell et al., Citation2012). IFNβ-1a and -1b therefore display differences in immunogenicity secondary to differences in glycosylation. The immunogenicity of IFNβ can also be enhanced by chemical modifications such as oxidation and deamidation, which can cause degradation (Hermeling et al., Citation2004). For example, oxidation-mediated aggregation has been reported to increase immunogenicity of IFNβ-1a in immune-tolerant transgenic mice compared with aggregates formed in the absence of oxidative stress (van Beers et al., Citation2011).
Other examples
In other examples, immune reactions against aggregates of injected human gamma-globulin were reported in the 1960s, with the production of antibodies specific for antigen either formed or revealed during aggregation (Ellis & Henney, Citation1969). Clinical studies of recombinant IFN-α2a for treatment of malignancies and viral diseases also reveal a strong correlation between immunogenicity and the presence of protein aggregation (Ryff, Citation1997). Additionally, persistent antibodies have been noted in patients with growth hormone deficiency treated with heavily-aggregated hGH, whereas transient antibodies were seen in patients given less aggregated forms (Moore & Leppert, Citation1980). Similarly, hGH preparations with lower aggregate concentrations of smaller sizes resulted in reduced immunogenicity in mice (Fradkin et al., Citation2009).
Factor VIII is a clotting factor that is deficient in patients with hemophilia A. Recombinant human factor VIII (rFVIII) is used to treat this condition; however, 15–30% of treated hemophilia patients develop ADA. Studies have indicated that aggregation of rFVIII provides a distinct antigen, or neo-epitope, to which an immune response is mounted (Josic et al., Citation1999; Purohit et al., Citation2006). Importantly, in this case endogenous protein does not display the neoepitope, so such antibodies lack neutralizing properties.
Omalizumab is a recombinant humanized anti-IgE mAb used to treat allergic asthma and rhinitis (Kim et al., Citation2010), ≈0.1% of patients treated with this mAb suffer an anaphylactic reaction. There is no clear mechanism behind this reaction, although the polysorbate excipient used in product formulation has been associated with hypersensitivity reactions (Price & Hamilton, Citation2007). Links have been made between adverse reactions in patients treated with omalizumab and those treated with Eprex®, which also contained a polysorbate excipient, as previously discussed.
Vaccines
In vaccine development, the induction of humoral immunity has been linked to protein aggregation (Wang et al., Citation2012). Thus, observations made in vaccine development may help to understand the role of aggregation in the unwanted immunogenicity of biotherapeutic products. In animal models oligomeric and multimeric forms of protein antigens have been shown to be more immunogenic than the monomeric forms (Denis et al., Citation2007; Qian et al., Citation2012; Rudra et al., Citation2010). For example, multimerization of papaya mosaic virus capsid protein as a carrier protein for hepatitis C virus is critical for immunogenicity of the vaccine in mice, which is absent with the monomeric form (Denis et al., Citation2007). This evidence supports the hypothesis that repetitive epitopes of protein aggregates can interact with immune cell receptors, leading to the breakdown of immune tolerance.
Increased immunogenicity of vaccine proteins is generally an advantage; however, the quality of immune response is also important. CD4+ TH cells polarize into distinct sub-sets: T-helper 2 (TH2) cells that activate naive B-cells to divide and secrete antibody, and T-helper-1 (TH1) cells that activate macrophages and stimulate cellular immunity. TH1 and TH2 cells have reciprocal antagonistic effects. Vaccines inducing Type 1 immunity were protective in animal models, whereas vaccines that stimulate Type 2 immunity could increase susceptibility to infection (Spellberg & Edwards, Citation2001). Oculorespiratory syndrome (ORS), associated with respiratory symptoms and conjunctivitis (Skowronski et al., Citation2003b), was recorded as an adverse effect in patients treated with an inactivated influenza vaccine (Babiuk et al., Citation2004). The implicated vaccine had been manufactured using a different viral splitting agent and was found to contain large aggregates of ∼500 unsplit virions, which were absent from competitor vaccines that did not provoke the adverse effect. A trend towards Type 2 polarization was also reported among vaccinated patients with ORS (Skowronski et al., Citation2003a). Experimental (mouse) studies have also shown that the strength and type of cellular immune response to the vaccine varies with formulation and extent of aggregation (Babiuk et al., Citation2004). Further investigation is required to understand how formulation could affect polarization of an immune response.
Screening for aggregation and immunogenicity
Due to the costly loss of protein and immunogenicity associated with protein aggregation, screening for aggregation is used in the development of biopharmaceuticals. More recently attention has focused on subvisible particles below 10 µm due to concerns over immunogenicity (Carpenter et al., Citation2009; Zoells et al., Citation2012). Analyzing protein aggregates can be challenging due to the unknown nature of the aggregates that may have formed in therapeutic protein formulations (den Engelsman et al., Citation2011). Protein aggregates are generally distinguished from protein monomers by mass balance or size; however, particles in the 1–100 µm size range are too small to be visible, but too large for size-exclusion chromatography (SEC) analysis, a standard technique for aggregate detection (Carpenter et al., Citation2009). Currently there is no single method available for detection of the whole size range of aggregates that may arise from bioprocessing (Kiese et al., 2008). Additionally, there is a lack of knowledge regarding the size and type of aggregates that can induce harmful ADA. Analytical methods differ with respect to measuring principle and the information that they provide, so a combination of techniques is necessary for characterization of aggregates. Some of the key techniques for analysis of protein particles and aggregates of different sizes are summarized in . Methods are discussed in greater detail in a previous review (den Engelsman et al., Citation2011).
Table 1. Methods for aggregate analysis in therapeutic protein development.
Immunogenicity testing
Regulatory agencies have recognized the importance of screening for protein aggregation and there is consequently increased pressure on the biotherapeutics industry arising from concerns over safety and efficacy. In order to demonstrate clinical safety and efficacy, immunogenicity testing is now a key component of biotherapeutic drug development. Clinical studies are also required under International Conference on Harmonization (ICH) guidelines to measure various ADA and characterize the ADA response. The formation of neutralizing antibodies can affect safety and efficacy, but non-neutralizing antibodies can also be a concern due to effects on half-life and biodistribution (Shankar et al., Citation2006).
The induction of ADA in animals and patients is a key endpoint concerning the immunogenicity of biotherapeutics. To study the immunogenicity of protein therapeutics, methods to detect the presence of and to characterize antibodies are required. A range of techniques exist which are useful for investigating the presence of antigen-specific antibody; these include immunoassays that can identify antibodies capable of binding to antigen and bioassays that can distinguish between neutralizing and non-neutralizing antibodies (Wadhwa & Thorpe, Citation2007). A more detailed survey is beyond the scope of this article.
Animal models
Animal models are a potentially useful approach to measure antibody responses to bio-therapeutics. The 2011 ICH S6 Guideline (pre-clinical safety evaluation of biotechnology-derived pharmaceuticals) describes the need for detection and characterization of antibodies in repeat dose studies using animal models. A relevant species must be used for in vivo studies, i.e. one in which the target epitope is expressed. Although non-clinical immunogenicity studies are required, immune responses are species-specific; therefore, induction is not entirely predictive of antibody formation in humans (Shankar et al., Citation2006; Wang et al., Citation2008). Species-specific immunogenicity is related to the lack of genetic diversity in animal models, as genetic diversity is implicated in immunogenicity (Brinks et al., Citation2011). Rodent models for immunogenicity testing are, therefore, less useful than animals that show a higher degree of homology with humans and more genetic diversity than inbred mouse strains, such as non-human primates; however, these are not widely used due to ethical constraints. Conventional non-transgenic animal models can be useful for highly-conserved proteins, but a lack of immune tolerance to human proteins limits their use for immunogenicity testing. These animal models can be useful for comparing the immunogenicity of two similar products, i.e. the immunogenicity of an originator and biosimilar product; this may not reflect the human situation but may provide a warning against advancement of a biosimilar if the immunogenicity profile observed differs to that of the originator.
Despite the limitations associated with the use of animals to predict immunogenicity, several transgenic animal models have been generated for this purpose. Transgenic mice are often the preferred in vivo model to predict immunogenicity as they are immune tolerant to the administered human protein (Hermeling et al., Citation2006; van Beers et al., Citation2010) and can be used to study the immunogenicity of biotherapeutic aggregates. For example, in a study by van Beers et al. (Citation2010), the IFNβ-1a aggregate percentage and extent of denaturation were shown to influence the ability of aggregates to break tolerance in transgenic mice. In these experiments, immune tolerant mice were immunized with IFNβ-1a formulations and antibody responses measured. Only non-covalently bound aggregates that retained some native epitopes were able to break tolerance resulting in a transient immune response, removal of aggregates prevented this breakdown of tolerance (van Beers et al., Citation2010). Additionally, mice expressing human MHC molecules can be used to compare antibody and T-cell responses to vaccines and protein therapeutics (de Groot & Martin, Citation2009). Use of animal models in immunogenicity testing is discussed more extensively in a recent review (Brinks et al., Citation2011).
In vitro assays
A number of in vitro techniques can also be used to assess the immunogenic potential of therapeutic proteins. These could be used to predict the risk of immunogenicity in a pre-clinical setting. The expression of APC-surface molecules differs following activation; for example, the expression of MHC (Class I and II), costimulatory molecules, and cytokine receptors is enhanced. Flow cytometry is an in vitro technique that can be used to determine differences in cell surface molecule expression indicative of APC maturation that may initiate T-cell responses (Gaitonde & Balu-Iyer, Citation2011). [3H]-Thymidine based T-cell proliferation assays are also useful tools to study the activation and proliferation of T-cells in the presence of antigen (Joubert et al., Citation2012). Additionally, the release of immunomodulatory cytokines can be characterized by enzyme-linked immunosorbent assay. This approach can be used to assess the quality of an induced immune response, as specific cytokines can be markers of TH1 (IL-12 and IFNγ) or TH2 immunity (IL-4 and IL-10). T-cells that respond to a particular epitope in vitro can be labeled with MHC Class II oligomers and sorted by flow cytometry, the phenotype of responsive T-cells can then be determined using intracellular cytokine staining (de Groot & Martin, Citation2009; Tobery et al., Citation2006). In addition to the assays described above, in silico techniques have been developed for the prediction of antigenicity by identification of potential T-cell epitopes (Tovey et al., Citation2011). In silico methods have been shown to successfully identify MHC Class II restricted epitopes within biotherapeutics (Koren et al., Citation2007). Knowledge of aggregation-prone regions may also help in the design and selection of biotherapeutic candidates and reduce aggregation concerns (Wang et al., Citation2009). For example, aggregation motifs that lack charge have been found in the light chain regions of mAbs including Erbitux® and Raptiva®. This computational approach could, therefore, be useful to screen biotherapeutic candidates early in drug development.
Concluding remarks
The development of unwanted immunogenicity against biotherapeutics poses significant clinical, scientific, and manufacturing challenges. A breakdown in tolerance following the formation of aggregates with repetitive epitopes seems to be an important mechanism by which ADA are induced. However, the precise immunological mechanisms remain poorly defined. There are still many unanswered questions regarding the immunogenicity of protein aggregates, including whether ADA formation is caused primarily by a breakdown of B-cell tolerance through direct interaction with BcR or a more classical mechanism involving APC and T-cell activation. It is also unknown what type or level of aggregation is required to induce an unwanted immune response. With these questions in mind, it is important to increase our understanding of mechanisms underlying protein aggregate immunogenicity; this may allow for more effective screening and improved manufacture to avoid aggregation-associated adverse events.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Abbas, A. K., Lohr, J., Knoechel, B., and Nagabhushanam, V. 2004. T-Cell tolerance and autoimmunity. Autoimmun. Rev. 3:471–475
- Acosta-Sampson, L., and King, J. 2010. Partially folded aggregation intermediates of human yD-, yC-, and yS-crystallin are recognized and bound by human αB-crystallin chaperone. J. Mol. Biol. 401:134–152
- Andrews, S. F., and Wilson, P. C. 2010. The anergic B-cell. Blood 115:4976–4978
- Andya, J. D., Hsu, C. C., and Shire, S. J. 2003. Mechanisms of aggregate formation and carbohydrate excipient stabilization of lyophilized humanized monoclonal antibody formulations. AAPS pharmSci. 5:21–31
- Arakawa, T., Kita, Y., and Carpenter, J. F. 1991. Protein-solvent interactions in pharmaceutical formulations. Pharm. Res. 8:285–291
- Avery, D. T., Bryant, V. L., Ma, C. S., et al. 2008. IL-21-induced isotype switching to IgG and IgA by human naive B-cells is differentially regulated by IL-4. J. Immunol. 181:1767–1779
- Babin, C., Majeau, N., and Leclerc, D. 2013. Engineering of papaya mosaic virus (PapMV) nanoparticles with a CTL epitope derived from influenza NP. J. Nanobiotech. 11:1477–3155
- Babiuk, S., Skowronski, D. M., De Serres, G., et al. 2004. Aggregate content influences the THl/TH2 immune response to influenza vaccine: Evidence from a mouse model. J. Med. Virol. 72:138–142
- Bachmann, M. F., and Zinkernagel, R. M. 1997. Neutralizing antiviral B-cell responses. Ann. Rev. Immunol. 15:235–270
- Bachmann, M. F., Rohrer, U. H., Kundig, T. M., et al. 1993. The influence of antigen organization on B-cell responsiveness. Science 262:1448–1451
- Baudouin, V., Crusiaux, A., Haddad, E., et al. 2003. Anaphylactic shock caused by immunoglobulin E sensitization after pre-treatment with chimeric anti-IL-2 receptor monoclonal antibody basiliximab. Transplantation 76:459–463
- Bennett, C. L., Luminari, S., Nissenson, A. R., et al. 2004. Pure red-cell aplasia and epoetin therapy. New Engl. J. Med. 351:1403–1408
- Boven, K., Knight, J., Bader, F., et al. 2005a. Epoetin-associated pure red cell aplasia in patients with chronic kidney disease: Solving the mystery. Nephrol. Dialysis Transplant. 20:33–40
- Boven, K., Stryker, S., Knight, J., et al. 2005b. The increased incidence of pure red cell aplasia with an Eprex formulation in uncoated rubber stopper syringes. Kidney Int. 67:2346–2353
- Bratko, D., Cellmer, T., Prausnitz, J. M., and Blanch, H. W. 2006. Effect of single-point sequence alterations on the aggregation propensity of a model protein. J. Am. Chem. Soc. 128:1683–1691
- Brinks, V., Jiskoot, W., and Schellekens, H. 2011. Immunogenicity of therapeutic proteins: The use of animal models. Pharm. Res. 28:2379–2385
- Carpenter, J. F., Randolph, T. W., Jiskoot, W., et al. 2009. Overlooking subvisible particles in therapeutic protein products: Gaps that may compromise product quality. J. Pharm. Sci. 98:1201–1205
- Casadevall, N., Nataf, J., Viron, B., et al. 2002. Pure red-cell aplasia and anti-erythropoietin antibodies in patients treated with recombinant erythropoietin. New Engl. J. Med. 346:469–475
- Cellmer, T., Bratko, D., Prausnitz, J. M., and Blanch, H. W. 2007. Protein aggregation in silico. Trends Biotech. 25:254–261
- Chennamsetty, N., Voynov, V., Kayser, V., et al. 2009. Design of therapeutic proteins with enhanced stability. Proc. Natl. Acad. Sci. USA 106:11937–11942
- Chi, E. Y., Krishnan, S., Randolph, T. W., and Carpenter, J. F. 2003. Physical stability of proteins in aqueous solution: Mechanism and driving forces in nonnative protein aggregation. Pharm. Res. 20:1325–1336
- Chou, D. K., Krishnamurthy, R., Randolph, T. W., et al. 2005. Effects of Tween 20 (R) and Tween 80 (R) on the stability of albutropin during agitation. J. Pharm. Sci. 94:1368–1381
- Cromwell, M. E., Hilario, E., and Jacobson, F. 2006. Protein aggregation and bioprocessing. AAPS J. 8:E572–E579
- Dasnoy, S., Dezutter, N., Lemoine, D., et al. 2011. High-throughput screening of excipients intended to prevent antigen aggregation at air-liquid interface. Pharm. Res. 28:1591–1605
- de Groot, A. S., and Martin, W. 2009. Reducing risk, improving outcomes: Bioengineering less immunogenic protein therapeutics. Clin. Immunol. 131:189–201
- den Engelsman, J., Garidel, P., Smulders, R., et al. 2011. Strategies for the assessment of protein aggregates in pharmaceutical biotech product development. Pharm. Res. 28:920–933
- Denis, J., Majeau, N., Acosta-Ramirez, E., et al. 2007. Immunogenicity of papaya mosaic virus-like particles fused to a hepatitis C virus epitope: Evidence for the critical function of multimerization. Virology 363:59–68
- DePalma, A. 2006. Improving stability while adding value. Gen. Eng. News 26:[online]. Available at: http://www.genengnews.com/gen-articles/improving-stability-while-adding-value/1259/. [last accessed 22 Apr 2013]
- Ellis, E. F., and Henney, C. S. 1969. Adverse reactions following administration of human gamma globulin. J. Allergy 43:45–54
- Everds, N. E., and Tarrant, J. M. 2013. Unexpected hematologic effects of biotherapeutics in nonclinical species and in humans. Toxicol. Pathol. 41:280–302
- Farrell, R. A., Marta, M., Gaeguta, A. J., et al. 2012. Development of resistance to biologic therapies with reference to IFNβ. Rheumatology 51:590–599
- Fink, A. L. 1998. Protein aggregation: Folding aggregates, inclusion bodies, and amyloid. Fold. Design 3:9–23
- Fradkin, A. H., Carpenter, J. F., and Randolph, T. W. 2009. Immunogenicity of aggregates of recombinant human growth hormone in mouse models. J. Pharm. Sci. 98:3247–3264
- Frokjaer, S., and Otzen, D. E. 2005. Protein drug stability: A formulation challenge. Nat. Rev. Drug Disc. 4:298–306
- Gaitonde, P., and Balu-Iyer, S. V. 2011. In vitro immunogenicity risk assessment of therapeutic proteins in preclinical setting. Drug Design Disc. Meth. Protocols 716:267–280
- Garbuzynskiy, S. O., Lobanov, M. Y., and Galzitskaya, O. V. 2010. FoldAmyloid: A method of prediction of amyloidogenic regions from protein sequence. Bioinformatics 26:326–332
- Gidalevitz, D., Huang, Z. Q., and Rice, S. A. 1999. Protein folding at the air-water interface studied with x-ray reflectivity. Proc. Natl. Acad. Sci. USA 96:326–332
- Hamada, H., Arakawa, T., and Shiraki, K. 2009. Effect of additives on protein aggregation. Curr. Pharm. Biotech. 10:400–407
- Harding, F. A., Stickler, M. M., Razo, J., and DuBridge, R. B. 2010. The immunogenicity of humanized and fully human antibodies residual immunogenicity resides in the CDR regions. mAbs 2:256–265
- Hari, S. B., Lau, H., Razinkov, V. I., et al. 2010. Acid-induced aggregation of human monoclonal IgG1 and IgG2: Molecular mechanism and the effect of solution composition. Biochemistry 49:9328–9338
- Haselbeck, A. 2003. Epoetins: Differences and their relevance to immunogenicity. Curr. Med. Res. Opin. 19:430–432
- Hermeling, S., Crommelin, D. J., Schellekens, H., and Jiskoot, W. 2004. Structure-immunogenicity relationships of therapeutic proteins. Pharm. Res. 21:897–903
- Hermeling, S., Schellekens, H., Crommelin, D. J., and Jiskoot, W. 2003. Micelle-associated protein in epoetin formulations: A risk factor for immunogenicity? Pharm. Res. 20:1903–1907
- Hermeling, S., Schellekens, H., Maas, C., et al. 2006. Antibody response to aggregated human interferon α2b in wild-type and transgenic immune tolerant mice depends on type and level of aggregation. J. Pharm. Sci. 95:1084–1096
- Horbett, T. A. 1988. Molecular-origins of the surface-activity of proteins. Prot. Eng. 2:172–174
- Hunziker, L., Recher, M., Macpherson, A. J., et al. 2003. Hyper-gamma-globulinemia and autoantibody induction mechanisms in viral infections. Nat. Immunol. 4:343–349
- Johnson, I. S. 1983. Human insulin from recombinant DNA technology. Science 219:632–637
- Joshi, O., Chu, L., McGuire, J., and Wang, D. Q. 2009. Adsorption and function of recombinant factor VIII at air-water interface in the presence of Tween 80. J. Pharm. Sci. 98:3099–3107
- Josic, D., Buchacher, A., Kannicht, C., et al. 1999. Degradation products of factor VIII which can lead to increased immunogenicity. Vox Sanguinis 77:90–99
- Joubert, M. K., Hokom, M., Eakin, C., et al. 2012. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J. Biol. Chem. 287:25266–25279
- Joubert, M. K., Luo, Q., Nashed-Samuel, Y., et al. 2011. Classification and characterization of therapeutic antibody aggregates. J. Biol. Chem. 286:25118–25133
- Katakam, M., and Banga, A. K. 1995. Aggregation of insulin and its prevention by carbohydrate excipients. PDA J. Pharm. Sci. Tech. 49:160–165
- Katakam, M., Bell, L. N., and Banga, A. K. 1995. Effect of surfactants on the physical stability of recombinant human growth hormone. J. Pharm. Sci. 84:713–716
- Kerwin, B. A. 2008. Polysorbates 20 and 80 used in the formulation of protein biotherapeutics: Structure and degradation pathways. J. Pharm. Sci. 97:2924–2935
- Kessler, M., Goldsmith, D., and Schellekens, H. 2006. Immunogenicity of biopharmaceuticals. Nephrol. Dialysis Transplant. 21:9–12
- Kiese, S., Papppenberger, A., Friess, W., and Mahler, H. C. 2008. Shaken, not stirred: Mechanical stress testing of an IgG1 antibody. J. Pharm. Sci. 97:4347–4366
- Kim, D. Y., and Yu, M. H. 1996. Folding pathway of human α1-antitrypsin: Characterization of an intermediate that is active but prone to aggregation. Biochem. Biophys. Res. Commun. 226:378–384
- Kim, H. L., Leigh, R., and Becker, A. 2010. Omalizumab: Practical considerations regarding the risk of anaphylaxis. Allergy Asthma Clin. Immunol. 6:1--9
- Koren, E., de Groot, A. S., Jawa, V., et al. 2007. Clinical validation of the “in silico” prediction of immunogenicity of a human recombinant therapeutic protein. Clin. Immunol. 124:26–32
- Kossovsky, N., Heggers, J. P., and Robson, M. C. 1987. Experimental demonstration of the immunogenicity of silicone protein complexes. J. Biomed. Mat. Res. 21:1125–1133
- Kouskoff, V., Lacaud, G., and Nemazee, D. 2000. T-Cell-independent rescue of B-lymphocytes from peripheral immune tolerance. Science 287:2501–2503
- Kreilgaard, L., Frokjaer, S., Flink, J. M., et al. 1998a. Effects of additives on the stability of recombinant human factor XIII during freeze-drying and storage in the dried solid. Arch. Biochem. Biophys. 360:121–134
- Kreilgaard, L., Jones, L. S., Randolph, T. W., et al. 1998b. Effect of Tween 20 on freeze-thawing- and agitation-induced aggregation of recombinant, human factor XIII. J. Pharm. Sci. 87:1597–1603
- Krishnan, S., and Raibekas, A. A. 2009. Multi-step aggregation pathway of human IL-1 receptor antagonist: Kinetic, structural, and morphological characterization. Biophys. J. 96:199–208
- Krishnan, S., Chi, E. Y., Webb, J. N., et al. 2002. Aggregation of granulocyte colony stimulating factor under physiological conditions: Characterization and thermodynamic inhibition. Biochemistry 41:6422–6431
- Lange, H., Zemlin, M., Tanasa, R. L., et al. 2008. Thymus-independent type 2 antigen induces a long-term IgG-related network memory. Mol. Immunol. 45:2847–2860
- Locatelli, F., Del Vecchio, L., and Pozzoni, P. 2007. Pure red-cell aplasia “epidemic” - Mystery completely revealed? Periton. Dialysis Intl. 27:303–307
- Maa, Y. F., and Hsu, C. C. 1997. Protein denaturation by combined effect of shear and air-liquid interface. Biotech. Bioeng. 54:503–512
- Mahler, H. C., Muller, R., Friess, W., et al. 2005. Induction and analysis of aggregates in a liquid IgG1-antibody formulation. Eur. J. Pharm. Biopharm. 59:407–417
- Manning, M. C., Chou, D. K., Murphy, B. M., et al. 2010. Stability of protein pharmaceuticals: An update. Pharm. Res. 27:544–575
- Moore, W. V., and Leppert, P. 1980. Role of aggregated human growth-hormone (hGH) in development of antibodies to hGH. J. Clin. Endocrinol. Metab. 51:691–697
- Narhi, L. O., Schmit, J., Bechtold-Peters, K., and Sharma, D. 2012. Classification of protein aggregates. J. Pharm. Sci. 101:493–498
- Ohashi, P. S., and DeFranco, A. L. 2002. Making and breaking tolerance. Curr. Opin. Immunol. 14:744–759
- Panitch, H., Goodin, D. S., Francis, G., et al. 2002. Randomized, comparative study of IFNβ-1a treatment regimens in MS – The EVIDENCE trial. Neurology 59:1496–1506
- Price, K. S., and Hamilton, R. G. 2007. Anaphylactoid reactions in two patients after omalizumab administration after successful long-term therapy. Allergy Asthma Proc. 28:313–319
- Purohit, V. S., Middaugh, C. R., and Balasubramanian, S. V. 2006. Influence of aggregation on immunogenicity of recombinant human factor VIII in hemophilia A mice. J. Pharm. Sci. 95:358–371
- Qian, F., Reiter, K., Zhang, Y. L., et al. 2012. Immunogenicity of self-associated aggregates and chemically crosslinked conjugates of the 42 kDa plasmodium falciparum merozoite surface protein-1. Plos One 7:e36996
- Radstake, T. R., Svenson, M., Eijsbouts, A. M., et al. 2009. Formation of antibodies against infliximab and adalimumab strongly correlates with functional drug levels and clinical responses in rheumatoid arthritis. Ann. Rheum. Dis. 68:1739–1745
- Ragheb, S., and Lisak, R. P. 2011. B-Cell-activating factor and autoimmune myasthenia gravis. Autoimm. Dis. 2011:939520
- Rosenberg, A. S. 2006. Effects of protein aggregates: An immunologic perspective. AAPS J. 8:501–507
- Ross, C., Clemmesen, K. M., Svenson, M., et al. Danish Multiple Sclerosis. 2000. Immunogenicity of IFNβ in multiple sclerosis patients: Influence of preparation, dosage, dose frequency, and route of administration. Ann. Neurol. 48:706–712
- Rudra, J. S., Tripathi, P. K., Hildeman, D. A., et al. 2010. Immune responses to coiled coil supra-molecular biomaterials. Biomaterials 31:8475–8483
- Ryff, J. C. 1997. Clinical investigation of the immunogenicity of IFNα2a. J. Interferon Cytokine Res. 17:29–33
- Sauerborn, M., Brinks, V., Jiskoot, W., and Schellekens, H. 2010. Immunological mechanism underlying the immune response to recombinant human protein therapeutics. Trends Pharmacol. Sci. 31:53–59
- Schaefer, J. V., and Plueckthun, A. 2012. Engineering aggregation resistance in IgG by two independent mechanisms: Lessons from comparison of Pichia pastoris and mammalian cell expression. J. Mol. Biol. 417:309–335
- Schellekens, H. 2010. The immunogenicity of therapeutic proteins. Disc. Med. 9:560–564
- Schernthaner, G. 1993. Immunogenicity and allergenic potential of animal and human insulins. Diabetes Care 16:155–165
- Schrodel, A., and de Marco, A. 2005. Characterization of the aggregates formed during recombinant protein expression in bacteria. BMC Biochem. 6:1--11
- Scott, D. W., and de Groot, A. S. 2010. Can we prevent immunogenicity of human protein drugs? Ann. Rheum. Dis. 69:72–76
- Serno, T., Carpenter, J. F., Randolph, T. W., and Winter, G. 2010. Inhibition of agitation-induced aggregation of an IgG-antibody by hydroxypropyl-β-cyclodextrin. J. Pharm. Sci. 99:1193–1206
- Shankar, G., Shores, E., Wagner, C., and Mire-Sluis, A. 2006. Scientific and regulatory considerations on the immunogenicity of biologics. Trends Biotech. 24:274–280
- Singh, S. K. 2011. Impact of product-related factors on immunogenicity of biotherapeutics. J. Pharm. Sci. 100:354–387
- Skowronski, D. M., Lu, H., Warrington, R., et al. 2003a. Does antigen-specific cytokine response correlate with the experience of oculo-respiratory syndrome after influenza vaccine? J. Infect. Dis. 187:495–499
- Skowronski, D. M., Strauss, B., De Serres, G., et al. 2003b. Oculo-respiratory syndrome: A new influenza vaccine-associated adverse event? Clin. Infect. Dis. 36:705–713
- Sluzky, V., Tamada, J. A., Klibanov, A. M., and Langer, R. 1991. Kinetics of insulin aggregation in aqueous-solutions upon agitation in the presence of hydrophobic surfaces. Proc. Natl. Acad. Sci. USA 88:9377–9381
- Soulas, P., Woods, A., Jaulhac, B., et al. 2005. Autoantigen, innate immunity, and T-cells cooperate to break B-cell tolerance during bacterial infection. J. Clin. Invest. 115:2257–2267
- Spellberg, B., and Edwards, J. E. 2001. Type 1 and Type 2 immunity in infectious diseases. Clin. Infect. Dis. 32:76–102
- Szomolanyi-Tsuda, E., and Welsh, R. M. 1998. T-Cell-independent antiviral antibody responses. Curr. Opin. Immunol. 10:431–435
- Tang, X. L., and Pikal, M. J. 2004. Design of freeze-drying processes for pharmaceuticals: Practical advice. Pharm. Res. 21:191–200
- Thirumangalathu, R., Krishnan, S., Ricci, M. S., et al. 2009. Silicone oil- and agitation-induced aggregation of a monoclonal antibody in aqueous solution. J. Pharm. Sci. 98:3167–3181
- Tobery, T. W., Dubey, S. A., Anderson, K., et al. 2006. A comparison of standard immunogenicity assays for monitoring HIV type 1 gag-specific T cell responses in ad5 HIV type 1 gag vaccinated human subjects. Aids Res. Hum. Retrov. 22:1081–1090
- Tovey, M. G., Legrand, J., and Lallemand, C. 2011. Overcoming immunogenicity associated with the use of biopharmaceuticals. Expert Rev. Clin. Pharmacol. 4:623–631
- Townsend, M. W., and Deluca, P. P. 1990. Stability of ribonuclease-A in solution and the freeze-dried state. J. Pharm. Sci. 79:1083–1086
- Treuheit, M. J., Kosky, A. A., and Brems, D. N. 2002. Inverse relationship of protein concentration and aggregation. Pharm. Res. 19:511–516
- van Beers, M. M., Sauerborn, M., Gilli, F., et al. 2010. Aggregated recombinant human IFNβ induces antibodies but no memory in immune-tolerant transgenic mice. Pharm. Res. 27:1812–1824
- van Beers, M. M. C., Sauerborn, M., Gilli, F., et al. 2011. Oxidized and aggregated recombinant human IFNβ is immunogenic in human IFNβ transgenic mice. Pharm. Res. 28:2393–2402
- Villalobos, A. P., Gunturi, S. R., and Heavner, G. A. 2005. Interaction of polysorbate 80 with erythropoietin: A case study in protein-surfactant interactions. Pharm. Res. 22:1186–1194
- Wadhwa, M., and Thorpe, R. 2007. Unwanted immunogenicity: Implications for follow-on biologicals. Drug Inform. J. 41:1–10
- Wang, J., Lozier, J., Johnson, G., et al. 2008. Neutralizing antibodies to therapeutic enzymes: Considerations for testing, prevention and treatment. Nat. Biotech. 26:901–908
- Wang, X., Das, T. K., Singh, S. K., and Kumar, S. 2009. Potential aggregation-prone regions in biotherapeutics. A survey of commercial monoclonal antibodies. mAbs 1:254–267
- Wang, W., Singh, S. K., Li, N., et al. 2012. Immunogenicity of protein aggregates-Concerns and realities. Int. J. Pharm. 431:1–11
- Weiss, W. F., Young, T. M., and Roberts, C. J. 2009. Principles, approaches, and challenges for predicting protein aggregation rates and shelf life. J. Pharm. Sci. 98:1246–1277
- Zoells, S., Tantipolphan, R., Wiggenhorn, M., et al. 2012. Particles in therapeutic protein formulations, Part 1: Overview of analytical methods. J. Pharm. Sci. 101:914–935