
Abstract
Bisphenol A (BPA) is a widely used monomer of polycarbonate plastics and epoxide resin that has been implicated in asthma pathogenesis when exposure occurs to the developing fetus. However, few studies have examined the relationship between perinatal BPA exposure and asthma pathogenesis in adulthood. This study used an isogenic mouse model to examine the influence of perinatal BPA exposure via maternal diet on inflammatory mediators associated with asthma in 6-month-old adult offspring by measuring bone marrow-derived mast cell (BMMC) production of lipid mediators (cysteinyl leukotrienes and prostaglandin D2), cytokines (interleukin [IL]-4, IL-5, IL-6, IL-13, and tumor necrosis factor [TNF]-α), and histamine. Global DNA methylation levels in BMMCs from adult offspring were determined to elucidate a potential regulatory mechanism linking perinatal exposure to mast cell phenotype later in life. Four BPA exposure doses were tested: low (50 ng BPA/kg diet, n = 5), medium (50 μg BPA/kg diet, n = 4), high (50 mg BPA/kg diet, n = 4), and control (n = 3). Following BMMC activation, increases in cysteinyl leukotriene (p < 0.01) and TNFα (p < 0.05) production were observed in all BPA-exposure groups, and increases in prostaglandin D2 (p < 0.01) and IL-13 (p < 0.01) production were observed in the high exposure group. Additionally, BMMCs from adult mice in all exposure groups displayed a decrease in global DNA methylation compared to control animals. Thus, perinatal BPA exposure displayed a long-term influence on mast cell-mediated production of pro-inflammatory mediators associated with asthma and global DNA methylation levels, suggesting a potential for mast cell dysregulation, which could affect pulmonary inflammation associated with allergic airway disease into adulthood.
Introduction
There is an accumulating body of literature supporting the developmental origins of adult asthma and other lung diseases (Duijts, Citation2012; Harding & Maritz, Citation2012; Henderson & Warner, Citation2012). For example, the worldwide prevalence of asthma, a chronic inflammatory disease of the airways characterized by wheezing, coughing, chest tightness, and airway obstruction (Murdoch & Lloyd, Citation2010), has been steadily increasing over the past 40 years (Anandan et al., Citation2010; Lai et al., Citation2009; To et al., Citation2012); current estimates suggest nearly 300 million individuals are diagnosed with asthma globally (Holgate et al., Citation2007; WHO, Citation2007). Furthermore, observational reports revealing that asthma prevalence is highest among industrialized countries suggest that one or more byproducts of industrialization present in the environment may be contributing to asthma pathogenesis, thus stressing the important role that environmental influences can have on disease status (Anandan et al., Citation2010; Asher, Citation2010; Masoli et al., Citation2004; To et al., Citation2012).
Bisphenol A (BPA), a monomer of polycarbonate plastics and epoxide resin, is a high production-volume chemical that has been implicated in asthma pathogenesis when exposure occurs to the developing fetus (Midoro-Horiuti et al., Citation2010; Nakajima et al., Citation2012). BPA routinely leaches into the food and water supply from consumer products including food and liquid storage containers, baby bottles, and linings of aluminum cans. While the main route of human exposure to BPA is through indigestion of contaminated food and water, possible exposure through inhalation and dermal routes may exist due to the presence of BPA in dental sealants and thermal paper (Marquet et al., Citation2011; Vandenberg et al., Citation2007). Human exposure to BPA is widespread with National Health and Nutrition Examination Survey (NHANES) data revealing detectable amounts of BPA in the urine of 95% of study participants (Calafat et al., Citation2005; Vandenberg et al., 2012a). Quantifications of BPA in infant cord blood (Vandenberg et al., Citation2007), amniotic fluid (Edlow et al., Citation2012), placenta (Schonfelder et al., Citation2002), and fetal liver tissues (Nahar et al., Citation2013) have indicated that fetal samples contain higher quantities of free BPA than seen in adult samples. The higher fetal burden of estrogenic BPA (Rogers et al., Citation2013) may result from the developing liver’s altered capacity for detoxification, enterohepatic recirculation, and elimination of xenobiotics (Ginsberg et al., Citation2004; Vandenberg et al., Citation2009). BPA is an endocrine-active compound that has been demonstrated to bind to several nuclear hormone receptors including estrogen receptors (ERs) (Routledge et al., Citation2000), thyroid hormone receptor (TR) (Zoeller et al., Citation2005), peroxisome proliferator-activated receptor-γ (PPAR-γ) (Kwintkiewicz et al., Citation2010), estrogen-related receptor-γ (ERR-γ) (Matsushima et al., Citation2007), and human pregnane X receptor (PXR) (Sui et al., Citation2012), as well as to G-protein coupled estrogen receptor-1 (GPER) (Dong et al., Citation2011; Pupo et al., Citation2012). Additionally, BPA has been demonstrated to modify the epigenome by changing DNA methylation profiles and thereby subsequently altering gene transcription (Kundakovic & Champagne, Citation2011; Rubin, Citation2011).
Asthma pathogenesis arises from a complex interplay of genetic susceptibility and environmental exposures (Holgate et al., Citation2007), resulting in abnormal inflammatory responses led by T-helper type 2 (TH2) lymphocytes. Mast cells play an unequivocal role in the pathogenesis of atopic asthma and other allergic diseases, and contribute to airway and lung tissue inflammation by secreting cytokines (tumor necrosis factor [TNF]-α, interleukin [IL]-4, IL-5, IL-6, and IL-13) and vasoactive agents such as histamine and lipid mediators (leukotrienes and prostaglandins). Observational epidemiologic studies indicate that mast cells are sensitive to activation or dysregulation by estradiol (E2) (Bonds & Midoro-Horiuti, Citation2013), and it has long been known that asthma prevalence is 2–3-times higher in women than in men (Leynaert et al., Citation2012; Vink et al., Citation2010). More recent studies have shown that peaks in E2 during ovulation (Thornton et al., Citation2012; Vrieze et al., Citation2003) or hormone replacement therapy (Barr et al., Citation2004; Dratva, Citation2010) are associated with worsened asthma symptoms in women. Additionally, E2 (Jensen et al., Citation2010; Zaitsu et al., Citation2007), progesterone (Jensen et al., Citation2010), and other xenoestrogens (Kennedy et al., Citation2012; Lee et al., Citation2011; Narita et al., Citation2007) are capable of activating human or rat mast cell lines in vitro in the absence of IgE cross-linking.
It has been suggested that heightened sensitivity to industrialization exposures, such as BPA, during critical windows of development may influence the pathogenesis of asthma or other allergic airway diseases in the exposed individual through altering important epigenetic mechanisms such as DNA methylation (Ahmed, Citation2000; Clayton et al., Citation2011; Teitell & Richardson, Citation2003). Perinatal exposures to BPA have resulted in altered TH2 allergic responses using the ovalbumin-induced asthma model in mice (Bauer et al., Citation2012; Midoro-Horiuti et al., Citation2010; Nakajima et al., Citation2012). Additionally, at μM levels, BPA has been demonstrated to increase the release of TNFα, IL-6, and histamine in both HMC-1 cells (human mast cell line) (Lee & Lim, Citation2010) and RBL-2H3 cells (rat mast cell line) (Lee et al., Citation2012). Using an isogenic mouse model, this study sought to determine if exposures to BPA through the maternal diet increase pro-inflammatory cytokine and lipid mediator production and alter global DNA methylation levels in bone marrow-derived mast cells (BMMCs) from adult mouse offspring. Interestingly, it was observed that cysteinyl leukotrienes (CysLTs), prostaglandin D2 (PGD2), TNFα, and IL-13 production were increased at 6 months-of-age following perinatal BPA exposure. A decrease in global DNA methylation levels in BMMCs from BPA-exposed animals was also observed. Thus, perinatal exposure to BPA results in enhanced pro-inflammatory mediator synthesis and altered DNA methylation in BMMCs from adult mice.
Materials and methods
Animals
Breeders were obtained from a University of Michigan Avy breeding colony maintained on a genetically identical C57BL/6 and C3H/HeJ background (Anderson et al., Citation2012; Waterland & Jirtle, Citation2003). At 6 weeks-of-age, virgin female breeders with an a/a genotype were randomly assigned to one of four modified phytoestrogen-free BPA-supplemented diets with 7% corn oil substituted for 7% soybean oil (Harland, Madison, WI): 50 ng BPA/kg diet (low dose, diet 09798), 50 μg BPA/kg diet (medium dose, diet 09797), 50 mg BPA/kg diet (high dose, diet 09518), or BPA-free control diet (diet 95092). All diet ingredients were supplied by Harland, except for BPA (National Toxicology Program standard). Female breeders were maintained on the assigned diet for 2 weeks before being paired with a heterozygous Avy/a male. Dams and offspring remained on the assigned diet throughout gestation and lactation until weaning on post-natal day 21. At weaning, all offspring were group-housed by sex and fed the BPA-free control diet and subsequently aged to 6 months. Offspring generated from the mating paradigm were 50% a/a and 50% Avy/a, although only a/a offspring were used in this study to avoid bias introduced by the Avy retro element, which induces ectopic Agouti expression and varies dramatically among isogenic Avy/a mice (Miltenberger et al., Citation1997; Waterland & Jirtle, Citation2003).
In this study, the control, 50 ng BPA/kg diet, 50 μg BPA/kg diet, and 50 mg BPA/kg diet groups each had 3, 5, 4, and 4 subjects, respectively. Due to experimental limitations, all animals in both the 50 ng BPA/kg diet group and the 50 μg BPA/kg diet group were female, while all animals in the 50 mg BPA/kg diet group were male, and animals in the control group were one male and two females. Additionally, offspring in the control, 50 ng BPA/kg diet, 50 μg BPA/kg diet, and 50 mg BPA/kg diet groups were generated from 1, 4, 2, and 3 litters, respectively.
All animals were housed in a University of Michigan animal facility in rooms maintained at 70 °C and a 25–30% relative humidity with a 12-h light–dark cycle. All mice had ad libitum access to assigned chow and filtered water throughout the study. Animals were treated according to National Institutes of Health guidelines for the use of experimental animals, and with approval of the University of Michigan Committee for the Use and Care of Animals.
Generation and culture of BMMC
Following euthanasia with CO2 inhalation, femurs were obtained from mice and lavaged with RPMI (Life Technologies, Invitrogen, Carlsbad, CA). Primary BMMC were generated by culturing bone marrow cells in RPMI containing 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin (Invitrogen) supplemented with 10 ng/ml murine IL-3 and 10 ng/ml murine stem cell factor (Sigma, St. Louis, MO) at 37 °C in 5% CO2. Throughout incubation, culture media and culture flasks were changed once weekly. After 4 weeks in culture, cells were transferred onto glass slides using a cytocentrifuge (STAT SPIN, Norwood, MA), and the mast cell phenotype was confirmed when 95% of the cells were positive for c-kit. During immuno-cytostaining, c-kit was probed using an anti-c-kit antibody produced in rabbit (Cell Signaling, Beverly, MA), secondarily probed using a goat anti-rabbit avidin biotin complex kit (Vector, Burlingame, CA), and visualized using a diaminobenzidine kit (DAB; Vector) according to manufacturer instructions.
Stimulation of BMMC for pro-inflammatory mediator production
Viable BMMC were pelleted by centrifugation, re-suspended in RPMI, enumerated using a hemocytometer, and plated in 96-well plates (Corning Life Sciences, Tewksbury, MA) at a concentration of 2.0 × 105 cells/well. BMMC were cultured with vehicle (0.001% ethanol) alone (as a control) or with 100 ng/ml anti-DNP IgE antibody (Sigma) for 1 h at 37 °C with 5% CO2 before the addition of 10 ng/ml DNP-BSA (Sigma) for 30 min to activate the cells via IgE cross-linking (Narita et al., Citation2007). After stimulation, cell culture media were collected and stored at −80 °C until analysis. The 30-min timepoint encompasses BMMC release of pre-formed mediators stored in granules, but may also capture mediators produced by de novo synthesis after activation by IgE cross-linking (Boyce, Citation2003).
CysLT and PGD2 determinations
Levels of CysLT and PGD2 (measured after methoximation [PGD2-MOX]) produced by mast cells following stimulation were determined using commercial enzyme immunoassay (EIA) kits (Cayman Chemical, Ann Arbor, MI), according to manufacturer instructions.
Cytokine determinations
Analyses of cytokines TNFα, IL-4, IL-5, IL-6, and IL-13 in cell culture supernatants were conducted by the University of Michigan Immunology Core Facility using commercial EIA kits (DuoSet, R&D Systems, Minneapolis, MN), according to manufacturer instructions.
Histamine determination
Analysis of histamine was conducted according to the protocol previously described by Zhao et al. (Citation2001). Briefly, 30 μl from collected supernatants were distributed on 384-well plates (Corning Life Sciences); 6 μl of 1 M NaOH and 1.5 μl of 10 mg/ml o-phthaldialdehyde (Sigma) prepared in methanol were added to each sample to induce histamine derivatization. After incubation at room temperature for 4 min, 5 μl of 3 M HCl were added to each well to halt histamine derivatization. Fluorescence was read at 530 nm (360 nm excitation, 450 nm emission) using a SpectraMax Gemini M2e fluorometer (Molecular Devices, Sunnyvale, CA). Values were expressed as percent histamine release of vehicle control (‘% histamine release’ in Figure 3).
Global DNA methylation determination
Unstimulated BMMC obtained from culture were collected by centrifugation, washed twice in cold PBS, and stored at −80 °C. Frozen cells were thawed, and DNA was extracted using a DNeasy Blood and Tissue Kit for purification of total DNA according to manufacturer instructions (Qiagen, Valencia, CA). Samples were processed by the automated QIAcube platform (Qiagen). Isolated DNA was quantified using a micro-volume nucleic acid spectrophotometer NanoDrop2000 (Thermo Fisher Scientific, Waltham, MA). Analysis of global DNA methylation was conducted on 50 ng of isolated sample using a commercial EIA kit that detects 5-methyl-cytosine (5-mC) (Epigentek, Farmingdale, NY), according to manufacturer instructions.
Statistical analyses
Data analyses were conducted using a one-way analysis of variance with a post-hoc Bonferroni test for separation of the means in Prism Graph Software (GraphPad Software, Inc., La Jolla, CA). In all cases, a p value of <0.05 was considered statistically significant.
Results
Perinatal BPA exposure increases adult BMMC CysLT and PGD2 levels after IgE cross-linking
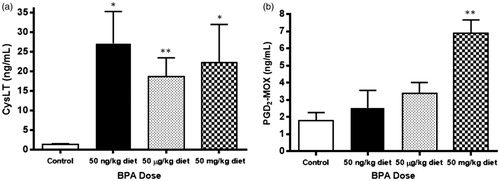
To test whether perinatal exposure to BPA up-regulates the production of pro-inflammatory lipid mediators known to contribute to the pathogenesis of allergic airway disease, BMMC secretion of eicosanoids CysLT and PGD2 following IgE cross-linking was assessed (). Following activation, 3–4-fold increases in the production of CysLT from the low (p = 0.013), medium (p = 0.005), and high (p = 0.036) BPA exposure groups compared to the control group were observed, with no differences between BPA exposure groups (). Additionally, a 2-fold increase in production of PGD2 in the high BPA exposure group (p = 0.009), without increased PGD2 levels in the low (p = 0.542) and medium (p = 0.120) BPA exposure groups, was observed ().
Figure 1. Levels of (a) CysLT and (b) PGD2 secreted after IgE cross-linking from BMMC of animals exposed perinatally to BPA. Concentrations of CysLT and PGD2 were determined from cell culture supernatants collected after 30 min of cellular activation. Bars represent mean (±SEM) for n = 3–5 mice per group. *p < 0.05 and **p < 0.01 compared to control.
Perinatal BPA exposure enhances adult BMMC IL-13 and TNFα production after IgE cross-linking
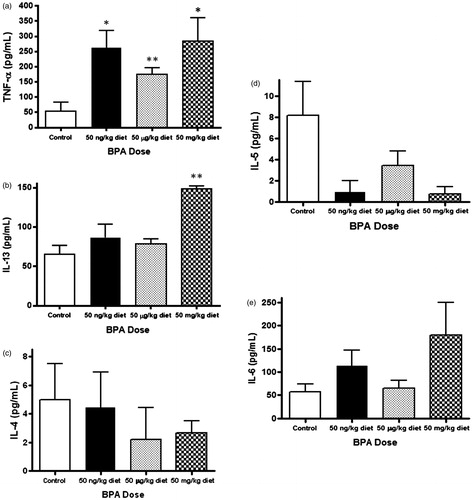
The impact of BPA exposure on IgE cross-linking-induced BMMC cytokine release was next assessed (). In these experiments, levels of TNFα, IL-4, IL-5, IL-6, and IL-13 were determined as these mast cell-derived cytokines contribute to allergic airway inflammation (Amin, Citation2012; Boyce, Citation2003; Bradding et al., Citation2006; Walls et al., Citation2001). IgE-mediated activation resulted in a 3–4-fold increase in BMMC release of TNFα from low (p = 0.018), medium (p = 0.008), and high (p = 0.019) BPA exposure groups compared with the control group, with no statistically significant differences between individual BPA exposure groups (). Furthermore, IL-13 release was also up-regulated in the high BPA exposure group (p = 0.0005) (). However, there were no differences in IL-4, IL-5, or IL-6 levels between the control group and BPA exposure groups following BMMC stimulation ().
Figure 2. Levels of (a) TNFα, (b) IL-13, (c) IL-4, (d) IL-5, and (e) IL-6 secreted after IgE cross-linking from BMMC of animals exposed perinatally to BPA. Concentrations of TNFα, IL-13, IL-4, IL-5, and IL-6 were determined from cell culture supernatants collected after 30 min of cellular activation. Bars represent mean (±SEM) for n = 3–5 mice per group. *p < 0.05 and **p < 0.01 compared to control.
Effect of perinatal BPA exposure on adult BMMC histamine release
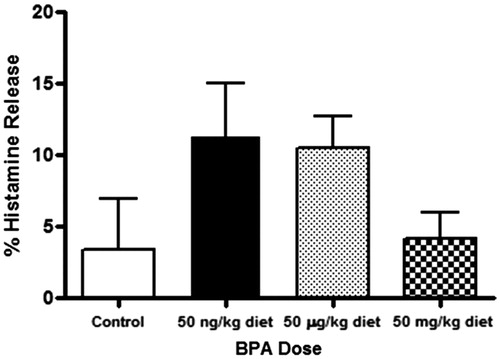
The effect of BPA exposure on IgE cross-linking-induced histamine release was measured (). Compared to controls, there was no difference is histamine release among the BMMC of BPA-exposed animals. However, mean % histamine release was ≈2-fold higher in the low (p = 0.213) and medium (p = 0.333) BPA groups, as compared to the control group.
Figure 3. Percentage of histamine release secreted after IgE cross-linking from BMMC of animals exposed perinatally to BPA, as compared to IgE-free control. Relative fluorescence units were determined from cell culture supernatants collected after 30 min of cellular activation. Bars represent mean (±SEM) for n = 3–5 mice per group.
Perinatal BPA exposure decreases global DNA methylation in adult BMMC
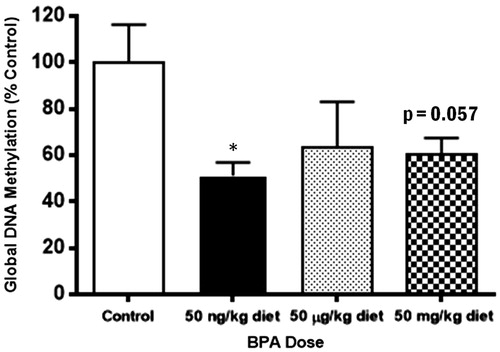
The observed increases in pro-inflammatory mediator secretion from BMMC of adult animals exposed to BPA perinatally suggest increased gene transcription of cytokines and proteins involved in the synthesis of eicosanoids, which may be regulated by DNA methylation changes (Richter et al., Citation2007). To test this hypothesis, the relative concentrations of 5-mC in DNA extracts from BMMC of BPA-exposed and control animals were determined by EIA. As shown in , global DNA methylation levels in extracts of BMMC from the BPA-exposed animals were decreased compared to controls, although statistical significance was observed only in the low BPA exposure group (p = 0.014). Near-significance was observed in the high BPA exposure group (p = 0.057), but not in the medium BPA exposure group (p = 0.232).
Figure 4. Decreased levels of global DNA methylation in BMMC from adult animals which had perinatal BPA exposure. DNA was isolated from cultured BMMC of adult animals, and 5-mC concentration was measured. Bars represent mean arbitrary OD units (±SEM) for n = 3–5 mice per group. *p < 0.05 compared to control.
Discussion
Since little is known regarding the long-term effects of perinatal BPA exposure on mast cell function, the current study measured mediator production from BMMC obtained from 6-month-old mice exposed perinatally to four doses of BPA (low, medium, high, and control) through the maternal diet. Enhanced lipid mediator synthesis (CysLT and PGD2) and cytokine release (TNFα and IL-13) following BMMC activation via IgE cross-linking for 30 min were observed. In association with these increases in pro-inflammatory mediator production, a decrease in global DNA methylation was observed. Thus, these results highlight the potential for persistent epigenetic modification by the endocrine-active monomer BPA, resulting in lasting consequences on mast cell-mediated pro-inflammatory lipid mediator and cytokine production in adulthood that could potentially worsen inflammation associated with allergic airway disease. Furthermore, the results from the current study align with previous reports demonstrating enhanced asthma pathogenesis in juvenile mice with perinatal BPA exposure (Midoro-Horiuti et al., Citation2010; Nakajima et al., Citation2012).
An important and novel finding of this study is that the increases in pro-inflammatory mediator production and changes in DNA methylation states were observed in cells obtained from adult animals that were exclusively exposed to BPA perinatally. Thus, effects of BPA exposure persisted for over 5 months following termination of exposure, supporting a role for early environmental exposures in sustained mast cell dysregulation of pro-inflammatory mediator production. Although the epigenome is vulnerable to environmentally-induced alterations throughout life, it is most susceptible during early development when the DNA synthetic rate is high and the elaborate DNA methylation patterns are being reprogrammed (Dolinoy et al., Citation2007b). Altered DNA methylation profiles have been demonstrated to be mitotically and meiotically heritable (Robertson, Citation2005), and several previous reports have demonstrated long-term effects from early exposures to BPA (Richter et al., Citation2007), especially pertaining to reproductive physiology (Edlow et al., Citation2012; Markey et al., Citation2005; Newbold et al., Citation2009). Additionally, there have been significant findings in newly focused epigenetic studies directed toward trans-generational outcomes in the F3 generation and beyond following an environmental exposure in the F0 generation (Skinner & Guerrero-Bosagna, Citation2009; Skinner et al., Citation2010). However, few in vivo studies have investigated long-term effects on mast cell function, or other immune functions for that matter, after perinatal BPA exposure from an epigenetic standpoint as the current study has done.
CysLT and PGD2 are arachidonic acid metabolites, with CysLT being generated via the 5-lipoxygenase (5-LO) pathway and PGD2 via cyclooxygenase-1 and -2 (COX1/2) pathways. Both are released from mast cells by degranulation as well as rapid de novo synthesis after cellular activation (Boyce, Citation2003). While data from this study indicate that perinatal BPA exposure resulted in increased CysLT and PGD2 release from BMMC of adult animals, a clear mechanism explaining increased CysLT and PGD2 release has yet to be determined. The observed global hypo-methylation in BMMC from BPA exposure groups compared to the control group suggests an overall increase in transcription of genes whose promoter regions were less methylated following exposure (Anderson et al., Citation2012; Dolinoy et al., Citation2007a); however, methylation changes to specific genes regulating CysLT and PGD2 production may or may not follow the global progression. Dysregulation of the mitogen-activated protein kinase (MAPK) pathway, which is upstream of where 5-LO and COX1/2 pathways converge, may explain the observed responses. Yi & Krieg (Citation1998) demonstrated that DNA demethylation resulted in increased c-Jun, p38, MAPK, and activator protein-1 (AP-1) activation. Additionally, Lee et al. (Citation2006) showed in head and neck cancer cell lines that silencing of suppressor of cytokine signaling-1 (SOCS-1) by DNA hyper-methylation resulted in increased downstream activation of signal transducer and activator of transcription 3 (STAT3) through activation of Janus kinases 1 and 2 (JAK1/2) and extra-cellular signal-regulated kinase (ERK). Future work examining gene-specific methylation changes to promoter regions of the proteins described here, both those that inhibit and promote lipid mediator and/or cytokine production, will be needed to identify the specific gene-regulation mechanisms that explain increased BMMC lipid mediator secretion following perinatal BPA exposure.
Following perinatal BPA exposure, the current study also observed an increase in BMMC release of the cytokines IL-13, which contributes to eosinophil recruitment and IgE production in the asthmatic lung, and TNFα, which increases airway hyper-responsiveness and sputum neutrophils (Chung & Barnes, Citation1999). However, increases in IL-4, -5, and -6 levels were not observed, suggesting that BPA can selectively promote mast cell-derived cytokine production. Cytokine release from mast cells activated by IgE cross-linking occurs through cellular degranulation as well as delayed (hours) de novo synthesis (Boyce, Citation2003; Xiang et al., Citation2001). Thus, cytokine concentrations measured in the current study likely represent release via degranulation only. The relative levels of cytokine concentrations reported here are important because possible alterations in expression of genes that regulate synthesis of cytokines are the same whether synthesis occurs in advance or de novo (Boyce, Citation2003). Future work will focus on investigating DNA methylation changes to promoter regions of the IL13 gene, which may explain the dose-dependent increase in BMMC IL-13 production. Recently, Yu et al. (Citation2012) demonstrated in a model of constitutive DNA methyltransferase 3 a (Dnmt3a) and 3 b (Dnmt3b) knockout (KO) mice that IL-13 cytokine production is modifiable depending on gene methylation state, with increased IL-13 production observed in KO mice compared to wild-type mice. Likewise, future work will examine the methylation state of the Tnf promoter to help explain BMMC increases in TNFα production. Several reports have described age-related or disease-related CpG methylation changes to the Tnf promoter (Gowers et al., Citation2011; Han et al., Citation2011; Wang et al., Citation2012), and Kamei et al. (Citation2010) observed that over-expression of Dnmt3a in white adipose tissue of mice fed a high-fat diet increased TNFα gene expression compared to wild-type mice fed a high-fat diet. In addition to the cytokines examined in this report, research should also further examine the T-helper cell type 1 (TH1) cytokine interferon (IFN)-γ and the TH2 cytokine IL-4. Yoshino et al. (Citation2004) showed that fetal exposure to BPA through maternal consumption of 300 μg BPA/kg BW/day from days 0 to 17 of gestation resulted in increased production of IFNγ and IL-4 in stimulated splenocytes. Likewise, Yan et al. (Citation2008) demonstrated similar results by showing that maternal exposure to 100 nM BPA in drinking water 2 weeks before mating and during the first week of gestation increased adult offspring production of IL-4 and IFNγ from stimulated splenocytes. Although the current study did not observe a difference in BMMC IL-4 production between BPA-exposed and control subjects, different responses in IL-4 levels across studies may be explained by variable BPA exposure windows, dietary composition, and/or relative amounts of IL-4 released by distinct cell types (Lacy & Stow, Citation2011).
It is important to note that limitations to the exposure model used in the current study are the number, litter distribution, and sex of animals included as test subjects. Animals observed in this study were generated from a larger heterozygous breeding scheme of Avy mice (Anderson et al., Citation2012, Citation2013), and were selected based on a/a genotype and age. Heterozygous Avy/a offspring were not included in the study group due to the inheritance of metabolic disorders, obesity, and tumorigenesis (Miltenberger et al., Citation1997; Morgan et al., Citation1999). Thus, the animals examined in this study represent skewed sex and litter distributions described in the Materials and methods section. In terms of pro-inflammatory mediator production from cells, some studies suggest sex differences in cellular production of TH2 cytokines, specifically TNFα, and CysLT. Spitzer (Citation1999) reported increased TNFα production from female rat alveolar macrophages and Kupffer cells following in vivo ethanol treatment compared to males. Bouman et al. (Citation2004) also reported that production of TNFα, IL-12, and IL-1β was increased in healthy males compared to healthy females, but suggested that higher cytokine quantity in males was the result of a higher initial number of circulating monocytes in the study participants. Similarly, Pergola et al. (Citation2008, Citation2011) reported a decrease in leukotriene production from peripheral monocytes, neutrophils, and whole blood of healthy males compared to healthy females, which was attributed to lower baseline expression of 5-LO in cells from males and inhibition of phospholipase D by 5α-dihydrotestosterone. In the current study, robust CysLT and TNFα up-regulation from BMMC in all BPA exposure groups was observed compared to the control group, with no significance between exposure groups. Thus, it is suspected that sex differences in cytokine production are minimal, if present, for CysLT and TNFα production in this model. On the other hand, possible mitigating influences of sex with IL-13 and PGD2 production cannot be ruled out. For these makers, significant IL-13 and PGD2 increases were observed only in the highest BPA exposure group, which is a group composed entirely of male animals. In contrast, although not statistically significant, mean histamine release was highest in the low and medium BPA exposure groups, which were comprised of all females, while histamine release in the high exposure group was comparable to control levels. Future research should examine possible baseline differences in cytokine, lipid mediator, and histamine production between sexes in mice and humans.
Ideally, sera BPA levels in dams or neonate offspring used in this study would be quantified to validate BPA dosing through the supplemented diets. However, the study design and limited subjects precluded these measurements. A study conducted by Jasarevic et al. (Citation2013) reported conjugated and unconjugated (free) BPA levels in sera of deer mice dams chronically (∼12 months) fed the same high BPA diet (50 mg BPA/kg diet) used in the current study. Conjugated BPA levels ranged from 1.6–157 ng/ml with a mean of ∼60 ng/ml, while free BPA levels ranged from 0.79–19.3 ng/ml with a mean of ∼5.5 ng/ml—values within the range seen in human exposures (Vandenberg et al., Citation2007). Comparatively, dams fed the control diet had conjugated or free BPA sera levels ranging from the limit of detection (0.1 ng/ml) to 0.79 ng/ml (Jasarevic et al., Citation2013). Those investigators also noted that, based on the linear response curves for BPA pharmacokinetics (Doerge et al., Citation2010, Citation2011; Taylor et al., Citation2011), dams chronically fed the medium or low BPA diets were expected to have free BPA sera levels below the limit of detection. BPA levels in the livers of 22-day-old offspring generated from this breeding scheme have been reported previously (Anderson et al., Citation2012). In that 2012 study, offspring exposed to the high BPA diet displayed a mean conjugated BPA level of 278 ng/g and a mean free BPA level of 164 ng/g. However, offspring in the medium and low exposure groups had much lower mean conjugated BPA (0.3 and 1.0 ng/g, respectively) and mean free BPA (1.8 and 1.8 ng/g, respectively) levels that did not differ from controls (conjugated: 0.6 ng/g, unconjugated 3.7 ng/g), but were within the range of human exposure.
Conclusions
While the debate regarding removal of BPA from consumer products persists, a strength of this study is its focus on levels of BPA relevant to human exposure using an exposure model that includes low (50 ng BPA/kg diet), medium (50 μg BPA/kg diet), and high (50 mg BPA/kg diet) doses, where the medium dose is the closest to the consumption levels seen in humans (Anderson et al., Citation2012, Citation2013). It is important to note that we observed significant increases in pro-inflammatory mediator secretion at the lowest BPA dose tested, which is well below the average human consumption levels. Altered biologic responses stemming from exposure to low doses of endocrine-active compounds, including BPA, have frequently been reported (Vandenberg et al., 2012b). Low-dose responses highlight an important concept: human exposure to small quantities of environmental products can have a profound impact on human health. The current report has established a novel association between perinatal BPA exposure through maternal diet and mast cell dysregulation in adulthood, as evidenced by increased CysLT, PGD2, TNFα, and IL-13 release from BMMC of adult offspring with perinatal BPA exposures. These responses occurred in conjunction with decreased global DNA methylation in BMMC, indicating a possible regulatory mechanism through altered DNA methylation profiles. Due to the important role of mast cells in the development of atopic asthma, BPA-induced mast cell dysregulation could have implications on asthma pathogenesis. Future studies focusing on the developmental origins of asthma will help to lay a better groundwork for reduction of environmental exposures to the developing fetus.
Declaration of interest
This work was supported by the NIH grants HL077417 (PM) and ES017524 (DCD), the Flight Attendant Medical Research Institute award CIA-103071 (PM), and the University of Michigan National Institutes of Environmental Health Sciences (NIEHS) Core Center (P30 ES017885). Support for EO was provided by Institutional Training Grant T32 ES007062.
Acknowledgements
We thank Joel Whitfield from the University of Michigan Immunology Core Facility for technical assistance.
References
- Ahmed, S. A. 2000. The immune system as a potential target for environmental estrogens (endocrine disrupters): A new emerging field. Toxicology 150:191–206
- Amin, K. 2012. The role of mast cells in allergic inflammation. Resp. Med. 106:9–14
- Anandan, C., Nurmatov, U., van Schayck, O. C. P., and Sheikh, A. 2010. Is the prevalence of asthma declining? Systematic review of epidemiological studies. Allergy 65:152–167
- Anderson, O. S., Nahar, M. S., Faulk, C., et al. 2012. Epigenetic responses following maternal dietary exposure to physiologically revelant levels of bisphenol A. Environ. Mol. Mutagen. 53:334–342
- Anderson, O. S., Peterson, K. E., Sanchez, B. N., et al. 2013. Perinatal bisphenol A exposure promotes hyperactivity, lean body composition, and hormonal responses across the murine life course. FASEB J. 24:1784–1792
- Asher, M. I. 2010. Recent perspectives on global epidemiology of asthma in childhood. Allergol. Immunopathol. (Madrid). 38:83–87
- Barr, R. G., Wentowski, C. C., Grodstein, F., et al. 2004. Prospective study of postmenopausal hormone use and newly diagnosed asthma and chronic obstructive pulmonary disease. Arch. Int. Med. 164:379–386
- Bauer, S. M., Roy, A., Emo, J., et al. 2012. The effects of maternal exposure to bisphenol A on allergic lung inflammation into adulthood. Toxicol. Sci. 130:82–93
- Bonds, R. S., and Midoro-Horiuti, T. 2013. Estrogen effects in allergy and asthma. Curr. Opin. Allergy Clin. Immunol. 13:92–99
- Bouman, A., Schipper, M., Heineman, M. J., and Faas, M. M. 2004. Gender difference in the non-specific and specific immune response in humans. Am. J. Reprod. Immunol. 52:19–26
- Boyce, J. A. 2003. The role of mast cells in asthma. Prostagl. Leukotr. Essent. Fatty Acids 69:195–205
- Bradding, P., Walls, A. F., and Holgate, S. T. 2006. The role of the mast cell in the pathophysiology of asthma. J. Allergy Clin. Immunol. 117:1277–1284
- Calafat, A. M., Kuklenyik, Z., Reidy, J. A., et al. 2005. Urinary concentrations of bisphenol A and 4-nonylphenol in a human reference population. Environ. Health Perspect. 113:391–395
- Chung, K. F., and Barnes, P. J. 1999. Cytokines in asthma. Thorax 54:825–857
- Clayton, E. M. R., Todd, M., Dowd, J. B., and Aiello, A. E. 2011. The impact of bisphenol A and triclosan on immune parameters in the US population NHANES 2003–2006. Environ. Health Perspect. 119:390–396
- Doerge, D. R., Twaddle, N. C., Vanlandingham, M., and Fisher, J. W. 2010. Pharmacokinetics of bisphenol A in neonatal and adult Sprague-Dawley rats. Toxicol. Appl. Pharmacol. 247:158–165
- Doerge, D. R., Twaddle, N. C., Vanlandingham, M., and Fisher, J. W. 2011. Pharmacokinetics of bisphenol A in neonatal and adult CD-1 mice: Inter-species comparisons with Sprague-Dawley rats and rhesus monkeys. Toxicol. Lett. 207:298–305
- Dolinoy, D. C., Huang, D., and Jirtle, R. L. 2007a. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. USA. 104:13056–13061
- Dolinoy, D. C., Weidman, J. R., and Jirtle, R. L. 2007b. Epigenetic gene regulation: Linking early developmental environment to adult disease. Reprod. Toxicol. 23:297–307
- Dong, S., Terasaka, S., and Kiyama, R. 2011. Bisphenol A induces a rapid activation of ERK1/2 through GPR30 in human breast cancer cells. Environ. Pollut. 159:212–218
- Dratva, J. 2010. Use of oestrogen oly hormone replacement therapy associated with increased risk of asthma onset in postmenopausal women. Evid. Based Med. 15:190–191
- Duijts, L. 2012. Fetal and infant origins of asthma. Eur. J. Epidemiol. 27:5–14
- Edlow, A. G., Chen, M., Smith, N. A., et al. 2012. Fetal bisphenol A exposure: Concentration of conjugated and unconjugated bisphenol A in amniotic fluid in the second and third trimesters. Reprod. Toxicol. 34:1–7
- Ginsberg, G., Slikker, W., Bruckner, J., and Sonawane, B. 2004. Incorporating children's toxicokinetics into a risk framework. Environ. Health Perspect. 112:272–283
- Gowers, I. R., Walters, K., Kiss-Toth, E., et al. 2011. Age-related loss of CpG methylation in the tumor necrosis factor promoter. Cytokine 56:792–797
- Han, Y. L, Guo, Z. Y., Fan, Y. C., and Wang, K. 2011. Hypomethylation of TNFα gene promoter in the patients with acute-on-chronic hepatitis B liver failure. Zhong. Shi Yan He Lin Chuang Bing Du Xue Za Zhi. 25:368–370
- Harding, R., and Maritz, G. 2012. Maternal and fetal origins of lung disease in adulthood. Semin. Fetal Neonat. Med. 17:67–72
- Henderson, A. J., and Warner, J. O. 2012. Fetal origins of asthma. Semin. Fetal Neonat. Med. 17:82–91
- Holgate, S. T., Davies, D. E., Powel, R. M., et al. 2007. Local genetic and environmental factors in asthma disease pathogenesis: Chronicity and persistence mechanisms. Eur. Respir. J. 29:793–803
- Jasarevic, E., Williams, S. A., Vandas, G. M., et al. 2013. Sex and dose-dependent effects of developmental exposure to bisphenol A on anxiety and spatial learning in deer mice (Peromyscus maniculatus bairdii) offspring. Horm. Behav. 63:180–189
- Jensen, F., Woudwyk, M., Teles, A., et al. 2010. Estradiol and progesterone regulate the migration of mast cells from periphery to the uterus and induce their maturation and degranulation. PLoS One 5:e14409
- Kamei, Y., Suganami, T., Ehara, T., et al. 2010. Increased expression of DNA methyltransferase 3a in obese adipose tissue: Studies with transgenic mice. Obesity 18:314–321
- Kennedy, R. H., Pelletier, J. H., Tupper, E. J., et al. 2012. Estrogen mimetic 4-tert-octylphenol enhances IgE-mediated degranulation of Rbl-2H3 mast cells. J. Toxicol. Environ. Health. 75:1451–1455
- Kundakovic, M., and Champagne, F. A. 2011. Epigenetic perspective on the developmental effects of bisphenol A. Brain Behav. Immun. 25:1084–1093
- Kwintkiewicz, J., Nishi, Y., Yanase, T., and Giudice, L. C. 2010. Peroxisome proliferator-activated receptor-γ mediates bisphenol A inhibition of FSH-stimulated IGF-1, aromatase, and estradiol in human granulosa cells. Environ. Health Perspect. 118:400–406
- Lacy, P., and Stow, J. L. 2011. Cytokine release from innate immune cells: Association with diverse membrane trafficking pathways. Blood 118:9–18
- Lai, C. K., Beasley, R., Crane, J., et al. 2009. Global variation in the prevalence and severity of asthma symptoms: Phase three of the International Study of Asthma and Allergies in Childhood (ISAAC). Thorax 64:467–483
- Lee, J., and Lim, K. T. 2010. Plant-originated glycoprotein (36 kDa) suppresses IL-4 and -10 in bisphenol A-stimulated primary cultured mouse lymphocytes. Drug Chem. Toxicol. 33:421–429
- Lee, J., Lee, S. J., and Lim, K. T. 2012. CTB glycoprotein (75kDa) inhibits IgE releasing, TNFα and IL-6 expressed by bisphenol A in vivo and in vitro. Food Chem. Toxicol. 50:2109–2117
- Lee, J., Oh, P. S., and Lim, K. T. 2011. Allergy-related cytokines (IL-4 and TNFα) are induced by di(2-ethylhexyl) phthalate and attenuated by plant-originated glycoprotein (75 kDa) in HMC-1 cells. Environ. Toxicol. 26:364–372
- Lee, T. L., Yeh, J., Van Waes, C., and Chen, Z. 2006. Epigenetic modification of SOCS-1 differentially regulates STAT3 activation in response to IL-6 receptor and epidermal growth factor receptor signaling through JAK and/or MEK in head and neck squamous cell carci-nomas. Mol. Cancer Ther. 5:8–19
- Leynaert, B., Sunyer, J., Carcia-Esteban, R., et al. 2012. Gender differences in prevealnce, diagnosis and incidence of allergic and non-allergic asthma: A population-based cohort. Thorax 67:625–631
- Markey, C. M., Wadia, P. R., Rubin, B. S., et al. 2005. Long-term effects of fetal exposure to low doses of the xenoestrogen bisphenol-A in the female mouse genital tract. Biol. Reprod. 72:1344–1351
- Marquet, F., Payan, J. P., Beydon, D., et al. 2011. In vivo and ex vivo percutaneous absorption of [14C]-bisphenol A in rats: A possible extrapolation to human absorption? Arch. Toxicol. 85:1035–1043
- Masoli, M., Fabian, D., Holt, S., and Beasley, R. 2004. The global burden of asthma: Executive Summary of the GINA Dissemination Committee report. Allergy 59:469–478
- Matsushima, A., Kakuta, Y., Teramoto, T., et al. 2007. Structural evidence for endocrine disruptor bisphenol A binding to human nuclear receptor ERR-γ. J. Biochem. 142:517–524
- Midoro-Horiuti, T., Tiwari, R., Watson, C. S., and Goldblum, R. M. 2010. Maternal bisphenol A exposure promotes the development of experimental asthma in mouse pups. Environ. Health Perspect. 118:273–277
- Miltenberger, R. J., Mynatt, R. L., Wilkinson, J. E., and Woychik, R. P. 1997. The role of the agouti gene in the yellow obese syndrome. J. Nutr. 127:1902S–1907S
- Morgan, H. D., Sutherland, H. G., Martin, D. I., and Whitelaw, E. 1999. Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet. 23:314–318
- Murdoch, J. R., and Lloyd, C. M. 2010. Chronic inflammation and asthma. Mutat. Res. 690:24–39
- Nahar, M. S., Liao, C., Kannan, K., and Dolinoy, D. C. 2013. Fetal liver bisphenol A concentrations and biotransformation gene expression reveal variable exposure and altered capacity for metabolism in humans. J. Biochem. Mol. Toxicol. 27:116–123
- Nakajima, Y., Goldblum, R. M., and Midoro-Horiuti, T. 2012. Fetal exposure to bisphenol A as a risk factor for the development of childhood asthma: An animal model study. Environ. Health. 11:1–7
- Narita, S., Goldblum, R. M., Watson, C. S., et al. 2007. Environmental estrogens induce mast cell degranulation and enhance IgE-mediated release of allergic mediators. Environ. Health Perspect. 115:48–52
- Newbold, R. R., Jefferson, W. N., and Padilla-Banks, E. 2009. Prenatal exposure to bisphenol A at environmentally relevant doses adversely affects the murine female reproductive tract later in life. Environ Health Perspect. 117:879–885
- Pergola, C., Dodt, G., Rossi, A., et al. 2008. ERK-mediated regulation of leukotriene biosynthesis by androgens: A molecular basis for gender differences in inflammation and asthma. Proc. Natl. Acad. Sci. USA. 105:19881–19886
- Pergola, C., Rogge, A., Dodt, G., et al. 2011. Testosterone suppresses phospholipase D, causing sex differences in leukotriene biosynthesis in human monocytes. FASEB J. 25:3377–3387
- Pupo, M., Pisano, A., Lappano, R., et al. 2012. Bisphenol A induces gene expression changes and proliferation effects through GPER in breast cancer cells and cancer-associated fibroblasts. Environ. Health Perspect. 120:1177–1182
- Richter, C. A., Birnbaum, L. S., Farabollini, F., et al. 2007. In vivo effects of bisphenol A in laboratory rodent studies. Reprod. Toxicol. 24:199–224
- Robertson, K. D. 2005. DNA methylation and human disease. Nat. Rev. Genet. 6:597–610
- Rogers, J. A., Metz, L., and Yong, V. W. 2013. Review: Endocrine disrupting chemicals and immune responses: A focus on bisphenol-A and its potential mechanisms. Mol Immunol. 53:421–430
- Routledge, E. J., White, R., Parker, M. G., and Sumpter, J. P. 2000. Differential effects of xenoestrogens on coactivator recruitment by estrogen receptor (ER)-α and ER-β. J. Biol. Chem. 275:35986–35993
- Rubin, B. S. 2011. Bisphenol A: An endocrine disruptor with widespread exposure and multiple effects. J. Steroid Biochem. Mol. Biol. 127:27–34
- Schonfelder, G., Wittfoht, W., Hopp, H., et al. 2002. Parent bisphenol A accumulation in the human maternal–fetal–placental unit. Environ. Health Perspect. 110:703–707
- Skinner, M. K., and Guerrero-Bosagna, C. 2009. Environmental signals and transgenerational epigenetics. Epigenomics 1:111–117
- Skinner, M. K., Manikkam, M., and Guerrero-Bosagna, C. 2010. Epigenetic trans-generational actions of environmental factors in disease etiology. Cell 21:214–222
- Spitzer, J. A. 1999. Gender differences in some host defense mechanisms. Lupus 8:380–383
- Sui, Y., Ai, N., Park, S. H., et al. 2012. Bisphenol A and its analogues activate human pregnane X receptor. Environ. Health Perspect. 120:399–405
- Taylor, J. A., vom Saal, F. S., Welshons, W. V., et al. 2011. Similarity of bisphenol A pharmaco-kinetics in rhesus monkeys and mice: Relevance for human exposure. Environ. Health Perspect. 119:422–430
- Teitell, M., and Richardson, B. 2003. DNA methylation in the immune system. Clin. Immunol. 109:2–5
- Thornton, J., Lewis, J., Lebrun, C. M., and Licskai, C. J. 2012. Clinical characteristics of women with menstrual-linked asthma. Respir. Med. 106:1236–1243
- To, T., Stanojevic, S., Moores, G., et al. 2012. Global asthma prevalence in adults: Findings from the cross-sectional world health survey. BMC Publ. Health 12:204–211
- Vandenberg, L. N., Chahoud, I., Heindel, J. J., et al. 2012a. Urinary, circulating, and tissue biomonitoring studies indicate wide-spread exposure to bisphenol A. Cien Saude Colet. 17:407–434
- Vandenberg, L. N., Colborn, T., Hayes, T. B., et al. 2012b. Hormones and endocrine-disrupting chemicals: Low-dose effects and nonmonotonic dose responses. Endocr. Rev. 33:378–455
- Vandenberg, L. N., Hauser, R., Marcus, M., et al. 2007. Human exposure to bisphenol A (BPA). Reprod. Toxicol. 24:139–177
- Vandenberg, L. N., Maffini, M. V., Sonnenschein, C., et al. 2009. Bisphenol A and the great divide: A review of controversies in the field of endocrine disruption. Endocrinol. Rev. 30:75–95
- Vink, N. M., Postma, D. S., Schouten, J. P., et al. 2010. Gender differences in asthma development and remission during transition through puberty: The TRacking Adolescents’ Individual Lives Survey (TRAILS) study. J. Allergy Clin. Immunol. 126:498–504
- Vrieze, A., Postma, D. S., and Kerstjens, H. A. M. 2003. Perimenstrual asthma: A syndrome without known cause or cure. J. Allergy Clin. Immunol. 112:271–282
- Walls, A. F., He, S., Buckley, M. G., and McEuen, A. R. 2001. Roles of the mast cell and basophil in asthma. Clin. Exp. Allergy 1:68–72
- Wang, B., Li, Y., Tan, Y., et al. 2012. Low-dose Cd induces hepatic gene hypermethylation, along with the persistent reduction of cell death and increase of cell proliferation in rats and mice. PLoS One 7:1–13
- Waterland, R. A., and Jirtle, R. L. 2003. Transposable elements: Targets for early nutritional effects on epigenetic gene regulation. Mol. Cell. Biol. 23:5293–5300
- World Health Organization (WHO). 2007. Global Surveillance, Prevention, and Control of Chronic Respiratory Diseases: A Comprehensive Approach. WHO: Geneva
- Xiang, Z., Block, M., Lofman, C., and Nilsson, G. 2001. IgE-mediated mast cell degranulation and recovery monitored by time-lapse photography. J. Allergy Clin. Immunol. 108:116–121
- Yan, H., Takamoto, M., and Sugane, K. 2008. Exposure to bisphenol A prenatally or in adulthood promotes TH2 cytokine production associated with reduction of CD4CD25 regulatory T-cells. Environ. Health Perspect. 116:514–519
- Yi, A. K., and Krieg, A. M. 1998. Rapid induction of mitogen-activated protein kinases by immune stimulatory CpG DNA. J. Immunol. 161:4493–4497
- Yoshino, S., Yamaki, K., Li, X., et al. 2004. Prenatal exposure to bisphenol A up-regulates immune responses, including T-helper-1 and T-helper-2 responses in mice. Immunology 112:489–495
- Yu, Q., Zhou, B., Zhang, Y., et al. 2012. DNA methyltransferase 3a limits the expression of IL-13 in T-helper-2 cells and allergic airway inflammation. Proc. Natl. Acad. Sci. USA. 109:541–546
- Zaitsu, M., Narita, S., Lambert, K. C., et al. 2007. Estradiol activates mast cells via a non-genomic estrogen receptor-alpha and calcium influx. Mol. Immunol. 44:1977–1985
- Zhao, Z. Z., Sugerman, P. B., Walsh, L. J., and Savage, N. W. 2001. A fluorometric microassay for histamine release from human gingival mast cells. J. Periodontal Res. 36:233–236
- Zoeller, R. T., Bansal, R., and Parris, C. 2005. Bisphenol-A, an environmental contaminant that acts as a thyroid hormone receptor antagonist in vitro, increases serum thyroxine, and alters RC3/neurogranin expression in the developing rat brain. Endocrinology 146:607–612