
Abstract
There is increasing evidence that the endogenous nitric oxide synthase (NOS) inhibitor asymmetric dimethyl-arginine (ADMA) is involved in the pathogenesis of chronic lung diseases. One important regulator of this molecule is the ADMA-metabolizing enzyme dimethyl-arginine dimethyl-aminohydrolase (DDAH). The objective of this study was to determine whether perturbation of the ADMA-DDAH pathway contributes to lung inflammation following exposure to cigarette smoke (CS). For these studies, wild-type and DDAH transgenic mice were sham or CS-exposed. Serum ADMA levels were determined by mass spectrometry. ADMA content and DDAH expression were also visualized in mouse lung tissue by immunohistochemistry. DDAH expression was determined by real-time quantitative PCR (qPCR). Inflammation was assessed by H&E staining and analyses of total cell counts and fluid tumor necrosis factor (TNF)-α levels (using ELISA) in lung lavage fluid. NF-κB binding activity in mouse lung epithelial (LA-4) cells was assessed by a transcription factor-binding assay. The results indicated that the concentration of serum ADMA was increased following exposure to CS, and this corresponded with increased ADMA content in bronchial epithelial cells in lung tissue. Total lung DDAH expression was significantly decreased in lung tissue and cultured LA-4 cells following CS exposure. Addition of exogenous ADMA increased CSE-mediated NF-κB binding activity and TNFα production in LA-4 cells more than 2-fold compared to that in CSE-exposed controls. CS-mediated lung inflammation was significantly attenuated in DDAH transgenic mice compared to in wild-type controls. These findings demonstrated that lung ADMA metabolism was altered in mice following CS exposure and suggested that ADMA played a role in CS-mediated inflammation through increasing the presence of inflammatory mediators in lung epithelial cells.
Introduction
Cigarette smoke is the primary risk factor for chronic obstructive pulmonary disease (COPD). CS-mediated inflammation plays a central role in the development of COPD (Angelis et al., Citation2014; Nyunoya et al., Citation2014). In addition to numerous other compounds, inhalation of CS also exposes the lung to high levels of free radicals present in the smoke. The exposure leads to the activation of endogenous oxidative processes, which in turn modulate the activity of redox-sensitive signal transduction pathways (Kou et al., Citation2011; Pacher et al., Citation2007). These pathways also include the transcription factor nuclear factor (NF)-κB (Tamimi et al., Citation2012).
One potential contributor of intracellular oxidant production is the uncoupling of nitric oxide synthase (NOS). NOS is ubiquitously expressed in the lung and present in both constitutively expressed and inducible isoforms. This enzyme produces nitric oxide (NO) from the substrate L-arginine. In conditions of limiting substrate availability, this molecule can be uncoupled to produce superoxide that combines with NO to form peroxynitrite. Elevated rates of tissue peroxynitrite production are associated with a number of respiratory diseases including COPD (Ichinose et al., Citation2000). Peroxynitrite generation is significantly elevated in sputum macrophages of COPD patients, which is negatively correlated with pulmonary function in these patients (Osoata et al., Citation2009).
One potential contributor to endogenous reactive species generation is asymmetric dimethyl-arginine (ADMA). ADMA is a naturally occurring analog of L-arginine and is a competitive inhibitor of all NOS isoforms. The bioavailability of L-arginine is determined by the relative concentrations of ADMA and L-arginine. L-Arginine deficiency leads to uncoupling of NOS and oxidant formation (Boger et al., Citation2000; Wells and Holian, Citation2007). It is established that elevated ADMA contributes to endothelial dysfunction (Raptis et al., Citation2014), pulmonary inflammation (Klein et al., Citation2010), and fibrosis (Wells et al., Citation2009). Lung ADMA levels have been associated with airway inflammation in both allergic asthma models (Ahmad et al., Citation2010; Klein et al., Citation2010) and in asthmatics (Scott et al., Citation2011). Further, circulating ADMA levels are altered in smokers (Sobczak et al., Citation2009; Wang et al., Citation2006; Zhang et al., Citation2006); however, the contribution of ADMA to the pathophysiology of chronic lung inflammation is unknown. Although the lung has been identified as a major source of ADMA (Bulau et al., Citation2007), the mechanisms controlling ADMA levels in inflammation are not well understood.
ADMA is generated by proteolytic cleavage of arginine-methylated proteins and metabolized by the enzyme dimethyl-arginine dimethyl-aminohydrolase (DDAH) (Ogawa et al., Citation1989). Two isoforms of DDAH have been cloned and characterized to date (Vallance and Leiper, Citation2004). DDAH I is the predominant isoform in the kidney and liver, while DDAH II is more highly expressed in the vasculature. Both isoforms are expressed in the lung (Bulau et al., Citation2007). Decreased DDAH pulmonary expression has been associated with elevated ADMA levels in a mouse model of allergic asthma (Ahmad et al., Citation2010). However, whether DDAH expression in the lung is altered following CS exposure has not yet been explored. Thus, we hypothesized that CS exposure contributed to non-allergic lung inflammation through perturbation of the ADMA-DDAH pathway. We utilized in vivo and in vitro models to study the effects of CS exposure on ADMA levels and DDAH expression, and explored the consequences of elevated ADMA on inflammatory mediators in mouse lung epithelial cells. Some of the results of these studies have been previously reported in the form of an abstract (see Wells et al., Citation2011).
Materials and methods
Animals and treatment
C57BL/6 and hDDAH transgenic (Tg) mice were originally obtained from Jackson Laboratory (Bar Harbor, ME) and subsequently bred and maintained in micro-isolator units at University of Nebraska Medical Center’s specific pathogen-free animal facility. Mice were allowed food and water ad libitum and were used experimentally at 7–14-weeks-of-age. All animals were handled in accordance with National Institutes of Health guidelines. The University of Nebraska Medical Center Institutional Animal Care and Use Committee approved this study.
Mice were exposed to either air or CS using the Teague small animal whole-body smoke exposure system (Model TE-10, Teague Enterprises, Davis, CA) (Bracke et al., Citation2010; Di Stefano et al., Citation2002; Elliott et al., Citation2006, Citation2007). In the longer exposure experiments, animals were presented with whole-body mainstream and sidestream cigarette smoke via inhalation from 3R4F research cigarettes (Tobacco Health Research Institute, University of Kentucky, Lexington, KY) for 2 h/day, 5 days/week, for 4 weeks (WT, n = 11–12; DDAH Tg, n = 7–8). Utilizing the Teague small animal cigarette smoke exposure system, it has been demonstrated that the smoke generated from two packs of cigarettes per day in rodents produces a similar level of cotinine (Gentry-Nielsen et al., Citation2004) and nicotine (Martins-Green et al., Citation2014) metabolites as measured in equivalent human smoking. This amount of cigarettes is the equivalent of smoking one to two packs of cigarettes a day. For the shorter exposure experiments, mice were exposed to cigarette smoke for 20 min, twice a day, for 4 days (WT, n = 3–4; DDAH Tg, n = 3). Control animals were sham-exposed to compressed air.
Determination of L-arginine and ADMA concentrations
The analysis of serum arginine and ADMA concentrations was conducted utilizing liquid chromatography (LC) mass spectrometry (MS) in multiple reactions monitoring mode (MRM assay). LC/MS/MS MRM analyses were performed employing a Q TRAP 4000 triple Quadruple mass spectrometer (AB Sciex, Foster City, CA) integrated with an Agilent 1200 series HPLC system. The separation of the molecules was carried out in reverse phase chromatography mode with a Thermo Fisher C 18 analytical column [50 × 2.1 mm (I.D.) Hypersil Gold column] packed with silica (1.9-µm particle size). The mobile phases included water containing 0.1% formic acid (mobile phase A) and methanol containing 0.1% formic acid (mobile phase B). Chromatography was performed at 25 °C with a flow rate of 0.35 ml/min. Initially, the column was equilibrated with 50:50 methanol:water. The samples (control, n = 12; smoke, n = 16) were loaded with the same solvent composition in order to reduce the solvent effect on analysis and to allow for the compounds co-elution. The gradient started at 2 min with 2% mobile phase A for 0.5 min and increased linearly to 95% mobile phase B:mobile phase A at 6 min, with subsequent re-equilibration with 50:50 mobile phase A:mobile phase B for 4 min. Nitrogen was used as the nebulizing and drying gas at 350 °C.
The Q trap 4000 mass spectrometer was operated in positive electrospray ionization mode (ESI+). The ion spray voltages were set at 5500 with GS1 and GS2 set at 30. Initially MRM transitions were developed for standard compounds by infusing the compounds with the same MS set-up. The following transitions were observed after fragmentation (CID): m/z 175.109-70.2 [collision energy (CE), 31 volts] for L-arginine (Sigma, St. Louis, MO) and m/z 203.234-70.1 (CE, 35 volts) for ADMA (>99% purity, Sigma). The LC/MS/MS MRM method was developed using the above-mentioned transition and the instrument was calibrated using L-arginine and ADMA standards. Stock solutions of L-arginine and ADMA were made and the calibrators treated exactly the same as the samples. Quantitative determinations were performed using Analyst 1.4.2 software automatically by calculating the area XIC of peaks of specific compound eluted and plotted against concentrations of each compound. Serum (50 µl) containing proteins were precipitated by adding with an equal volume of methanol and then stored at 4 °C for 30 min. The precipitated protein was removed by centrifugation at 8000 x g and the collected supernatant was appropriately diluted with the loading solvent used for LC/MS/MS analysis. Peak areas for individual compounds in the biological samples were calculated as in the protocol used for standard compounds; concentrations were derived from the standard curve.
Histology and immunostaining
Three mice per group were utilized for all histological observations. Twenty-four hours following the final CS exposure, mice were euthanized by overdose injection with sodium pentobarbital. Lungs were removed and inflation-fixed through the trachea with 10% formaldehyde-phosphate-buffered saline (PBS, pH 7.4) solution, processed, embedded in paraffin blocks, serially-sectioned at 7-µm, and mounted onto slides. Sections were then either stained with H&E or for anti-ADMA, DDAH I or DDAH II expression. For immunohistochemical (IHC) detection of ADMA and of DDAH I and II, sections were deparaffinized, hydrated, and washed with PBS. Before staining, endogenous peroxidase activity was inhibited using Peroxo-block (Invitrogen, Carlsbad, CA). Slides were then incubated for 30 min with a 1:1500 dilution of ADMA, 1:300 dilution of anti-DDAH I, or 1:200 of anti-DDAH II (Santa Cruz Biotechnology, Santa Cruz, CA). Bound antibodies were revealed using an immunoperoxidase kit (Vector Laboratories, Burlingame, CA). Control sections were stained with secondary antibody only. All sections were counterstained with hematoxylin (Fisher Scientific, Pittsburgh, PA). The amount of DDAH I and II staining in the airway epithelium was quantified using Image-Pro® Plus by analyzing a minimum of 25 high-powered fields of view (Media Cybernetics, Inc., Betheseda, MD).
RNA isolation and cDNA synthesis
RNA extraction was performed using TRIzol reagent (Invitrogen). RNA quantity and quality were evaluated using a Nanodrop spectrophotometer (Thermo Scientific, Wilmington, DE); all RNA had an A260/A280 ratio of 1.8–2.0. cDNA was synthesized by a reverse transcription reaction as follows: 1 µg isolated RNA and 2.5 µM random hexamers (Applied Biosystems, Carlsbad, CA) were initially incubated at 70 °C for 10 min followed by a 2 min incubation on ice. Subsequently, PCR buffer, 500 µM dNTP mix, and 10 mM DTT (Invitrogen) were added to the reaction mixture and the sample further incubated at room temperature for 5 min. Finally, 200 U of Superscript II were added to the reaction mixture and the incubation was performed in successive stages (i.e. 5 min at room temperature, 50 min at 42 °C, and 15 min at 70 °C). The cDNA mixture was then diluted 1:3 in water and stored at −20 °C.
Quantitative PCR
Quantitative PCR was performed on the cDNA using the following reaction: 1X Taqman master mix and mouse DDAH I primer/probe (Applied Biosystems, Mm01319453_m1) or DDAH II primer/probe (Applied Biosystems, Mm00516769_g1) and ribosomal RNA primer and probe mix in 25 µl reactions in a 96-well plate. Reactions were performed in duplicate and each experiment was repeated at least three times. The plate was placed in an ABI Prism 7500 Sequence detection system (Applied Biosystems). Reactions underwent 2 min at 50 °C, 10 min at 95 °C, and then 40 cycles of 15 s at 95 °C and 1 min at 60 °C. Ribosomal RNA primers and probes (Applied Biosystems) were used as an endogenous control. All data was reported as fold-change from control. A minimum of 17 mice samples was used (control, n = 17; smoke, n = 22), while three-to-four samples were analyzed for the cell culture experiments.
Cell culture
LA-4 cells were obtained from the American Type Culture Collection (Manassas, VA) and maintained in Ham’s modified F-12 medium supplemented with 15% fetal bovine serum (Hyclone, Logan, UT) and antibiotic/antimycotic solution (Media Tech, Herndon, VA) at 37 °C in a humidified atmosphere with 5% CO2–95% air. Cells were seeded at ∼50% confluence and used when they reached 95–100% confluence.
Cells were treated in serum-free media with 10% cigarette smoke extract (CSE), 100 µM ADMA, and/or 5 mM N-acetyl cysteine (NAC; Sigma). CSE was produced by bubbling the smoke from two non-filtered 3R4F research cigarettes through 25 ml of sterile saline. Cigarettes burned for 6 min. CSE was freshly generated prior to each treatment and administered within 30 min, except in the aged CSE experiments where the CSE was aged at room temperature for 24 h.
Cellular NF-κB activation
Activation of NF-κB was determined as previously described (Klein et al., Citation2010) with a commercially available ELISA kit (NF-κB Transcription Factor Assay Kit, Cayman Chemical, Ann Arbor, MI) according to the manufacturer instructions (n = 4/group). The Nuclear Extraction Kit from Cayman Chemical was used to isolate nuclear extracts by following manufacturer directions. Results were normalized to protein concentrations as determined by a Bradford assay.
Determination of TNFα levels
Lung fluid from the first 1.0-ml aliquot and media collected from cell culture experiments were assayed for cytokines with a mouse TNFα Duo-set ELISA kit (R&D Systems, Inc., Minneapolis, MN) according to manufacturer protocols. Samples were used undiluted (n = 4/group). Data are expressed as pg/ml of retrieved culture supernatant. The level of sensitivity of the kit was 31.25 pg TNFα/ml.
Collection of lung lavage cells and fluid
To prepare for lung lavage, mice were euthanized by a lethal intraperitoneal injection of pentobarbital sodium (2.5 mg) 24 h following the final CS exposure. The lungs were then lavaged with four 1.0-ml aliquots of cold PBS. Recovery was 80–90% for all animals. Cells were collected, pooled, and counted on a Coulter Particle Counter (Beckman Coulter, Hialeah, FL).
Measurement of nitrite concentrations
Greiss reagents (1% sulfanilamide and 0.1% napthylethylenediamine in 5% phosphoric acid; Ricca Chemical, Pocomoke, MD) were used to measure the nitrite concentrations in the lavage fluid (WT, n = 9–15; DDAH Tg, n = 9) as previously described (Wells et al., Citation2009). Calibration curves were made with NaNO2 (Sigma) dissolved in PBS.
Statistical analysis
Means ± SE were calculated for all samples and, except where otherwise noted, p values were calculated using an unpaired t-test or a one-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison test. All data were expressed as means ± SE. A p value <0.05 was accepted as statistically significant. A Grubb’s test was performed to detect outliers.
Results
ADMA is elevated following 4-week CS exposure
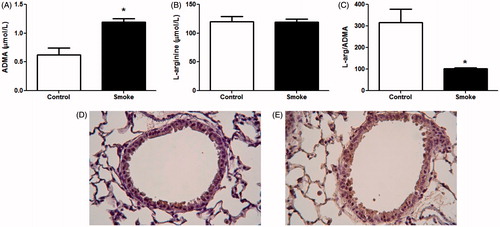
Using an established 4-week CS exposure model (Bracke et al., Citation2010), this study first determined whether circulating L-arginine and ADMA levels were altered following sub-chronic exposure to CS. Quantification of L-arginine and ADMA by LC-MS confirmed that circulating ADMA levels were nearly 2-fold greater following a 4-week exposure to CS (). There was no change in serum L-arginine levels (), resulting in a significantly lower circulating L-arginine/ADMA ratio (). IHC with an antibody that recognized both free and protein-incorporated ADMA was performed to determine localization of ADMA in lung tissue. In air-exposed animals, there was staining predominantly in bronchial epithelial cells (). In smoke-exposed mice, staining intensity was increased in the bronchial epithelium and throughout the alveolar spaces (). Although it was not possible to determine whether the increased staining intensity for ADMA in the smoke-exposed lungs was due to elevated free or protein-incorporated ADMA, these immunohistochemical findings were consistent with the LC-MS data demonstrating elevated free ADMA in serum.
Figure 1. ADMA levels following 4-week exposure to CS. Serum and lungs were collected after 4 weeks of smoke exposure. Serum (A) ADMA and (B) L-arginine levels were determined by LC-MS (control, n = 12; smoke, n = 16). (C) Ratio of L-arginine to ADMA levels in serum. Data shown are means ± SE; *p < 0.001 vs control animals. ADMA levels in lung tissue were visualized using IHC. Lung sections of (D) control and (E) smoke-exposed mice were stained with antibody that recognizes both free and protein-incorporated forms of ADMA. Magnification 40×. Both the LC-MS data and IHC data show elevated ADMA after smoke exposure.
CS exposure decreased DDAH expression in vivo
Free intracellular ADMA is metabolized by DDAH (Ogawa et al., Citation1989). To determine whether increased circulating ADMA was associated with altered ADMA metabolism by DDAH in the lung following CS exposure, lung DDAH mRNA levels were measured in whole lung homogenates using qPCR. The results indicated that mRNA levels of DDAH I were significantly lower in lungs of CS-exposed mice (). Pulmonary expression levels of DDAH II mRNA were not significantly lower following exposure to CS.
Figure 2. Lung DDAH expression following 4-week exposure to CS. Cell DDAH I and DDAH II expression was visualized by IHC. Lung sections of (A and C) control and (B and D) smoke-exposed mice were stained with antibodies to (A and B) DDAH I or (C and D) DDAH II. Magnification 40×. (E) DDAH I and DDAH II mRNA expression levels were determined in whole lung homogenates of control and smoke-exposed mice using real-time qPCR (control, n = 17; smoke, n = 22). Data shown are means ± SE. *p < 0.05 vs control animals. (F) The amount of DDAH I and DDAH II staining was quantified as the percentage positive (brown) stained vs total cell counterstain (purple) for each field. The data illustrates that DDAH protein levels are lower in CS-exposed mice.
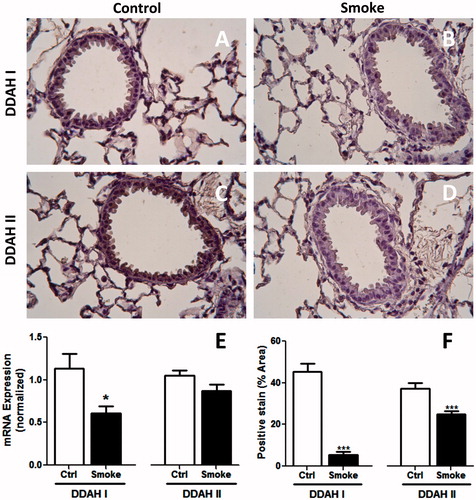
Both isoforms of DDAH are expressed in lung tissue (Bulau et al., Citation2007); however, relative expression of these isoforms in specific cell types has not been well established. Thus, this study next explored DDAH expression in lung tissue sections by IHC. Protein expressions of both DDAH I and II were observed in bronchial epithelial and alveolar cell types (). Exposure to CS led to overall decreased staining for both DDAH isotypes, particularly in the airway epithelium (). This decrease was quantified in . Taken together, these findings indicated that decreased metabolism by DDAH in the lung epithelium may have contributed to increased serum ADMA levels following CS exposure.
Effect of CSE on DDAH RNA expression in vitro is reversed by N-acetylcysteine (NAC)
To establish whether CS affects DDAH expression in lung epithelial cells in vitro, this study utilized the mouse LA-4 lung epithelial cell line. We have previously shown this cell line has measurable NOS activity and is responsive to inflammatory cytokine stimulation (Wells and Holian, Citation2007). Here, LA-4 cells were exposed to either freshly prepared or aged CSE (10%) for 6 h. Exposure to fresh, but not aged, CSE significantly decreased both DDAH I () and DDAH II () expressions. These findings provided further support for the conclusion that CS altered ADMA metabolism through effects on DDAH expression.
Figure 3. Effects of CSE on DDAH expression in mouse lung epithelial cells in vitro. Cultured LA-4 cells were treated with either fresh or aged (24 h) 10% CSE for 6 h in serum-free media and mRNA expression for (A) DDAH I and (B) DDAH II was determined using real-time qPCR (n = 3/group). Cells were treated with CSE and/or 5 mM NAC for 6 h in serum-free media and mRNA expression for (C) DDAH I and (D) DDAH II was determined using real-time qPCR (n = 4/group). Data shown are means ± SE. *p < 0.05 vs control cells. Fresh CSE decreased DDAH I and II. NAC was able to block this effect.
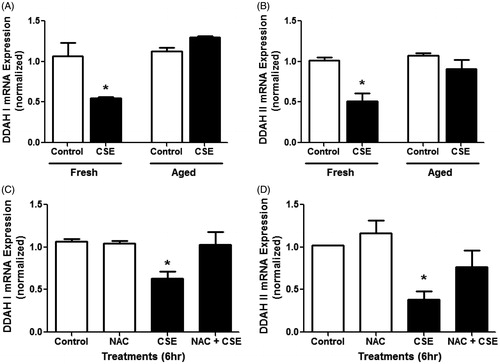
The lack of effect of aged CSE on DDAH expression implicated volatile components of CSE in the regulation of DDAH expression. Thus, this study next determined whether the anti-oxidant NAC would attenuate the effects of fresh CSE on DDAH expression. For both DDAH I and DDAH II, treatment with 5 mM NAC alone had no effect on DDAH expression (). Co-treatment with both CSE and NAC, however, reversed the effects of CSE on DDAH I and II expressions, suggesting a role for oxidants in CSE-mediated down-regulation of DDAH.
ADMA in combination with CSE increases NF-κB binding activity and TNFα production in vitro
In addition to oxidative stress, NF-κB mediated pro-inflammatory responses to CS play a key role in the pathogenesis of chronic inflammatory lung diseases (Di Stefano et al., Citation2002; MacNee, Citation2005; Rahman and MacNee, Citation1998; Tamimi et al., Citation2012). Our laboratory previously found that ADMA induced oxidative and nitrosative stress in lung epithelial cells (Klein et al., Citation2010). Furthermore, we showed that, in mouse lung epithelial cells, ADMA significantly increased NF-κB binding activity in the presence of inflammatory cytokines. To examine whether ADMA also enhanced NF-κB binding activity following CSE, LA-4 cells were exposed to fresh CSE and/or ADMA (100 µM), and the nuclear NF-κB binding activity was then measured (). The results indicated that ADMA or CSE alone had little effect on NF-κB binding activity at 24 h. However, when combined, there was a significant increase in NF-κB binding activity.
Figure 4. Effects of ADMA and CSE on NF-κB activity and TNFα production in mouse lung epithelial cells. LA-4 cells treated with 100 µM ADMA ±10% CSE in serum-free media for ∼24 h. (A) Relative NF-κB binding activity in nuclear extracts was determined (n = 4/group). (B) TNFα levels in the media were also assessed (n = 4/group). Data shown are means ± SE. *p < 0.05. This data indicates that ADMA in combination with CSE increases both NF-κB activity and TNFα production in vitro.
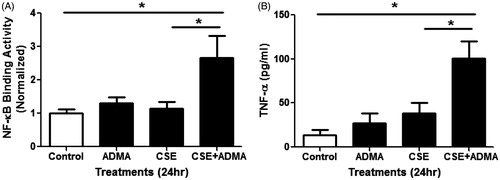
NF-κB regulates several inflammatory cytokines including TNFα and is increased in the airway epithelium of smokers compared to non-smokers (Di Stefano et al., Citation2002). To determine whether the effects of ADMA on NF-κB activity leads to enhanced cytokine production by epithelial cells, we measured TNFα production following exposure to CSE and/or ADMA (100 µM). Consistent with our findings for NF-κB binding activity, the combination of CSE and ADMA significantly increased TNFα production by these cells (). These findings suggest that ADMA may contribute to CS-mediated pulmonary inflammation through activation of pro-inflammatory mediators in lung epithelial cells.
Mice over-expressing DDAH I were protected from CS-mediated lung inflammation
To confirm the role of elevated ADMA in CS-mediated lung inflammation in vivo, transgenic mice carrying the human transgene to DDAH I (DDAH Tg) were employed. These mice have a ≈2-fold reduction of circulating endogenous ADMA while their L-arginine levels are unchanged (Dayoub et al., Citation2003). In a previous study, we confirmed that pulmonary expression of DDAH I is ≈9-fold higher than wild-type (WT) littermate controls (Klein et al., Citation2010). That study also ascertained that lower endogenous ADMA levels protected against lung inflammation in an allergic asthma model. The DDAH Tg mice and their WT littermates were exposed to CS for 4 weeks, as described in the Methods.
In the study here, inflammation was qualitatively assessed histologically. H&E-stained sections revealed that, in contrast to control WT () and DDAH Tg () mice, there was mild inflammation around small airways in the CS-exposed WT mice (). However, little inflammation was observed in the CS-exposed DDAH Tg mice (). To quantitatively assess inflammatory cell infiltrates in the airway, total cell numbers and TNFα levels in lung lavage fluid were determined. WT mice exposed to CS had ≈50% more lavage cells compared to their air-exposed counterparts (). Relative numbers of monocytic and granulocytic cell types were counted. Approximately 98% of the cells were macrophages. Thus, a significant increase in macrophages primarily accounted for the elevated cell numbers (data not shown). Lavage cell numbers in CS-exposed DDAH Tg mice were indistinguishable from those in unexposed WT littermate controls.
Figure 5. CS-mediated lung inflammation in DDAH transgenic mice. Lung inflammation was assessed in lung tissue. Lung sections of (A and B) wild-type (WT) and (C and D) hDDAh Tg mice were H&E stained, and inflammation was visualized following (A and C) air or (B and D) 4-week CS exposure. Original magnification = 10×; increased magnification = 40×. Total cells in whole lung lavage were counted following (E) 4-week (WT, n = 11–12; DDAH Tg, n = 7–8) and (G) 4 days (WT, n = 6; DDAH Tg, n = 5) smoke exposures. TNFα levels in BAL fluid were also assessed in both the (F) 4-week (WT, n = 8–9; DDAH Tg, n = 7–8) and (H) the 4 day (WT, n = 3–4; DDAH Tg, n = 3) models. Data shown are means ± SE. *p < 0.05 vs WT control, Ψp < 0.05 vs WT smoke.
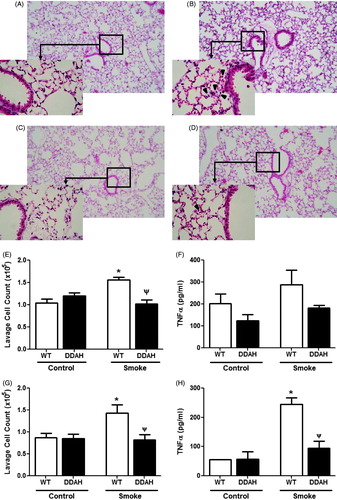
TNFα levels in lavage fluid were not increased in the WT longer (4-week) CS-exposed mice compared to WT unexposed controls (). Therefore, we employed a second, shorter CS exposure model in which lavage TNFα levels have been shown to be elevated (Churg et al., Citation2002, Citation2003; Shen et al., Citation2014). Similar to the 4-week exposure model, DDAH Tg mice were protected from inflammation as measured by inflammatory cell infiltrates in lavage fluid (). Consistent with this, TNFα, while significantly increased in the CS-exposed WT mice, was indistinguishable from unexposed WT and DDAH Tg mice. These results show that mice with lower endogenous ADMA levels are less susceptible to CS-induced lung inflammation compared to their WT littermates.
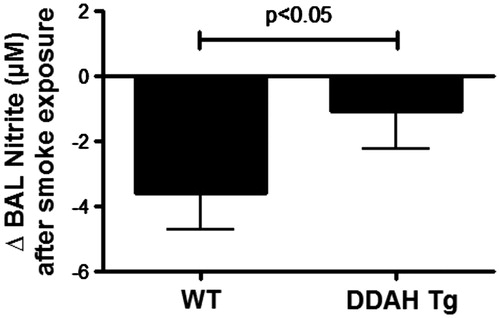
As an inhibitor of NOS, these lower levels of ADMA should affect the levels of nitric oxide production. To confirm this, the total nitrite level in the lavage fluid of the 4-week mice was measured (). There was a CS-induced decrease in nitrite levels in the WT mice (Control = 12.9 ± 3.3 µM vs Smoke = 9.2 ± 2.2 µM). This decrease was significantly less in the DDAH Tg CS-exposed mice (Control = 9.1 ± 1.4 µM vs Smoke = 8.0 ± 2.6 µM). Consequently, ADMA levels are affecting NOS and nitric oxide production.
Figure 6. CS-mediated lung lavage nitrate levels in DDAH transgenic mice. Nitric oxide production was measured by nitrite formation in lung lavage fluid. Lung lavage (BAL) of wild-type (WT, n = 9–15) and hDDAh transgenic mice (DDAH Tg, n = 9) was assayed for total nitrite content following cigarette smoke exposure for 4 weeks. The cigarette smoke-induced decrease in nitrite levels was significantly less in DDAH Tg mice compared to WT mice. Data shown are means ± SE. *p < 0.05 DDAH Tg vs WT control.
Discussion
Emerging evidence indicates that ADMA plays a role in the pathogenesis of chronic lung diseases (reviewed in Zakrzewicz and Eickelberg, Citation2009). Our laboratory and others have demonstrated that ADMA is not only elevated in allergic inflammation (Ahmad et al., Citation2010; Scott et al., Citation2011; Wells et al., Citation2007), but that elevated ADMA also potentiates inflammation in allergic inflammation models (Klein et al., Citation2010). However, the association between ADMA levels and the pathophysiology of other chronic respiratory diseases such as COPD remain unclear. Thus, we utilized a mouse model of CS-mediated lung inflammation to determine whether CS exposure leads to alterations in the ADMA-DDAH pathway. The salient findings of this study are: (1) circulating ADMA levels are increased in mice following sub-chronic CS exposure; (2) CS exposure decreases DDAH expression in vivo and in vitro; (3) ADMA synergizes with CSE to increase nuclear NF-κB activity and TNFα production in mouse lung epithelial cells; and (4) transgenic mice with increased DDAH expression and low circulating ADMA levels are protected from CS-mediated inflammation.
The present finding that sub-chronic exposure to CS is associated with significantly higher serum ADMA levels in mice is consistent with two studies in human subjects. In a small study, Zhang et al. (Citation2006) reported that plasma ADMA levels were nearly 2-fold higher in otherwise healthy smokers compared to healthy non-smokers. In a larger study, there was a modest association between smoking and increased ADMA levels (Wang et al., Citation2006). However, the association between increased ADMA and smoking in humans is still controversial (Sobczak et al., Citation2009). In a large, cross-sectional study in elderly men with hypercholesterolemia, Eid et al. (Citation2004) found an association between smoking and decreased circulating ADMA levels. Clearly establishing a link between smoking and lower ADMA levels in this study is complicated, however, by the high frequency of other cardiovascular disease risk factors and the potential impact of medications such as statins, which have been shown to lower ADMA levels (Lu et al., Citation2004; Vladimirova-Kitova and Deneva-Koycheva, Citation2012). It is likely that plasma ADMA concentrations are regulated by multiple factors that may differ between healthy and disease states. Thus, the effect of cigarette smoke exposure on ADMA levels is undoubtedly complex and additional larger studies in otherwise healthy current and non-smokers will be necessary to establish whether smoking itself leads to altered circulating ADMA levels.
Although evidence supports that methylarginine metabolism by the pulmonary system significantly contributes to circulating ADMA levels (Bulau et al., Citation2007), the source of increased serum ADMA in our model is uncertain. The IHC staining localization of free and protein-incorporated ADMA in control lungs suggested that the majority of ADMA was contained in airway epithelial cells. In contrast, ADMA appeared more diffuse in the lungs of mice exposed to CS, indicating a change in tissue distribution. However, the qualitative assessments conducted in this study did not allow identification of specific cell types contributing to higher circulating ADMA in CS-exposed mice. Additional studies will be necessary to pinpoint which pulmonary cell types are the major contributors to increased circulating ADMA levels under pathophysiological conditions.
Synthesis and metabolism of endogenous methylarginines are highly regulated. Although there have been reports of ADMA regulation through the alanine-glyoxylate aminotransferase 2 (AGXT2) alternate pathway (Rodionov et al., Citation2010), several groups have demonstrated that DDAH I is the critical enzyme for intracellular degradation of ADMA (Bulau et al., Citation2007; Hu et al., Citation2011; Pope et al., Citation2009). In the 4-week CS exposure model here, there was a significant decrease in overall DDAH I mRNA expression in the lung following smoke exposure. IHC staining revealed lower DDAH I expression in both the bronchial epithelium and alveolar compartment. A similar decrease in DDAH II staining was observed by IHC; however, total lung tissue DDAH II mRNA levels were not significantly different following CS exposure. This may be due to decreased expression of DDAH II in a sub-set of pulmonary epithelial cells or a lower sensitivity of qPCR for detecting a change in total lung tissue. Nonetheless, in vitro exposure of lung epithelial cells to CSE resulted in decreased expression of both DDAH I and DDAH II, thus indicating that CS exposure affected both isoforms. These findings were consistent with decreased DDAH I expression in rabbit cavernous tissue following in vivo exposure to CSE (Imamura et al., Citation2007) and supported the notion of decreased pulmonary DDAH expression leading to elevated ADMA in our model. The demonstration that mice over-expressing DDAH were protected from CS-mediated inflammation underscored the importance of this molecule in CS-mediated inflammation.
ADMA is derived from the proteolysis of proteins containing methylated arginine residues. Protein-arginine methylation is catalyzed by a family of enzymes termed protein-arginine methyltransferases (PRMT) (Paik and Kim, 1968). Intracellular protein hydrolysis releases free ADMA. In exchange for L-arginine and other cationic amino acids, ADMA is transported out of cells through cationic amino acid transporters (CAT) (Baylis, Citation2006; Closs et al., Citation1997; Welch and Wilcox, Citation1997) where it can have effects at sites distal from its production. Although we are not aware of any studies demonstrating an effect of CS on PRMT expression or function, Zhang et al. (Citation2006) reported a significant reduction in the expression of CAT1 in human endothelial cells following a brief exposure to 10% CSE. Further studies will be necessary to determine whether CS exposure affects pulmonary expression of PRMT or CAT in our sub-chronic CS exposure model.
In human endothelial cells, reduced DDAH activity following CSE exposure is attenuated by the addition of the anti-oxidant NAC (Zhang et al., Citation2006). The finding here that CSE-mediated decreased DDAH expression in mouse lung epithelial cells was reversed by NAC was consistent with this previous observation and argues for a common mechanism in these two different cell types. Cigarette smoke is complex and, although this study did not attempt to identify the specific compound(s) responsible for the effects on DDAH expression, the finding here that the effect of decreased DDAH expression was lost following aging of the smoke suggested volatile components in CSE may regulate DDAH expression. Indeed, the protein kinase C-mediated cigarette smoke stimulation of pro-inflammatory cytokines in airway epithelium requires the presence of volatile acetaldehyde in the smoke (Wyatt et al., Citation2000). DDAH activity and levels have previously been shown to be oxidant-sensitive (Forbes et al., Citation2008; Sousse et al., Citation2011). However, to the best of our knowledge, this was the first demonstration that DDAH mRNA expression in lung epithelial cells was affected by oxidative stress.
The finding here that CS-mediated inflammation was attenuated in DDAH transgenic mice that have low endogenous levels of ADMA suggested that ADMA may have an effect on production of inflammatory mediators in the lung. Our data also show that, in the wild-type mouse, CS-mediated reduction in DDAH coincides with reduced NO, likely through the action of ADMA inhibition of NOS. We have previously shown that, in the presence of inflammatory cytokines, ADMA enhances NF-κB activity in LA-4 cells (Klein et al., Citation2010). In the present study, it was shown that, in combination with CSE, ADMA not only increased NF-κB activity in these cells, but also induced the production of TNFα after 24 h. This effect was not seen at earlier timepoints, suggesting that any ADMA-mediated generation of oxidants may have contributed to increased activation of NF-κB and production of TNFα in this cell line (Loukili et al., Citation2010). Indeed, the role of ADMA in enhancing NF-κB activity is also supported by the finding that elevated NO is capable of inhibiting NF-κB binding to DNA (Matthews et al., 1996). These findings indicate that elevated ADMA may contribute to inflammation by enhancing inflammatory mediator production by epithelial cells and increasing the recruitment of inflammatory cells in response to CS exposure.
Conclusions
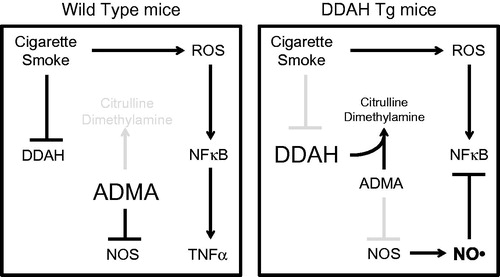
Based on the present findings, we propose a model for the contribution of the ADMA-DDAH pathway in the inflammatory effects of CS in the lung (). In this model, CSE cause a reduced expression of DDAH I and II in lung epithelial cells. Because DDAH is not converting ADMA into citrulline dimethylamine, smoke exposure could lead to increased ADMA. Elevated ADMA inhibits NOS activity that then enhances CS-mediated inflammation by allowing for increased NF-κB activity and the release of inflammatory mediators such as TNFα by airway epithelial cells.
Figure 7. Model for the ADMA/DDAH pathway in CS-mediated inflammation. In this model, CSE reduces DDAH I and II expression in lung epithelial cells, which leads to increased ADMA. Elevated ADMA inhibits NOS, which enhances CS-mediated inflammation allowing for increased NF-κB activity and the release of inflammatory mediators such as TNFα by airway epithelial cells.
In summary, our data support the hypothesis that ADMA plays a role in the pathology of CS-mediated inflammation. Although additional studies will be necessary to confirm these findings in humans, this study provided further evidence that modulation of pulmonary ADMA metabolism may present a novel therapeutic approach for the treatment of inflammation associated with chronic lung diseases such as COPD.
Declaration of interest
This work was supported by UNMC College of Public Health Dean’s Mentored Research Fund (SMW), R00HL088550 (SMW) from the National Heart, Lung, and Blood Institute (NHLBI), a component of the National Institutes of Health (NIH), and U54OH010162 (TAW) from the National Institute for Occupational Safety and Health, part of the Centers for Disease Control. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NHLBI or NIH.
Related Research Data
References
- Ahmad, T., Mabalirajan, U., Ghosh, B., and Agrawal, A. 2010. Altered asymmetric dimethyl-arginine metabolism in allergically inflamed mouse lungs. Am. J. Respir. Cell Mol. Biol. 42:3–8
- Angelis, N., Porpodis, K., Zarogoulidis, P., et al. 2014. Airway inflammation in chronic obstructive pulmonary disease. J. Thoracic Dis. 6:S167–172
- Baylis, C. 2006. Arginine, arginine analogs, and nitric oxide production in chronic kidney disease. Nat. Clin. Pract. Nephrol. 2:209–220
- Boger, R. H., Bode-Boger, S. M., Tsao, P. S., et al. 2000. An endogenous inhibitor of nitric oxide synthase regulates endothelial adhesiveness for monocytes. J. Am. Coll. Cardiol. 36:2287–2295
- Bracke, K. R., Dentener, M. A., Papakonstantinou, E., et al. 2010. Enhanced deposition of low-molecular-weight hyaluronan in lungs of cigarette smoke-exposed mice. Am. J. Respir. Cell Mol. Biol. 42:753–761
- Bulau, P., Zakrzewicz, D., Kitowska, K., et al. 2007. Analysis of methyl-arginine metabolism in the cardiovascular system identifies the lung as a major source of ADMA. Am. J. Physiol. 292:L18–24
- Churg, A., Dai, J., Tai, H., et al. 2002. Tumor necrosis factor-α is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am. J. Respir. Crit. Care Med. 166:849–854
- Churg, A., Wang, R. D., Tai, H., et al. 2003. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via TNFα release. Am. J. Respir. Crit. Care Med. 167:1083–1089
- Closs, E. I., Basha, F. Z., Habermeier, A., and Forstermann, U. 1997. Interference of L-arginine analogues with L-arginine transport mediated by the y+ carrier hCAT-2B. Nitric Oxide Biol. Chem. 1:65–73
- Dayoub, H., Achan, V., Adimoolam, S., et al. 2003. Dimethyl-arginine dimethylaminohydrolase regulates NO synthesis: Genetic and physiological evidence. Circulation 108:3042–3047
- Di Stefano, A., Caramori, G., Oates, T., et al. 2002. Increased expression of NF-κB in bronchial biopsies from smokers and patients with COPD. Eur. Resp. J. 20:556–563
- Eid, H. M., Arnesen, H., Hjerkinn, E. M., et al. 2004. Relationship between obesity, smoking, and the endogenous nitric oxide synthase inhibitor, asymmetric dimethyl-arginine. Metab. Clin. Exp. 53:1574–1579
- Elliott, M. K., Sisson, J. H., West, W. W., and Wyatt, T. A. 2006. Differential in vivo effects of whole cigarette smoke exposure versus cigarette smoke extract on mouse ciliated tracheal epithelium. Exp. Lung Res. 32:99–118
- Elliott, M. K., Sisson, J. H., and Wyatt, T. A. 2007. Effects of cigarette smoke and alcohol on ciliated tracheal epithelium and inflammatory cell recruitment. Am. J. Respir. Cell Mol. Biol. 36:452–459
- Forbes, S. P., Druhan, L. J., Guzman, J. E., et al. 2008. Mechanism of 4-HNE mediated inhibition of hDDAH-1: Implications in no regulation. Biochemistry 47:1819–1826
- Gentry-Nielsen, M. J., Vander Top, E., Snitily, M. U., et al. 2004. A rat model to determine the biomedical consequences of concurrent ethanol ingestion and cigarette smoke exposure. Alcoholism: Clin. Exp. Med. 28:1120–1128
- Hu, X., Atzler, D., Xu, X., et al. 2011. Dimethyl-arginine dimethylaminohydrolase-1 is the critical enzyme for degrading the cardiovascular risk factor asymmetrical dimethyl-arginine. Arterio-sclerosis Thrombosis Vasc. Biol. 31:1540–1546
- Ichinose, M., Sugiura, H., Yamagata, S., et al. 2000. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am. J. Respir. Crit. Care Med. 162:701–706
- Imamura, M., Waseda, Y., Marinova, G.V., et al. 2007. Alterations of NOS, arginase, and DDAH protein expression in rabbit cavernous tissue after administration of cigarette smoke extract. Am. J. Physiol. 293:R2081–2089
- Klein, E., Weigel, J., Buford, M. C., et al. 2010. Asymmetric dimethyl-arginine potentiates lung inflammation in a mouse model of allergic asthma. Am. J. Physiol. 299:L816–825
- Kou, Y. R., Kwong, K., and Lee, L. Y. 2011. Airway inflammation and hypersensitivity induced by chronic smoking. Resp. Physiol. Neurobiol. 178:395–405
- Loukili, N., Rosenblatt-Velin, N., Rolli, J., et al. 2010. Oxidants positively or negatively regulate nuclear factor-κB in a context-dependent manner. J. Biol. Chem. 285:15746–15752
- Lu, T. M., Ding, Y. A., Leu, H. B., et al. 2004. Effect of rosuvastatin on plasma levels of asymmetric dimethyl-arginine in patients with hypercholes-terolemia. Am. J. Cardiol. 94:157–161
- MacNee, W. 2005. Pulmonary and systemic oxidant/anti-oxidant imbalance in chronic obstructive pulmonary disease. Proc. Am. Thor. Soc. 2:50–60
- Martins-Green, M., Adhami, N., Frankos, M., et al. 2014. Cigarette smoke toxins deposited on surfaces: Implications for human health. PLoS ONE. 9:e86391
- Matthews, J., Botting, C., Panico, M., et al. 1996. Inhibition of NF-κB binding by nitric oxide. Nucl. Acids Res. 24:2236–2242
- Nyunoya, T., Mebratu, Y., Contreras, A., et al. 2014. Molecular processes that drive cigarette smoke-induced epithelial cell fate of the lung. Am. J. Respir. Cell Mol. Biol. 50:471–482
- Ogawa, T., Kimoto, M., and Sasaoka, K. 1989. Purification and properties of a new enzyme, NG, NG-dimethyl-arginine dimethylaminohydrolase, from rat kidney. J. Biol. Chem. 264:10205–10209
- Osoata, G. O., Hanazawa, T., Brindicci, C., et al. 2009. Peroxynitrite elevation in exhaled breath condensate of COPD and its inhibition by fudosteine. Chest 135:1513–1520
- Pacher, P., Beckman, J. S., and Liaudet, L. 2007. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 87:315–424
- Paik, W. K., and Kim, S. 1968. Protein methylase I. Purification and properties of the enzyme. J. Biol. Chem. 243:2108–2114
- Pope, A. J., Karrupiah, K., Kearns, P. N., et al. 2009. Role of dimethyl-arginine dimethylaminohydrolases in the regulation of endothelial nitric oxide production. J. Biol. Chem. 284:35338–35347
- Rahman, I., and MacNee, W. 1998. Role of transcription factors in inflammatory lung diseases. Thorax 53:601–612
- Raptis, V., Georgianos, P. I., Sarafdis, P. A., et al. 2014. Elevated asymmetric dimethyl-arginine is associated with oxidant stress aggravation in patients with early stage autosomal dominant polycystic kidney disease. Kidney Blood Pressure Res. 38:72–82
- Rodionov, R. N., Murray, D. J., Vaulman, S. F., et al. 2010. Human alanine-glyoxylate aminotransferase 2 lowers asymmetric dimethyl-arginine and protects from inhibition of nitric oxide production. J. Biol. Chem. 285:5385–5391
- Scott, J. A., North, M. L., Rafii, M., et al. 2011. Asymmetric dimethyl-arginine is increased in asthma. Am. J. Respir. Crit. Care Med. 184:779–785
- Shen, L. L., Liu, Y. N., Shen, H. J., et al. 2014. Inhalation of glycopyrronium inhibits cigarette smoke-induced acute lung inflammation in a murine model of COPD. Int. Immunopharmacol. 18:358–364
- Sobczak, A., Goniewicz, M. L., and Szoltysek-Boldys, I. 2009. ADMA and SDMA levels in healthy men exposed to tobacco smoke. Atherosclerosis 205:357–359
- Sousse, L. E., Yamamoto, Y., Enkhbaatar, P., et al. 2011. Acute lung injury-induced collagen deposition is associated with elevated asymmetric dimethyl-arginine and arginase activity. Shock 35:282–288
- Tamimi, A., Serdarevic, D., and Hanania, N. A. 2012. The effects of cigarette smoke on airway inflammation in asthma and COPD: Therapeutic implications. Resp. Med. 106:319–328
- Vallance, P., and Leiper, J. 2004. Cardiovascular biology of the asymmetric dimethyl-arginine: Dimethyl-arginine dimethylaminohydrolase pathway. Arteriosclerosis Thrombosis Vasc. Biol. 24:1023–1030
- Vladimirova-Kitova, L. G., and Deneva-Koycheva, T. I. 2012. The effect of simvastatin on asymmetric dimethyl-arginine and flow-mediated vasodilation after optimizing the LDL level: A randomized, placebo-controlled study. Vasc. Pharmacol. 56:122–130
- Wang, J., Sim, A. S., Wang, X. L., et al. 2006. Relations between plasma asymmetric dimethyl-arginine (ADMA) and risk factors for coronary disease. Atherosclerosis 184:383–388
- Welch, W. J., and Wilcox, C. S. 1997. Macula densa arginine delivery and uptake in the rat regulates glomerular capillary pressure. Effects of salt intake. J. Clin. Invest. 100:2235–2242
- Wells, S., Buford, M., and Holian, A. 2007. Asymmetric dimethyl-arginine (ADMA) exacerbates airway hyper-responsiveness and inflammation in murine models of asthma. Am. J. Respir. Crit. Care Med. 175:A204
- Wells, S., Klein, E., Weigel, J., and Wyatt, T. 2011. Asymmetric dimethyl-arginine (ADMA) potentiates cigarette smoke-induced pulmonary inflammation in mice. Am. J. Respir. Crit. Care Med. 183:A3257
- Wells, S. M., and Holian, A. 2007. Asymmetric dimethyl-arginine induces oxidative and nitrosative stress in murine lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 36:520–528
- Wells, S. M., Buford, M. C., Migliaccio, C. T., and Holian, A. 2009. Elevated asymmetric dimethyl-arginine alters lung function and induces collagen deposition in mice. Am. J. Respir. Cell Mol. Biol. 40:179–188
- Wyatt, T. A., Schmidt, S. C., Rennard, S. I., et al. 2000. Acetaldehyde-stimulated PKC activity in airway epithelial cells treated with smoke extract from normal and smokeless cigarettes. Proc. Soc. Exp. Biol. Med. 225:91–97
- Zakrzewicz, D., and Eickelberg, O. 2009. From arginine methylation to ADMA: A novel mechanism with therapeutic potential in chronic lung diseases. BMC Pulm. Med. 9:5
- Zhang, W. Z., Venardos, K., Chin-Dusting, J., and Kaye, D. M. 2006. Adverse effects of cigarette smoke on NO bioavailability: Role of arginine metabolism and oxidative stress. Hypertension 48:278–285