
Abstract
Interleukin-23 (IL-23) is a regulator of cellular immune responses involved in controlling infection and autoimmune diseases. Strong evidence has shown that IL-23 plays a role in the maintenance of immune responses by influencing the proliferation and survival of IL-17-producing T-helper (TH)-17 cells. The critical role of the IL-23/TH17 axis in immune-mediated diseases has emerged from different studies. It has also been seen that polymorphisms in the IL-23 receptor (IL-23R) gene might influence IL-23 responses. Interestingly, a functional single nucleotide polymorphism (SNP) in the IL-23 receptor gene (IL-23R; rs11209026, 1142 G wild-type A reduced function, Arg381Gln, R381Q) seems to confer a measure of protection against development of inflammatory bowel disease (IBD; Crohn’s disease, ulcerative colitis), ankylosing spondylitis, rheumatoid arthritis, psoriasis, thyroiditis, recurrent spontaneous abortion and asthma, suggesting that a perturbation in the IL-23 signaling pathway is likely to be relevant to the pathophysiology of these diseases. The aim of this review was to provide an evaluation of what is currently known about the protective role of R381Q variant in IL-23R gene in immune-based diseases.
IL-23 (Interleukin IL-23)
Interleukin (IL)-23 is a novel cytokine formed via the binding of a shared IL-12p40 subunit to a p19 protein. IL-12p40 is mainly produced by activated myeloid cells as well as epithelial and endothelial cells (Parham et al. Citation2002). The p19 subunit was initially discovered while looking for sequences related to the IL-6 family of cytokines (by searching sequence databases with structure-based alignment tools). This subunit has a four-helix bundle structure, is closely related to the IL-12 p35 subunit and is expressed predominately by activated dendritic cells and phagocytic cells. Following exposure to pathogen-derived molecules that bind toll-like receptors, IL-23 is secreted primarily by activated dendritic cells, monocytes and macrophages (Oppmann et al. Citation2000). IL-23 plays a role in the maintenance of immune responses by controlling T-cell memory function and by influencing the proliferation and survival of IL-17- producing T-helper (TH)-17 cells (Frucht Citation2002; Bettelli et al. Citation2007). Furthermore, IL-23 can shape TH1-immunity via CD3+CD56+ T-cells, through the production of interferon (IFN)-γ early in the immune response (van de Vosse et al. Citation2004). The most likely interpretation of the accumulating set of data is that IL-23 plays a more important role than IL-12 in chronic inflammation. Some research has shown that IL-23 can induce increases in IL-6 production and that this may account for the tissue injury characteristic of inflammatory bowel disease (IBD) (Fuss et al. Citation2006).
Crohn’s disease (CD) and ulcerative colitis (UC) are two related, relapsing and chronic inflammatory disorders of the gastrointestinal tract, commonly denoted as IBD. Although the precise etiology of IBD remains unknown, both animal models and human studies have pointed out a substantial genetic contribution to disease susceptibility. Several chromosomal regions have been in linkage with IBD susceptibility in genome-wide scan analysis and subsequent association studies further identified specific genetic variants associated with IBD (Oliver et al. Citation2007). In 2001 the CARD15 (caspase-activating recruitment domain-15)/NOD2 (nucleotide-binding oligomerization domain 2 encoding cytosolic proteins that enable recognition of peptidoglycans) gene on chromosome 16 was identified as the first susceptible gene for IBD, specifically in CD. A meta-analysis of 37 studies of NOD2/CARD15 variants estimated that homozygous carriage of NOD2 mutant alleles conferred a 17.1-fold increased risk of developing CD, whereas heterozygous carriage increased the CD risk CD 2.4-fold. Although several studies have confirmed this link, NOD2/CARD15 accounts for < 20% of all CD cases (Venegas et al. Citation2008). Genetic analyses have linked IBD to specific genetic variants, the IBD5 haplotype (spanning the organic cation transporters SLC22A4 and SLC22A5 and other genes) on chromosome 5q31. The identification that the gene CARD15 or NOD2 is important in the pathogenesis of CD has re-focused attention on the role of the innate immune system in IBD. IL-12 and IL-23 are critical immunoregulatory cytokines that bridge innate and adaptive immunity, playing an important role in intestinal inflammatory pathologies (Venegas et al. Citation2008).
IL-23 is known to be a regulator of cellular immune responses involved autoimmune diseases such as IBD (McGovern & Powrie, Citation2007), psoriasis (Torti & Feldman, Citation2007) and rheumatoid arthritis (RA; Kim et al. Citation2007). In humans, IL-23 is over-expressed in clinical samples of psoriasis (Lee et al. Citation2004), CD (Schmidt et al. Citation2005) and ankylosing spondylitis (Wang et al. Citation2009). Becker et al. (Citation2006) showed that blocking the p40 subunit of either IL-12 or IL-23, but not blocking the p19 unit (Ferguson et al. Citation2010), was effective in reducing disease symptoms in chemically-induced colitis models. They suggested that there is a pre-disposition to IL-23-induced chronic inflammation in the terminal ileum. It has also been shown that IL-23, and not IL-12, was an important promoter of chronic joint inflammation (Murphy et al. Citation2003).
IL-23 receptor (IL-23R)
The IL-23 receptor (IL-23R) gene is located on chromosome 1 (1p31). The effects of IL-23 on T-cells are mediated via a receptor complex consisting of an IL-12Rβ1 and a specific IL-23R chain. The latter protein is comprised of an extracellular domain, a single trans-membrane domain and a 252 amino acid cytoplasmic domain. IL-23 and IL-12 signal through a common IL-12Rβ1 chain complemented by the IL-23R and the IL-12Rβ2 (Oppmann et al. Citation2000; Trinchieri et al. Citation2003). IL23R is most highly expressed on activated T-cells – particularly of the TH17 subtype and on natural killer (NK) cells and at lower levels on monocytes, macrophages and dendritic cells (Parham et al. Citation2002). IL23R is a key player in the proliferation and survival of TH17 cells, which are critical for host defense against bacterial, fungal and viral infections at mucosal surfaces, whereas membrane expression patterns of the IL-23R chain are still undefined. IL-23R transcripts are, however, found in bone marrow and in various T-cell subsets (Parham et al. Citation2002). Factors increasing IL23R mRNA expression include IL-23 itself, IL-6, IL-21, transforming growth factor (TGF)-β and TH17 cell activation. Among the determinants that decrease transcriptional activity of IL23R is the TH1-type cytokine IFNγ that simultaneously increases IL12Rβ2 mRNA expression (Lacher et al. Citation2010).
IL-23/IL-17 axis: Role of TH17 cells
The expression of IL-17 characterizes a subset of CD4+ helper T-cells, i.e. TH17 cells. In healthy individuals, ≈1% of CD4+ T-cells in the peripheral blood are TH17 cells. In mice, many cytokines are required and act in a co-ordinated way to induce TH17 differentiation. TH17 cells express the master transcription factor RORC (RAR-related orphan receptor C) encoded by RORγt (RAR-related orphan receptor-γ transcription factor) gene and surface markers CCR6, IL-23R, CD161; they differentiate in the presence of TGFβ1 and at least one pro-inflammatory cytokine such as IL-1β, -6, -21 or -23 (Bettelli et al. Citation2008; Korn et al. Citation2009). In addition to IL-17A, IL-17F and IL-26, TH17 cells produce cytokines shared with other TH cell subsets, such as IL-22 and IFNγ (Volpe et al. Citation2008; Boniface et al. Citation2010). TH17 cells are critical for host defense against bacterial, fungal and viral infections at mucosal surfaces. TH17 cells also play a pathogenic role in autoimmune diseases. Only marginal increases in the number of these cells are detected in peripheral blood of patients with autoimmune diseases. However, together with IL-17+CD8+ (TC17) cells, TH17 cell numbers can dramatically increase in a pathologic microenvironment, where TH17 and TC17 cells can secrete high amounts of IL-17 (Annunziato et al. Citation2007; Cosmi et al. Citation2008; Kleinschek et al. Citation2009; Kryczek et al. Citation2009). Thus, TH17 cells might be actively recruited to/expanded in the site of pathologic damage and their localization could be important for TH17 cell-associated pathology.
The TH17 pathway has been linked to the pathogenesis of several autoimmune diseases, including psoriasis, experimental allergic encephalomyelitis, psoriasis, multiple sclerosis, rheumatoid arthritis and IBD. The proportion of TH17 cells in the peripheral blood and decidua was significantly higher in unexplained RSA (recurrent spontaneous abortion) patients compared to in normal pregnant women (Wilke et al. Citation2011).
IL-23R signaling pathway in TH17 cells
Binding of IL-23 to IL-23R complex leads to IL-23-dependent gene expression. On activation by IL-23, IL23R signals through a JAK/STAT pathway. IL23R associates constitutively with JAK2 (Pidasheva et al. Citation2011). JAK proteins generally associate through their 4.1, ezrin, radixin, moesin (FERM) domains with membrane proximal regions of cytokine receptors. In a ligand-dependent manner, STAT3 (mainly), STAT1, STAT4 and STAT5 can be activated by IL-23 (Parham et al. Citation2002). On ligand binding, JAK2 phosphorylates IL23R at Tyr705, recruiting STAT3 to the receptor complex, where it is further phosphorylated by JAK2 (Schindler & Plumlee Citation2008). Phosphorylated STAT3 homodimerizes and translocates to the nucleus where it triggers downstream expression of cytokines, including IL-17A, IL-17F, IL-22 and IL-21 in TH17 cells (reviewed in Altshuler et al. (Citation2008) and Ouyang et al. (Citation2008)). Thus, IL-23 plays a critical role in TH17 responses and production of the lineage defining the cytokine IL-17A (Acosta-Rodriguez et al. Citation2007; Wilson et al. Citation2007; Manel et al. Citation2008).
As well as functioning as a mediator of cytokine production in TH17 pathway cells, several studies also suggested that IL23R is a key player in the proliferation and survival of TH17 cells (Oppmann et al. Citation2000; Harrington et al. Citation2005). Studies in intestinal tissue have shown that IL-17F and IL-22 mRNA expression (induced via IL-23 signaling) are significantly increased in inflamed colonic lesions in CD compared to in healthy subjects with no inflamed biopsies and that IL-22 is associated with a higher expression of inflammatory mediators (Brand et al. Citation2006; Seiderer et al. Citation2008).
Role of IL-23/IL-17 in pathogenesis of autoimmune diseases
Initial studies suggested that IL-23 is involved in the generation of the described TH17 cells. However, other studies have shown that IL-23 alone does not influence the de novo differentiation of naive T-cells into TH17 cells in vitro and it is only efficient in inducing the proliferation of committed IL-17 producing effector and memory T-cells (Hue et al. Citation2006; Tonel et al. Citation2010). Strong evidence for the importance of the IL-23/IL-17 axis in immune-mediated diseases has emerged from genetics studies. The role of IL-23/IL-17 axis in the development of inflammatory diseases has been fully elucidated (Hue et al. Citation2006; Ahern et al. Citation2010). IL-23 and TH17 cytokines act co-ordinately to influence the balance between tolerance and immunity in the intestine (Capon et al. Citation2007) ().
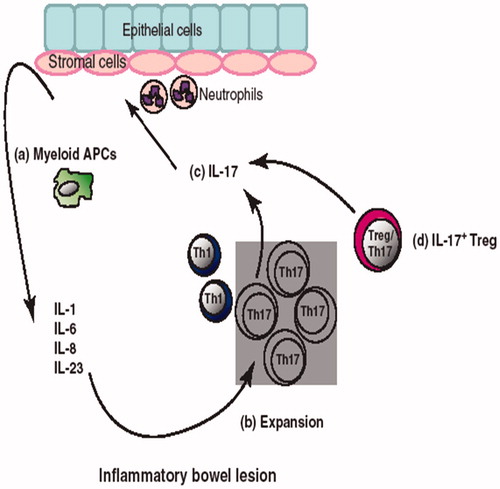
Figure 1. IL-17+ cells and IBD. (a) High amounts of inflammatory cytokines are found in the IBD environment. These cytokines are produced by a variety of cells including myeloid APC, stromal cells, epithelial cells and neutrophils. (b) Myeloid APC along with IL-1, IL-6 and IL-23 lead to TH17 cell expansion. (c) TH17 cells further induce epithelial and stromal cells to express more pro-inflammatory cytokines, promote neutrophil recruitment and accelerate local inflammation. (d) Treg/TH17 (IL-17+Foxp3+) cells are also found in IBD environments. Treg/TH17 cells suppress adaptive T-cell immunity, but also secrete inflammatory cytokines. Thus, these cells are termed as inflammatory Treg cells.
One of the most robust genetic findings is the association of a variant in the IL23R gene with CD and UC, psoriasis (Capon et al. Citation2007; Cargill et al. Citation2007) and ankylosing spondylitis (Rueda et al. Citation2008). Some studies have shown that selectively targeting IL-23 prevents autoimmune models of multiple sclerosis (Chen et al. Citation2006) and in a clinically-relevant model of psoriasis (Tonel et al. Citation2010). In mice, lack of IL-23 makes them resistant to experimental models of arthritis and multiple sclerosis (Cua et al. Citation2003; Murphy et al. Citation2003). Two monoclonal antibodies neutralizing the p40 chain (i.e. ustekinumab and briakinumab) – and, hence, blocking both IL-12 and IL-23 activity – have been developed and demonstrated to have a clinical benefit in early phase trials, as well as toward efficacy in a sub-population of patients with CD (Mannon et al. Citation2004; Leonardi et al. Citation2008).
Recent studies also reveal that the IL-23/IL-17 pathway is involved in the pathogenesis of asthma, especially in severe asthma and steroid-resistant asthma, which may feature neutrophil-dominant inflammation. Blocking IL23/IL-17 pathways using monoclonal antibodies has been reported to be successful in the reduction in skin and airway inflammation in animal models (Guan et al. Citation2012).
R381Q polymorphisms in the IL-23R gene
Polymorphisms in the IL-23R chain may influence IL-23 responses. The frequency of a functional single nucleotide polymorphism (SNP) in the IL-23 receptor gene (IL-23R; rs11209026, 1142 G wild-type A reduced function, Arg381Gln, R381Q) is significantly higher among healthy controls than in patients, suggesting a protective effect of the rare allele from immune-mediated chronic inflammation. The SNP rs11209026 encodes an amino acid change (Arg381Gln) in the protein product and has functional consequences; thus, R381Q is a causal variant (Ferguson et al. Citation2010). This variant is a non-coding variant together with other genetic markers located in the IL23R gene (in exon 9 and coding for IL23R intra-cytoplasmic tail) and in its downstream intergenic region was identified as exerting a potent protective effect against IBD susceptibility. Therefore, a change in the highly-conserved Arg381 for Gln381 might have functional consequences in the IL-23R transducing pathway, modifying interactions between IL-23R and its associated JAK2 kinase. This might lead to a reduction in the cellular response to IL-23 and could explain the protective effect of this infrequent allele in IBD pathogenesis (Oliver et al. Citation2007).
The R381Q variant is an uncommon non-R381Q polymorphism which occurs at a frequency of 0–17%, depending on the population. The associated SNP, consisting in a guanine (G) to adenine (A) substitution at DNA level, results in an arginine (R) to glutamine (Q) substitution in position 381 (R381Q) within the cytoplasmic domain of the IL-23R. By virtue of its location, this polymorphism could potentially interfere with either surface localization of the IL23R or signal transduction; it is unlikely to interfere with ligand binding (Chaligné et al. Citation2008; Lerch-Bader et al. Citation2008).
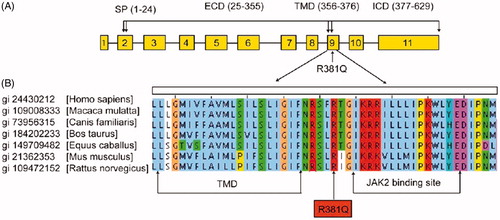
The R381Q polymorphism is located between the trans-membrane domain and putative JAK2 binding site in the cytoplasmic portion of IL-23R protein and is absolutely conserved across different species (). Recent studies have shown that untransformed polyclonal IL-23 RQ381+ T-cell lines had a decreased number of IL-23R+ cells and, thus, reduced responsiveness to IL-23 as compared to IL-23R R381 T-cell lines (Pidasheva et al. Citation2011). A allele of the IL-23R R381Q gene variant suggested that its protective effect against autoimmune disease was driven by an impairment of TH17 cell effector functions, i.e. IL-17A production, rather than of TH17 cell differentiation (Mosayebian et al. Citation2015). The R381Q allele conferred protection against IBD (including CD and UC) (Duerr et al. Citation2006), psoriasis (Capon et al. Citation2007), ankylosing spondylitis (Rueda et al. Citation2008), Graft-vs-Host (GVH) disease after marrow transplantation (Elmaagacli et al. Citation2008), rheumatoid arthritis, recurrent spontaneous abortion (RSA) or asthma (Abdollahi et al. Citation2015; Mosayebian et al. Citation2015).
Figure 2. The arginine at position 381 of the IL23R is absolutely conserved across different species. (a) Map of IL23R gene. (b) Sequence alignment of IL23R protein sequences from different species (adapted from Pidasheva et al. Citation2011).
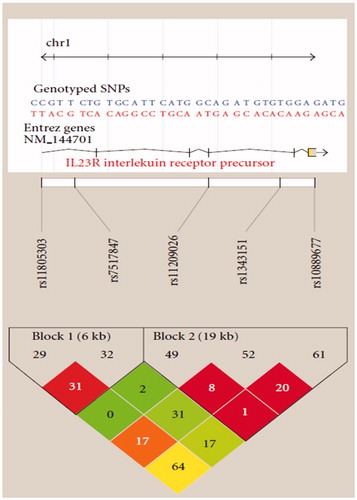
Other studies indicated the decreased STAT3 activation in primary CD8+ T-cells from R381Q IL23R carriers and cells transfected with R381Q IL23R (). The decreased IL-17 production in TH17 CD4+ and CD8+ T-cells from individuals carrying the disease-protective R381Q IL23R variant relative to WT IL23R carriers is consistent with the general contribution of IL-17 to inflammation (Abraham & Cho Citation2009). The IL-23R Linkage disequilibrium (LD) plot is shown in .
Figure 3. IL-23R Linkage disequilibrium (LD) plot. Two haplotype blocks were constructed for the IL23R (adapted from Ferguson et al. Citation2010).
Genetic variability in the IL23R gene
Variants in IL-23R gene can associate with some diseases. Duerr et al. (Citation2006) reported on an association between Crohn’s Disease (CD, a chronic inflammatory disease of gastrointestinal tract) and variants in the IL-23R gene (Duerr et al. Citation2006; Lakatos et al. Citation2008). A study by Yu et al. (Citation2012) showed that the IL-23R rs7530511 and rs11805303 variants were associated with UC susceptibility in a Chinese Han population. The IL-23R rs7530511 variant allele was a protective factor for UC, while rs11805303 was significantly associated with an increased risk for UC development. The Yu et al. study demonstrated that patients with the IL-23R haplotypes who also carry the IL-17A risk haplotypes had a higher susceptibility to UC. This indicated that gene–gene interactions between IL23R and IL17A might occur. A study from Magyari et al. (Citation2008) reported that the IL-23R gene variants rs10889677 C/A and rs2201841 T/C appeared to increase susceptibility to CD. However, the rs10889677 and rs2201841 in that study had no significant relationship with UC susceptibility. Ferguson et al. (Citation2010) detected five common SNP in the IL-23R gene (e.g. rs11805303, rs7517847, rs1343151, rs11209026, rs10889677) in CD patients from a New Zealand population and found that rs1343151 and/or rs7517847 variants strongly reduced the risk of developing CD at both the allelic and genotype level. Nevertheless, Yu et al. (Citation2012) did not find an apparent association between these SNP in IL-23R and UC susceptibility, but the rs11805303 and rs7517847 could form three haplotypes with TT (with two thymine bases) to increase UC susceptibility.
Glas et al. (Citation2007) analyzed 10 IL-23R SNP (including nine SNP in our studies) in a large German IBD cohort. Those authors found the rs1004819 was the major IL-23R variant associated with CD in that cohort, while the rs11209026 IL23R variant was a protective marker for CD and UC. Another study examined associations between the IL23R gene and IBD susceptibility. The authors analyzed eight genetic variants spanning the IL23R gene and its downstream intergenic region that were significantly associated with IBD in a North American population. Interestingly, several IL23R genetic variants were found to exert an independent and strong protective effect against IBD susceptibility in a Spanish population, confirming a contribution of the IL23R gene to IBD (Oliver et al. Citation2007). The different associations of the IL23R intergenic region polymerphisms in different populations might account for the genetic heterogeneity in LD patterns and ancestral haplotype backgrounds between different populations.
Interestingly, a protective IL23R haplotype (AAAC) including the Gln381 allele together with three alleles of IL23R variants (e.g., rs100489629, rs11209026, rs10889677) flanking exon 10 were observed. Therefore, it might be hypothesized this protective haplotype could affect IL23R splicing, leading to a higher expression of soluble isoforms, inhibiting IL-23 pro-inflammatory activities, thereby exerting a protective effect in IBD (Oliver et al. Citation2007).
Over the years, it became clear that different variants of the IL-23R also played roles in a number of autoimmune diseases like rheumatoid arthritis (Hamdy et al. Citation2015), psoriasis, ankylosing spondylitis, multiple sclerosis, psoriatic arthritis, psoriasis vulgaris, autoimmune thyroid disease, and Graves’ ophthalmopathy. This suggested to some that perturbations in the IL-23 signaling pathway were likely relevant to the pathophysiology of these inflammation-based diseases (Safrany & Melegh Citation2009). Interestingly, in other pathologies, such as systemic sclerosis, systemic lupus erythematosus, Sjogren’s syndrome or myocardial infarct, the same effect was not seen. It needs to be noted that those results were very controversial, both in terms of the various polymorphisms and also in population specificity.
Gene–gene interactions among the IL23/IL17 genes
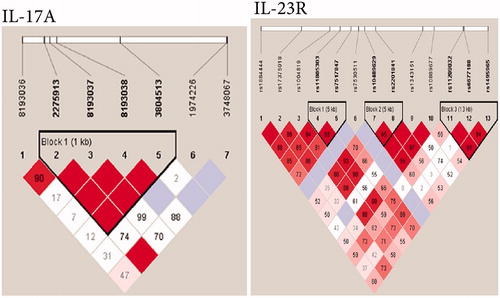
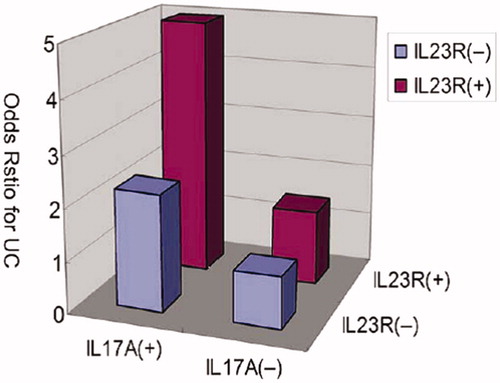
In one survey, the linkage disequilibrium (LD) patterns of the SNP were used to identify three LD blocks from the SNP of IL-23R and one LD block in the SNP of IL-17A (). To determine whether any specific haplotype would confer a higher risk or protection for UC, specific and global-haplotype tests of association were done. The most common haplotype (i.e. TT) of IL-23R, formed with rs11805303 and rs7517847, was observed to be a risk haplotype, with a frequency of 60% in UC patients and 50% in controls. The rare haplotype (GGTT) of IL-17A, formed with four SNP (e.g. rs2275913, rs8193037, rs8193038, rs3804513), was also a risk haplotype, with a frequency of 3.3% in UC patients and 0.7% in controls. Another two haplotypes of IL-23R were both protective factors; one was tagged by the rs10489629-A and rs2201841-T alleles and the frequency of this haplotype was 0.7% in UC patients and 3.7% in controls. Another rare haplotype was formed with three wild-type alleles (GTG) of rs11209032, rs6677188 and rs1495965, the frequency of which was 1.1% in UC patients and 4.1% in controls.
Figure 4. Linkage disequilibrium (LD) patterns of region around IL23R and IL17A genes in a Chinese Han population. (a) IL23R. (b) IL17A. Numbers indicate extent of D’ between two SNP and in the dark area which have no digital respective D’ = 1. Dark color indicates strong connection. Three linkage disequilibrium blocks were found in IL-23R and one in IL-17A (adapted from Yu et al. Citation2012).
In the analysis of gene–gene interactions of the IL23/IL17 genes, the two risk haplotypes of IL-23R and IL-17A together were combined; the analyses found a significant increase in UC risk in IL-23R block1 Haplotyp3, when the risk haplotype of IL-17A, Haplotyp4, was also present (p = 0.004). The interaction between these two risk haplotypes was statistically significant (p = 0.014). Further, the risk haplotypes of IL-23R and IL-17A synergistically increased the UC risk (), suggesting there was a gene–gene interaction between IL-23R and IL-17A haplotypes in the IL-23/IL-17 pathway that affected the pathogenesis of UC (Yu et al. Citation2012). The statistical interaction for CD-associated variants in NOD2 with IL-12B and IL-23R was also investigated. The three NOD2 SNPs (rs2066844, rs2066845 and rs2066847) were classified as homozygous wild-type (d/d), heterozygous carrier (d/D) or homozygous mutant carrier, including compound heterozygotes (D/D), as previously identified in Ferguson et al. (Citation2010).
Figure 5. Increase of ulcerative colitis risk with the haplotypes of IL23R and IL-17A. Relative risk haplotypes were plotted and demonstrate association between IL-23R block1 haplotype3 and IL-17A haplotype4 (p = 0.014 for interaction) (adapted from Yu et al. Citation2012).
Gene–gene interactions among the IL23 and NOD2/CARD15 genes
In a German study, beside R381Q, the intronic IL23R variant rs1004819 showed the strongest association (without any epistatic interaction) with either the NOD2/CARD15 or the SLC22A4/A5 genes. This variant was also associated with ileal involvement. The association between autophagy 16-like 1 (ATG16L1) rs2241880 on 2q37 and CD was identified through a genome-wide association study of 19 779 non-synonymous SNPs. ATG16L1 encodes a small coiled-coil protein which interacts with ATG5 and ATG12 to form a 350 kDa multimeric complex that plays a crucial role in the bulk degradation or autophagy of cytoplasmic proteins and organelles. ATG16L1 is expressed in the colon, small bowel, intestinal epithelial cells, leukocytes and spleen. Multiple splice variants have been reported; however, no significant difference in expression was observed in CD vs control tissues. Expression of ATG16L1 was independent of the rs2241880 genotype (Lakatos et al. Citation2008).
IL23R and ATG16L1 susceptibility variants and IBD (CD and UC)
One study showed an ATG16L1 variant (rs2241880) was associated with a 38% increase in risk for Crohn’s disease for high mutational load, whereas IL-23R variant R381Q decreased the risk by 54% for high mutational load (Grigoras et al. Citation2015).
A recent meta-analysis of three CD GWA (3230 patients and 4829 controls) and replication studies (3664 independent patients) confirmed the associations between IL23R and ATG16L1 and CD, in addition to identifying a number of other susceptibility genes (Barrett et al. Citation2008). Importantly, their study analyzed different intronic variants of IL23R, rs11465804, ATG16L1 and rs3828309, compared with our analysis of coding variants. The interpretation of their replication of IL23R in the context of a previous study was also complicated by the fact that they considered the risk associated with the major allele. However, their reported combined OR of 1.28 in case-control and 1.3 in family based replication between ATG16L1 rs3828309 and CD is very similar to the combined OR of 1.33 generated in Cotterill et al.’s (Citation2010) study. This was not surprising considering the strong linkage disequilibrium across ATG16L1 and reflected a tight homogeneity across studies investigating ATG16L1 and CD. The Barrett et al. (Citation2008) analysis also incorporated studies of non-European ancestry to broadly establish the relevance of these variants. That study showed that IL23R and ATG16L1 altered susceptibility to CD and the effects were consistent across all populations of European ancestry; however, only ATG16L1 was relevant to IBD in the Asian population. IL23R and ATG16L1 susceptibility variants were also genotyped.
Cotterill et al. (Citation2010) performed replication and meta-analyses; the meta-analysis for IL23R (p.R381Q, rs11209026) derived data from 26 studies including a total of 12 991 patients and 14 598 controls and confirmed the protective effect of the minor allele (n = 26, OR = 0.41; 95% CI = 0.37–0.46). Data from 25 studies were included in the ATG16L1 meta-analysis, which had combined totals of 11 909 patients and 15 798 controls. The ATG16L1 variant (p.T300A, rs2241880) increased the risk of CD (n = 25, OR = 1.33; 95% CI = 1.28–1.39). Consistent with a majority of other studies, those authors did not find an association between ATG16L1 variants and UC. However, the lack of replication of an association between IL23R with UC in this study probably reflects a lack of statistical power to detect this more modest effect. A comprehensive meta-analysis of CARD15 variants has established a robust association with an increased risk of CD. In the meta-analysis presented here for IL23R, with data from nearly 13 000 patients and 14 500 controls, the combined OR of 0.41 supports a significant protective effect of the c.1142G→A minor allele for CD. The majority of studies were consistent with respect to overall risk, except for one of a Jewish CD population that found no association (but had a small sample size and wide CIs); therefore, appropriately, in both analytical models, this study had a small study weight. Data from ≈ 12 000 patients and ≈16 000 controls were included in the ATG16L1 meta-analysis confirming that the ATG16L1 c.1338A→G minor allele is a susceptibility factor for CD (OR = 1.34). The ATG16L1 variant was not associated with CD in studies of Italian, Brazilian and Jewish-Canadian CD populations, but these had overlapping risk estimates and small sample sizes and each lacked adequate power to definitively detect a positive association.
In a study in a Greek population, the polymorphisms R702W, G908R and 3020insC in the: CARD15 gene; NOD2 gene; ATG16L1 (autophagy-related 16-like 1) gene; and R381Q in the IL23R gene were assessed in 110 childhood-onset CD, 364 adult-onset CD and 539 healthy individuals. The rare Q allele R381Q polymorphism was under-represented in both pediatric and adult CD cases; no differences were observed between the childhood and adult cohorts (Gazouli et al. Citation2010).
The CTLA4 gene may also be considered a plausible candidate for a genetic association with IBD. Its product, the cytotoxic T-lymphocyte-associated protein 4 (CTLA4), is a T-cell suppressor that plays an essential role in the function of the CD25+CD4+ regulatory cells that control the process of intestinal inflammation. Three CTLA4 variants (e.g. CT60, JO31 and JO27-1) clearly, albeit moderately, were found to be associated with a decreased risk for CD in subjects carrying minor alleles of R381Q within the IL23R gene (G/A and A/A), while no effect was observed in the IL-23R wild type homozygotes (Hradsky et al. Citation2010).
R381Q polymorphism: Mechanisms of action
Recent studies have shown that a functional single-nucleotide polymorphism (SNP) (Arg381Gln; R381Q; rs11209026; 1142 G wild-type A reduced function) in the IL-23R gene (in exon 9) led to decreased IL-23-dependent IL-17 production (Guan et al. Citation2012). The disease-protective R381Q IL-23R may result in a loss of function, in primary human CD4+ and CD8+ T-cells, leading to decreases in IL-23/TH17 pathway cytokine production () or a gain of function, which would lead to enhanced microbial clearance.
Figure 6. Summary of results obtained in study using genotype-selectable normal donors is depicted. IL23RQ381 donors had a significant decrease in IL23R+ T-cells, leading to decreased IL-23-induced STAT3 phosphorylation. A decrease in STAT3 signaling in T-cells like TH17 could then modulate the extent/duration of the response in the host, leading to decreased secretion of pro-inflammatory cytokines like IL-17 and IL-22 in the gut. This could help explain the protective effect of R381Q variant in CD and other autoimmune disorders (adapted from Pidasheva et al. Citation2011).
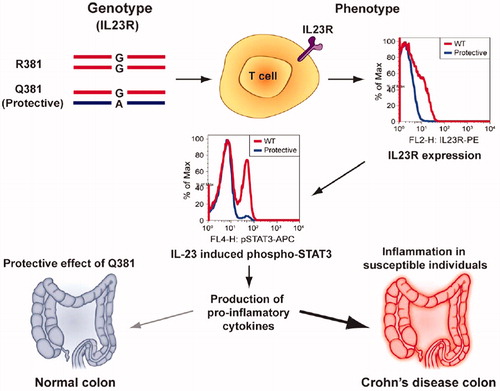
Indeed, a survey by Di Meglio and colleagues (Di Meglio et al. Citation2011). suggested that IL-23R RQ381 exerted its protective effects by attenuation of IL-23-induced TH17 functions (IL-17A production) without interfering with TH17 differentiation (Pidasheva et al. Citation2011). Further, Sarin et al. (Citation2011) showed that CD4+CD45RO+ and CD8+ T-cells from healthy R381Q IL-23R carriers had decreased IL-23-dependent IL-17 and IL-22 production relative to WT (wild-type; G allele) cells. Several mechanisms might contribute to these observations, including a reduced capacity of IL-23RQ381 to activate STAT proteins due to an impaired association of JAK2 proteins with the cytoplasmic tail of the receptor. As a result, R381Q CD8+ and TH17 CD4+ T-cells displayed decreased IL-23- and STAT3-dependent expansion, STAT3 phosphorylation and STAT3 activation compared with WT cells (Sarin et al. Citation2011) ().
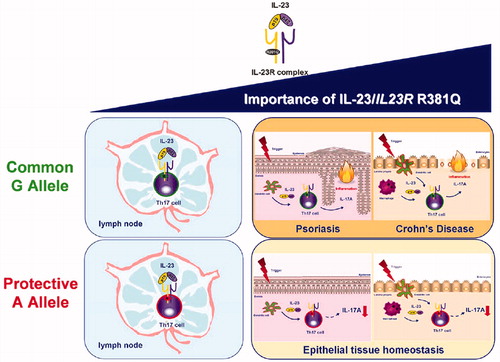
This SNP was also associated with a lower percentage of circulating TH17 and TC17 cells. Abdollahi et al. (Citation2015) reported that the number and activity of both circulating TH17 cells and in vitro (ex vivo)-differentiated TH17 cells did not differ between G and A allele carriers. This finding supported the belief for a major role for IL-23 in generation of TH17 cell effector responses during tissue inflammation (rather than in systemic inflammation) and demonstrated the importance of IL-23 in mediating TH17 effector responses in humans. Those results also provided support for a critical role for the IL-23/IL- 23R signaling in generating pathogenic TH17 responses. Further, those studies provided an explanation for the protective role of R381Q in autoimmune disorders and further supported the hypothesis that blocking the IL-23 pathway could lead to improvements in hosts with autoimmune disorders like CD or psoriasis (Sandborn et al. Citation2008) ().
Figure 7. Importance of IL-23 and IL23R R381Q gene variant in TH17 cell function. The IL-23/IL-23R axis plays a pivotal role in both TH17 differentiation in the lymph nodes and TH17 cell effector functions in peripheral tissues, with the latter becoming increasingly relevant, especially in the context of IL-23-induced pathologies, like psoriasis and Crohn’s disease. This protective effect against autoimmune disease is driven through impairment of TH17 cell effector functions, i.e. IL-17A production, rather than TH17 differentiation (adapted from Di Meglio et al. 2011).
Raymond et al. (Citation2015) investigated another mechanism of function for this SNP. Results of that study showed the R381Q variant promoted expression of a soluble IL-23R-encoding mRNA species. In fact, the R381Q polymorphism altered the IL-23R α-chain mRNA splicing and favored exon 9 skipping by reducing the binding of the splicing enhancer SF2. This enhanced expression of the Δ9 mRNA consequently diminished IL-23 signaling. Multiple splice forms of the human IL23R transcript exist and one, Δ9, encodes a soluble form of the receptor. Thus, a presence of an R381Q variant increases the expression of the soluble form of IL23R mRNA (which, in turn, then functions as a decoy receptor) and lowers the ability of a host to develop a TH17 phenotype upon IL-23 stimulation (Raymond et al. Citation2015).
Role for R381Q variants in IBD (CD and UC)
Some studies have analyzed the frequency of R381Q in inflammatory diseases wherein TH17 cells have an important role in the adverse inflammation; the majority of such studies done were related to the effect of R381Q in IBD (particularly CD) (). One study reported frequencies of 6% and 25% for R381Q in, respectively, CD patients and control subjects, illustrating that its presence appeared to confer some degree of protection (Venegas et al. Citation2008). Those investigators also studied the frequency of IL-23R R381Q gene variant in patients with CD in a non-Jewish European population. The results showed the frequency of CD patients having the A allele was 1.9%, while it was 7% for controls. Another study of CD in an Ashkenazi Jewish (AJ) population indicated a rare IL-23R R381Q variant was more frequent among AJ controls than among AJ CD patients (7% vs 2%, respectively), indicating a significant reduction in CD risk for carriers (Venegas et al. Citation2008; Yu et al. Citation2012). The association of this particular variant SNP with a decreased risk of CD has also been confirmed in pediatric populations (Dubinsky et al. Citation2007) and in a large UK database studied by the Wellcome Trust (see Burton et al. Citation2007). There are different frequencies of R381Q variants in populations of patients with various immune-mediated diseases and, thus, in control groups in different studies. For example, in the above-noted CD study, the difference (6% vs 25%) seemed to suggest a closer link or relevance.
Research on pediatric IBD (CD and UC) patients revealed an association between the presence of an R381Q variant and IL-23R transcriptional activity in colonic tissues (Lacher et al. Citation2010). The prevalence of this SNP was lower in CD patients, but not in UC patients, when compared with control subjects. Lakatos et al. (Citation2008) showed that the frequency of the A allele in UC patients was 2.7%, while in controls it was 4.7%. Although allele frequency in patients was lower than in controls in their study, the difference was not statistically significant.
A study based in New Zealand associated IL-23R Arg381Gln with CD and possibly also UC (Roberts et al. Citation2007). However, the authors reported no significant association between IL-23R genotype and IBD phenotypes and also that the association was only seen in subjects who did not carry variant alleles in NOD2 (nucleotide binding oligomerization domain). The authors did not consider other SNP or potential interactions. Positive results of varying quality and significance have also been reported from other populations, including subjects from Scotland, Continental Europe, North America, Brazil and Israel (Büning et al. Citation2003; Libioulle et al. Citation2007; Roberts et al. Citation2007; Baldassano et al. Citation2007; Baptista et al. Citation2008; Weersma et al. Citation2008). No associations were seen in a Japanese study (Yamazaki et al. Citation2007); those authors did not find any positive association between IL23R gene polymorphisms and CD in Japanese populations. It is likely that these disparate findings could be attributed to distinct ethnic differences of genetic backgrounds of CD that have been reported previously between Japanese and other populations for other genes. It should be noted that the different results in allele frequencies between the studies might also be explained by large regional and ethnic differences in genotypes, by the broad spectrum of clinical phenotypes of patients with CD and by the relatively small numbers of cases included in most studies.
Role for R381Q variant in non-gastrointestinal autoimmune diseases
Some research has been performed to investigate the potential role for R381Q variants in non-gastrointestinal autoimmune diseases. A study to assess variants in the IL23R and STAT3 genes in Croatian populations revealed no association with Hashimoto’s thyroiditis (Stevic et al. Citation2014). That suggested to the authors that IL-23R/STAT3 polymorphisms affecting TH17 signaling efficiency were not major determinants for risk of Hashimoto’s thyroiditis, at least in Croatians. Previous studies examining ankylosing spondylitis also indicated that the A allele frequency of R381Q was lower in spondylitis patients than in healthy controls (Rahman et al. Citation2008; Duan et al. Citation2012).
Rheumatoid arthritis (RA) is a chronic progressive systemic inflammatory autoimmune disease of the joints that can lead to long-term joint damage and disability. A major aim of the research by Hamdy et al. (Citation2015) was to investigate the association between three SNP in the IL23R gene (rs11209026, rs2201841, rs10889677) and RA in Egyptian patients. The results showed the AA genotype of the rs11209026 (Arg381Gln) SNP was significantly associated with an increased susceptibility to RA (p = 0.001). However, there were no differences with regard to genotypes and of rs7517847 and rs17375018 between patients with RA and normal controls. Those results suggest that IL-23 receptor AA genotype variant of rs11209026 would contribute to RA etiology; consequently, it might be a genetic marker for RA. Another study reported that a R381Q polymorphism modulated IL-17A expression in patients with RA (Hazlett et al. Citation2012). Those authors suggested that, in patients with the 381Gln allele, higher IL-23 concentrations might have been needed to produce IL-17A concentrations similar to those in patients with a 381Arg allele.
Recurrent spontaneous abortion (RSA) is defined as the occurrence of three or more sequential spontaneous abortions before the 20th week of gestation (Arjmand & Samadi Citation2016). Human TH17 cells producing IL-17 may play a major role in rejecting conceptus antigens and, therefore, may be harmful to the maintenance of pregnancy. The proportions of TH17 (CD4+IL-17A+) cells in the peripheral blood and decidua were found to be significantly higher in unexplained RSA subjects than in normal pregnant women (Tavasolian et al. Citation2014). The pro-inflammatory features of IL-23 have been linked with TH17 cell responses through the expansion and maintenance of TH17 cells (Abdollahi et al. Citation2015). In that study, the association between R381Q variant and recurrent spontaneous abortion was investigated and a significant difference was found between the case and control group subjects with regard to a presence of the R381Q variant. Specifically, 2% of RSA patients had the R381Q variant, while the control value was 7.5%. That was the first report on an association between polymorphisms in the IL-23R gene (i.e. as related to the R381Q variant specifically) and RSA.
A separate study was done to determine the presence and prevalence of IL-23R gene mutations in asthma, a chronic inflammatory disease of the airways. Previous studies have shown that the populations of TH17 cells were more frequent in the circulation of asthmatic patients than in normal controls (Hellings et al. Citation2003; Oda et al. Citation2005). This suggested that IL-17A and IL-17F (cytokines secreted by TH17 cells) recruited neutrophils into the airways and bronchi of animal models of asthma. IL-17, secreted by the TH17 cells, has a major role in stimulating bronchial fibroblasts, epithelial cells and smooth muscle cells and inducing the expression of a variety of cytokines and chemokines that are important for granulopoiesis and neutrophil recruitment (Hellings et al. Citation2003; Oda et al. Citation2005). This led to the hypothesis that R381Q polymorphisms in the IL-23 receptor might be a pre-disposing genotype for asthma. presents a compilation of association studies related to R381Q polymorphisms.
Table 1. Summary of association study results related to IL-23R rs11209026.
A decrease in IL-23R-expressing TH17 and TC17 subsets in R381Q healthy carriers reflects the cumulative effects of altered IL-23 function. Thus far, there have been no reports that R381Q carriers are at increased risk for infectious complications. It remains possible that, under certain conditions (e.g. high levels of TH17-differentiating cytokines or specific microbial infections), R381Q IL-23R carriers may be able to mount normal TH17 responses. The present findings of decreased IL-23R function associated with the R381Q IL-23R protective allele highlight a potentially desirable selective functional alteration that balances adequate defense with protection against excessive inflammation.
Conclusions
Probable protective effects of R381Q polymorphism against inflammatory-based diseases may lead us towards developing potential therapeutic approaches that interfere with IL-23R functions in patients with immune disorders. Insights gleaned from such “gene-to-function” studies could be translated into designing more efficient/cost-effective clinical trials for immune-targeted drug therapy in immune mediated diseases.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Abdollahi E, Tavasolian F, Ghasemi N, Mirghanizadeh SA, Azizi M, Ghoryani M, Samadi M. 2015. Association between lower frequency of R381Q variant (rs11209026) in IL-23 receptor gene and increased risk of recurrent spontaneous abortion (RSA). J Immunotoxicol. 12:317–321
- Abraham C, Cho J. 2009. IL‐23/TH17 pathways and inflammatory bowel disease. Inflamm Bowel Dis. 15:1090–1100.
- Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. 2007. IL-1β and -6, but not TGFβ are essential for the differentiation of IL-17-producing human T-helper cells. Nat Immunol. 8:942–949.
- Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, Powrie F. 2010. IL-23 drives intestinal inflammation through direct activity on T-cells. Immunity. 33:279–288.
- Altshuler D, Daly MJ, Lander ES. 2008. Genetic mapping in human disease. Science. 322:881–888.
- Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Filì L, Ferri S, Frosali F. 2007. Phenotypic and functional features of human TH17 cells. J Exp Med. 204:1849–1861.
- Arjmand F, Samadi M. 2016. Association of 14-bp insertion/deletion polymorphism of HLA-G gene with idiopathic recurrent miscarriages in infertility center patients in Yazd, Iran. J Immunotoxicol. 13:249–254.
- Baldassano RN, Bradfield JP, Monos DS, Kim CE, Glessner JT, Casalunovo T, Frackelton EC, Otieno FG, Kanterakis S, Shaner JL. 2007. Association of variants of the IL–23 receptor gene with susceptibility to pediatric Crohn’s disease. Clin Gastroenterol Hepatol. 5:972–976.
- Baptista ML, Amarante H, Picheth G, Sdepanian VL, Peterson N, Babasukumar U, Lima HC, Kugathasan S. 2008. CARD15 and IL23R influences Crohn's disease susceptibility but not disease phenotype in a Brazilian population. Inflamm Bowel Dis. 14:674–679.
- Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM. 2008. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 40:955–962.
- Becker C, Dornhoff H, Neufert C, Fantini M, Wirtz S, Huebner S, Nikolaev A, Lehr H, Murphy A, Valenzuela D. 2006. IL-23 cross-regulates IL-12 production in T-cell-dependent experimental colitis. J Immunol. 177:2760–2764.
- Bettelli E, Korn T, Oukka M, Kuchroo VK. 2008. Induction and effector functions of TH17 cells. Nature. 453:1051–1057.
- Bettelli E, Oukka M, Kuchroo VK. 2007. TH17 cells in the circle of immunity and autoimmunity. Nat Immunol. 8:345–350.
- Boniface K, Blumenschein WM, Brovont-Porth K, McGeachy MJ, Basham B, Desai B, Pierce R, McClanahan TK, Sadekova S, de Waal Malefyt R. 2010. Human TH17 cells comprise heterogeneous subsets including IFNγ-producing cells with distinct properties from the TH1 lineage. J Immunol. 185:679–687.
- Borgiani P, Perricone C, Ciccacci C, Romano S, Novelli G, Biancone L, Petruzziello C, Pallone F. 2007. IL-23R Arg381Gln is associated with suscepti–bility to Crohn’s disease but not with phenotype in an Italian population. Gastroenterology. 133:1049–1051.
- Brand S, Beigel F, Olszak T, Zitzmann K, Eichhorst ST, Otte J-M, Diepolder H, Marquardt A, Jagla W, Popp A. 2006. IL-22 is increased in active Crohn’s disease and promotes pro-inflammatory gene expression and intestinal epithelial cell migration. Am J Physiol. 290:G827–G838.
- Büning C, Genschel J, Weltrich R, Lochs H, Schmidt H. 2003. The IL‐25 gene located in the inflammatory bowel disease (IBD) 4 region: No association with inflammatory bowel disease. Eur J Immunogen. 30:329–333.
- Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, Kwiatkowski DP, McCarthy MI, Ouwehand WH, Samani NJ. 2007. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 447:661–678.
- Capon F, di Meglio P, Szaub J, Prescott NJ, Dunster C, Baumber L, Timms K, Gutin A, Abkevic V, Burden AD. 2007. Sequence variants in the genes for IL-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Human Genet. 122:201–206.
- Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, Matsunami N, Ardlie KG, Civello D, Catanese JJ. 2007. A large-scale genetic association study confirms IL-12B and leads to identification of IL23R as psoriasis-risk genes. Am J Human Genet. 80:273–290.
- Chaligné R, Tonetti C, Besancenot R, Roy L, Marty C, Mossuz P, Kiladjian J-J, Socié G, Bordessoule D, Le Bousse-Kerdilès M-C. 2008. New mutations of MPL in primitive myelo-fibrosis: only MPL W515 mutations promote G1/S-phase transition. Leukemia. 22:1557–1566.
- Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, Blumenschein W, Churakovsa T, Low J, Presta L. 2006. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 116:1317–1326.
- Cosmi L, de Palma R, Santarlasci V, Maggi L, Capone M, Frosali F, Rodolico G, Querci V, Abbate G, Angeli R. 2008. Human IL-17-producing cells originate from a CD161+CD4+ T-cell precursor. J Exp Med. 205:1903–1916.
- Cotterill L, Payne D, Levison S, McLaughlin J, Wesley E, Feeney M, Durbin H, Lal S, Makin A, Campbell S. 2010. Replication and meta-analysis of 13,000 cases defines the risk for IL-23 receptor and autophagy-related 16-like 1 variants in Crohn’s disease. Canad J Gastroenterol. 24:297–305.
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T. 2003. IL-23 rather than IL-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 421:744–748.
- Di Meglio P, Di Cesare A, Laggner U, Chu C-C, Napolitano L, Villanova F, Tosi I, Capon F, Trembath RC, Peris K. 2011. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PloS one. 6:e17160.
- Duan Z, Pan F, Zeng Z, Zhang T, Wang S, Li G, Xu S, Xu J, Zhang L. 2012. IL-23 receptor genetic polymorphisms and ankylosing spondylitis susceptibility: a meta-analysis. Rheumatol Intl. 32:1209–1214.
- Dubinsky MC, Wang D, Picornell Y, Wrobel I, Katzir L, Quiros A, Dutridge D, Wahbeh G, Silber G, Bahar R. 2007. IL‐23 receptor (IL‐23R) gene protects against pediatric Crohn's disease. Inflamm Bowel Dis. 13:511–515.
- Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A. 2006. A genome-wide association study identifies IL-23R as an inflammatory bowel disease gene. Science. 314:1461–1463.
- Elmaagacli A, Koldehoff M, Landt O, Beelen D. 2008. Relation of an IL-23 receptor gene polymorphism to graft-versus-host disease after hematopoietic-cell transplantation. Bone Marrow Transplant. 41:821–826.
- Ferguson LR, Han DY, Fraser AG, Huebner C, Lam WJ, Morgan AR. 2010. IL23R and IL12B SNPs and haplotypes strongly associate with Crohn's disease risk in a New Zealand population. Gastroenterol Res Practice. 2010:1–12.
- Fraser Cummings J, Ahmad T, Geremia A, Beckly J, Cooney R, Hancock L, Pathan S, Guo C, Cardon LR, Jewell DP. 2007. Contribution of the novel inflammatory bowel disease gene IL23R to disease susceptibility and phenotype. Inflamm Bowel Dis. 13:1063–1068.
- Frucht DM. 2002. IL-23: a cytokine that acts on memory T-cells. Science Signaling. 2002:pe1.
- Fuss IJ, Becker C, Yang Z, Groden C, Hornung RL, Heller F, Neurath MF, Strober W, Mannon PJ. 2006. Both IL‐12p70 and IL‐23 are synthesized during active Crohn's disease and down‐regulated by treatment with anti‐IL‐12 p40 monoclonal antibody. Inflamm Bowel Dis. 12:9–15.
- Gazouli M, Pachoula I, Panayotou I, Mantzaris G, Chrousos G, Anagnou NP, Roma-Giannikou E. 2010. NOD2/CARD15, ATG16L1, and IL23R gene polymorphisms and childhood-onset of Crohn’s disease. World J Gastroenterol. 16:1753.
- Glas J, Seiderer J, Wetzke M, Konrad A, Torok H-P, Schmechel S, Tonenchi L, Grassl C, Dambacher J, Pfennig S. 2007. rs1004819 is the main disease-associated IL23R variant in German Crohn's disease patients: Combined analysis of IL23R, CARD15, and OCTN1/2 variants. PLoS One. 2:e819.
- Grigoras CA, Ziakas PD, Jayamani E, Mylonakis E. 2015. ATG16L1 and IL23R variants and genetic susceptibility to Crohn's Disease: Mode of inheritance based on meta-analysis of genetic association studies. Inflamm Bowel Dis. 21:768–776.
- Guan Q, Ma Y, Aboud L, Weiss C, Qing G, Warrington R, Peng Z. 2012. Targeting IL‐23 by employing a p40 peptide‐based vaccine ameliorates murine allergic skin and airway inflammation. Clin Exp Allergy. 42:1397–1405.
- Hamdy G, Darweesh H, Khattab EA, Fawzy S, Fawzy E, Sheta M. 2015. Evidence of association of IL-23 receptor gene polymorphisms with Egyptian rheumatoid arthritis patients. Human Immunol. 76:417–420.
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. 2005. IL-17-producing CD4+ effector T-cells develop via a lineage distinct from the T-helper Type 1 and 2 lineages. Nat Immunol. 6:1123–1132.
- Hazlett J, Stamp L, Merriman T, Highton J, Hessian P. 2012. IL-23R rs11209026 poly-morphism modulates IL-17A expression in patients with rheumatoid arthritis. Genes Immun. 13:282–287.
- Hellings PW, Kasran A, Liu Z, Vandekerckhove P, Wuyts A, Overbergh L, Mathieu C, Ceuppens JL. 2003. IL-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol. 28:42–50.
- Hradsky O, Dusatkova P, Lenicek M, et al. 2010. The CTLA4 variants may interact with the IL23R-and NOD2-conferred risk in development of Crohn's disease. BMC Med Genet. 11:91.
- Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, Maloy KJ. 2006. IL-23 drives innate and T-cell-mediated intestinal inflammation. J Exp Med. 203:2473–2483.
- Kim H-R, Kim H-S, Park M-K, Cho M-L, Lee S-H, Kim H-Y. 2007. The clinical role of IL-23p19 in patients with rheumatoid arthritis. Scand J Rheumatol. 36:259–264.
- Kleinschek MA, Boniface K, Sadekova S, Grein J, Murphy EE, Turner SP, Raskin L, Desai B, Faubion WA, de Waal Malefyt R. 2009. Circulating and gut-resident human TH17 cells express CD161 and promote intestinal inflammation. J Exp Med. 206:525–534.
- Korn T, Bettelli E, Oukka M, Kuchroo VK. 2009. IL-17 and TH17 Cells. Ann Rev Immunol. 27:485–517.
- Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, Huang E, Finlayson E, Simeone D, Welling TH. 2009. Phenotype, distribution, generation, and functional and clinical relevance of TH17 cells in human tumor environments. Blood. 114:1141–1149.
- Lacher M, Schroepf S, Helmbrecht J, Von Schweinitz D, Ballauff A, Koch I, Lohse P, Osterrieder S, Kappler R, Koletzko S. 2010. Association of the IL‐23 receptor gene variant rs11209026 with Crohn’s disease in German children. Acta Paediatr. 99:727–733.
- Lakatos PL, Szamosi T, Szilvási A, Molnar E, Lakatos L, Kovacs A, Molnár T, Altorjay I, Papp M, Tulassay Z. 2008. ATG16L1 and IL23 receptor (IL23R) genes are associated with disease susceptibility in Hungarian CD patients. Digest Liver Dis. 40:867–873.
- Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, Dhodapkar M, Krueger JG. 2004. Increased expression of IL-23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. 199:125–130.
- Leonardi CL, Kimball AB, Papp KA, Yeilding N, Guzzo C, Wang Y, Li S, Dooley LT, Gordon KB, Investigators PS. 2008. Efficacy and safety of ustekinumab, a human IL-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomized double-blind placebo-controlled trial (PHOENIX 1). Lancet. 371:1665–1674.
- Lerch-Bader M, Lundin C, Kim H, Nilsson I, von Heijne G. 2008. Contribution of positively charged flanking residues to the insertion of trans-membrane helices into the endoplasmic reticulum. Proc Natl Acad Sci USA. 105:4127–4132.
- Libioulle C, Louis E, Hansoul S, Sandor C, Farnir F, Franchimont D, Vermeire S, Dewit O, De Vos M, Dixon A. 2007. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 3:e58.
- Magyari L, Melyaregu G, Melegh B. 2008. Susceptibility genetic variants in Hungarian morbus Crohn and ulcerative colitis patients. Orvosi Hetilap. 150:81–88.
- Manel N, Unutmaz D, Littman DR. 2008. Differentiation of human TH17 cells requires TGFβ and induction of the nuclear receptor RORγt. Nat Immunol. 9:641–649.
- Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, Dolin B, Goodman N, Groden C, Hornung RL. 2004. Anti-IL-12 antibody for active Crohn's disease. New Engl J Med. 351:2069–2079.
- McGovern D, Powrie F. 2007. The IL23 axis plays a key role in the pathogenesis of IBD. Gut. 56:1333–1336.
- Mosayebian A, Meshkat R, Ghasemi R, Samadi M. 2015. Association between IL-23 receptor R381Q gene polymorphism and asthma. Iran J Allergy Asthma Immunol. (in press).
- Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. 2003. Divergent pro-and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 198:1951–1957.
- Oda N, Canelos PB, Essayan DM, Plunkett BA, Myers AC, Huang S-K. 2005. IL-17F induces pulmonary neutrophilia and amplifies antigen–induced allergic response. Am J Respir Crit Care Med. 171:12–18.
- Oliver J, Rueda B, Lopez-Nevot MA, Gómez–García M, Martín J. 2007. Replication of an association between IL23R gene polymorphism with inflammatory bowel disease. Clin Gastroenterol Hepatol. 5:977–981.
- Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K. 2000. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 13:715–725.
- Ouyang W, Kolls JK, Zheng Y. 2008. Biological functions of TH17 cell effector cytokines in inflammation. Immunity. 28:454–467.
- Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F. 2002. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rβ1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 168:5699–5708.
- Pidasheva S, Trifari S, Phillips A, Hackney JA, Ma Y, Smith A, Sohn SJ, Spits H, Little RD, Behrens TW. 2011. Functional studies on the IBD susceptibility gene IL23R implicate reduced receptor function in the protective genetic variant R381Q. PLoS One. 6:e25038.
- Rahman P, Inman RD, Gladman DD, Reeve JP, Peddle L, Maksymowych WP. 2008. Association of IL‐23 receptor variants with ankylosing spondylitis. Arthritis Rheum. 58:1020–1025.
- Raymond YY, Brazaitis J, Gallagher G. 2015. The human IL-23 Receptor rs11209026 A allele promotes the expression of a soluble IL-23R-encoding mRNA apecies. J Immunol. 194:1062–1068.
- Roberts RL, Gearry RB, Hollis Moffatt JE, Miller AL, Reid J, Abkevich V, Timms KM, Gutin A, Lanchbury JS, Merriman TR. 2007. IL23R R381Q and ATG16L1 T300A are strongly associated with Crohn's disease in a study of New Zealand Caucasians with inflammatory bowel disease. Am J Gastroenterol. 102:2754–2761.
- Rueda B, Orozco G, Raya E, Fernandez-Sueiro JL, Mulero J, Blanco FJ, Vilches C, González-Gay MA, Martin J. 2008. The IL23R Arg381Gln non-synonymous polymorphism confers susceptibility to ankylosing spondylitis. Ann Rheum Dis. 67:1451–1454.
- Safrany E, Melegh B. 2009. Functional variants of the IL-23 receptor gene in non-gastrointestinal autoimmune diseases. Cur Med Chem. 16:3766–3774.
- Sandborn WJ, Feagan BG, Fedorak RN, Scherl E, Fleisher MR, Katz S, Johanns J, Blank M, Rutgeerts P, Group UCsDS. 2008. A randomized trial of Ustekinumab, a human IL-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn's disease. Gastroenterology. 135:1130–1141.
- Sarin R, Wu X, Abraham C. 2011. Inflammatory disease protective R381Q IL23 receptor polymorphism results in decreased primary CD4+ and CD8+ human T-cell functional responses. Proc Natl Acad Sci USA. 108:9560–9565.
- Schindler C, Plumlee C, editors. 2008. Inteferons open the JAK-STAT pathway. In: Seminars in cell and developmental biology. Amsterdam: Elsevier. p 311–318.
- Schmidt C, Giese T, Ludwig B, Mueller-Molaian I, Marth T, Zeuzem S, Meuer SC, Stallmach A. 2005. Expression of IL‐12‐related cytokine transcripts in inflammatory bowel disease: elevated IL‐23p19 and IL‐27p28 in Crohn's disease but not in ulcerative colitis. Inflamm Bowel Dis. 11:16–23.
- Seiderer J, Elben I, Diegelmann J, Glas J, Stallhofer J, Tillack C, Pfennig S, Jürgens M, Schmechel S, Konrad A. 2008. Role of the novel TH17 cytokine IL‐17F in inflammatory bowel disease (IBD): up-regulated colonic IL‐17F expression in active Crohn's disease and analysis of the IL17F p. His161Arg polymorphism in IBD. Inflamm Bowel Dis. 14:437–445.
- Stevic MS, Stefanic M, Tokic S, Glavas-Obrovac L, Mihaljevic S, Karner I. 2014. Pilot study of variants of the IL-23R and STAT3 genes reveals no association with Hashimoto’s thyroiditis in the Croatian population. Endocrine Res. 39:164–167.
- Tavasolian F, Abdollahi E, Samadi M. 2014. Association of the IL4R single-nucleotide polymorphism I50V with recurrent spontaneous abortion (RSA). J Assisted Repro Genet. 31:851–856.
- Tonel G, Conrad C, Laggner U, Di Meglio P, Grys K, McClanahan TK, Blumenschein WM, Qin J-Z, Xin H, Oldham E. 2010. Cutting edge: A critical functional role for IL-23 in psoriasis. J Immunol. 185:5688–5691.
- Torti DC, Feldman SR. 2007. Interleukin-12, interleukin-23, and psoriasis: current prospects. J Am Acad Dermatol. 57:1059–1068.
- Tremelling M, Cummings F, Fisher SA, Mansfield J, Gwilliam R, Keniry A, Nimmo ER, Drummond H, Onnie CM, Prescott NJ. 2007. IL23R variation determines susceptibility but not disease phenotype in inflammatory bowel disease. Gastroenterology. 132:1657–1664.
- Trinchieri G, Pflanz S, Kastelein RA. 2003. The IL-12 family of heterodimeric cytokines: new players in the regulation of T-cell responses. Immunity. 19:641–644.
- van de Vosse E, Hoeve MA, Ottenhoff TH. 2004. Human genetics of intracellular infectious diseases: molecular and cellular immunity against mycobacteria and salmonellae. Lancet Infect Dis. 4:739–749.
- Venegas M, Beltrán CJ, Álvarez L, Castro A, Torres T, Leal AD, Lahsen FM, Hermoso MA, Quera R. 2008. IL-23R Arg381Gln polymorphism in Chilean patients with inflammatory bowel disease. Eur Cytokine Netw. 19:190–195.
- Volpe E, Servant N, Zollinger R, Bogiatzi SI, BarHupé P, Barillot E, Soumelis V. 2008. A critical function for TGFβ, IL-23, and pro-inflammatory cytokines in driving and modulating human TH17 responses. Nat Immunol. 9:650–657.
- Wang X, Lin Z, Wei Q, Jiang Y, Gu J. 2009. Expression of IL-23 and IL-17 and effect of IL-23 on IL-17 production in ankylosing spondylitis. Rheumatol Intl. 29:1343–1347.
- Weersma RK, Zhernakova A, Nolte IM, Lefebvre C, Rioux JD, Mulder F, van Dullemen HM, Kleibeuker JH, Wijmenga C, Dijkstra G. 2008. ATG16L1 and IL23R are associated with inflammatory bowel diseases but not with celiac disease in the Netherlands. Am J Gastroenterol. 103:621–627.
- Wilke CM, Bishop K, Fox D, Zou W. 2011. Deciphering the role of TH17 cells in human disease. Trends Immunol. 32:603–611.
- Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F. 2007. Development, cytokine profile and function of human IL-17-producing helper T-cells. Nat Immunol. 8:950–957.
- Yamazaki K, Onouchi Y, Takazoe M, Kubo M, Nakamura Y, Hata A. 2007. Association analysis of genetic variants in IL23R, ATG16L1 and 5p13. 1 loci with Crohn’s disease in Japanese patients. J Human Genet. 52:575–583.
- Yu P, Shen F, Zhang X, Cao R, Zhao X, Liu P, Tu H, Yang X, Shi R, Zhang H. 2012. Association of single nucleotide polymorphisms of IL23R and IL-17 with ulcerative colitis risk in a Chinese Han population. PLoS One. 7:e44380.