
Abstract
Prostaglandin E2 (PGE2) is a potent physiological suppressor of liver fibrosis. Because the anti-ulcer drug rebamipide can induce the formation of endogenous PGE2, this study investigated the potential effects of rebamipide on development of a hepatic fibrosis that was inducible by carbon tetrachloride (CCl4). Groups of Wistar rats received intraperitoneal (IP) injections of CCl4 (0.45 ml/kg [0.72 g CCl4/kg]) over the course of for 4 weeks. Sub-sets of CCl4-treated rats were also treated concurrently with rebamipide at 60 or 100 mg/kg. At 24 h after the final treatments, liver function and oxidative stress were indirectly assessed. The extent of hepatic fibrosis was evaluated using two fibrotic markers, hyaluronic acid (HA) and pro-collagen-III (Procol-III); isolated liver tissues underwent histology and were evaluated for interleukin (IL)-10 and PGE2 content. The results indicated that treatment with rebamipide significantly inhibited CCl4-induced increases in serum ALT and AST and also reduced oxidative stress induced by CCl4. Fibrotic marker assays revealed that either dose of rebamipide decreased the host levels of Procol-III and HA that had become elevated due to the CCl4. At the higher dose tested, rebamipide appeared to be able to permit the hosts to have a normal liver histology and to minimize any CCl4-induced collagen precipitation in the liver. Lastly, the use of rebamipide was seen to be associated with significant increases in liver levels of both PGE2 and the anti-inflammatory cytokine IL-10. Based on these findings, it is concluded that rebamipide can retard hepatic fibrosis induced by CCl4 and that this effect may, in part, be mediated by an induction of PGE2 and IL-10 in the liver itself.
Keywords:
Introduction
Liver fibrosis, a serious health problem, is considered an end-result of chronic liver diseases, regardless of etiology (Bataller Citation2005). Usually, chronic liver diseases progress from inflammation to fibrosis and, eventually, to cirrhosis (Brenner Citation2009). Hepatic inflammation helps to drive fibrogenesis by causing increases in leukocyte-hepatic stellate cells (HSC) interactions, activity of cytokine networks and formation of small (pro-inflammatory) molecules such as reactive oxygen species (ROS) (Maher Citation2001). HSC are perisinusoidal cells located in the sub-endothelial space between hepatocytes and sinusoidal endothelial cells (Sangiovanni et al. Citation2006).
Following liver injury, these stellate cells undergo activation and convert from quiescent Vitamin A-rich cells into proliferative, fibrogenic, contractile myofibroblasts (Friedman Citation2000; Muhanna et al. Citation2008). In this regard, inflammatory mediators released from apoptotic hepatocytes, sinusoidal endothelium cells and leukocytes – especially those responsible for cellular immunity such as hepatic macrophages (Kupffer cells), natural killer (NK) cells and lymphocytes – are often responsible for the initiation of HSC activation in response to the injury (Fischer et al. Citation2002).
Kupffer cells play a pivotal role in driving inflammation to fibrosis. These cells secrete a large number of pro-inflammatory and fibrogenic mediators that promote inflammation and fibrosis, including transforming growth factor (TGF)-β, interleukin (IL)-1β, IL-8 and platelet-derived growth factor (PDGF) (Ramachandran and Iredale Citation2012). Moreover, Kupffer cells can also contribute to local increases in oxidative stress (Otogawa et al. Citation2007) that, in turn, promotes fibrogenesis through increases in expression of TGFβ (Poli Citation2000). Conversely, in a normal homeostatic mechanism, these same cells also begin to secrete IL-10 (Knolle et al. Citation1995; Wang et al. Citation2003) and prostaglandin (PG)-E2 (Dieter et al. Citation2004), both of which help to retard fibrosis progression. These anti-fibrotic effects of IL-10 is thought to be mediated by an inhibition of Type 1 collagen expression and a down-regulation of the expression of pro-fibrogenic cytokine-like TGFβ1 and tumor necrosis factor (TNF)-α (Zhang et al. Citation2004).
Prostaglandin E2 (PGE2) plays a predominant role in liver pathophysiology. It is one of the most important mediators produced during inflammatory responses (Breyer et al. Citation2001). PGE2 is considered a potent suppressor of liver fibrosis (Dieter et al. Citation2004). It also plays an important role in regulation of different cytokines and growth factors involved in liver fibrosis. It has been reported that PGE2 suppresses the release of IL-1, TNFα and endothelin-1 and induces the release of IL-10 and nitric oxide (NO) by Kupffer cells (Dieter et al. Citation1999).
Rebamipide is an anti-ulcer drug that acts via induction of endogenous prostaglandins (Arakawa et al. Citation1998). Moreover, it has been reported that rebamipide induces expression of the PGE2 receptors EP4 and EP2 in gastric mucosa (Suetsugu et al. Citation2000). Rebamipide also processes an anti-inflammatory activity that may be related to its ability to inhibit nuclear factor (NF)-κB signaling (Li et al. Citation2015), suppression of release of IL-8, IL-1β, IL-6 and TNFα, reduction in expression of a marker of inflammatory cell infiltration, myeloperoxidase (Choe et al. Citation2010; Fukuda et al. Citation2014; Funahashi et al. Citation2014), as well as induction of expression of IL-10 (Arakaki et al. Citation2014).
Moreover, rebamipide has a potential role in decreasing oxidative stress (Hahm et al. Citation1998) in that it can contribute to overall reductions in lipid peroxidation of cell membranes. The drug does this by aiding in the scavenging of hydroxyl radicals and other ROS (Hahm et al. Citation1997, Citation1998). In neutralizing ROS, many other anti-oxidants/drugs have been shown to induce anti-fibrotic effects (Cohen-Naftaly and Friedman Citation2011; Rosenbloom et al. Citation2013). The basis for how such activities are anti-fibrotic is that – apart from a well known ability to activate pathways that leads to increased expression of pro-inflammatory proteins/products – ROS generated by NADPH oxidase also can serve as second messengers for pro-fibrogenic factor signal transduction in HSC (de Minicis and Brenner Citation2007).
Other pharmacology studies have suggested that, beyond affecting the presence of pro-fibrotic ROS, rebamipide also acts against two important modulators of liver fibrosis, i.e. PGE2 and IL-10. Even with this knowledge, very limited data is available in the literature about the potential impact/utility of rebamipide as a therapeutic anti-fibrotic agent. Moreover, mechanisms of action for this drug against liver fibrosis (or general fibrosis) remain unclear. Given this gap in information, the present study was undertaken to first evaluate the impact of rebamipide on CCl4-induced hepatic fibrosis and, then, the possible role that changes in expression of PGE2 and IL-10 could have had in any observed effect.
Materials and methods
Drugs and chemicals
Carbon tetrachloride (CCl4) was purchased from El Phraanh (Cairo, Egypt). All other chemicals used herein were obtained from the El-Gomhouria Company for Trading Chemicals and Medical Appliances (Cairo, Egypt) and were of the highest quality/analytical grade. Rebamipide (as powder) was purchased from Sigma (St. Louis, MO). For these studies, the drug was suspended in 0.2% methylcellulose-saline solution (at a level of 20 mg drug/ml). For treatments, this suspension was then diluted and given via oral gavage at doses of 60 mg/kg/day (Sakurai et al. Citation2005) or 100 mg/kg/day (Diao et al. Citation2012). Controls received corn oil and 0.2% methylcellulose-saline solution by gavage.
Animals
Wister rats (male, ≈150 g, 6-weeks-of-age) were selected for use in this study. Rats were obtained from the National Research Center (Dokki, Giza, Egypt) and housed in a pathogen-free facility in cages with sawdust bedding. The facilities were maintained at 25 [±2]°C, with an ≈50% relative humidity and a 12-h light:dark cycle. All rats had ad libitum access to standard rodent chow and filtered water. The Ethics Committee of Tanta University approved all protocols involving animal use in these studies.
Experimental protocol
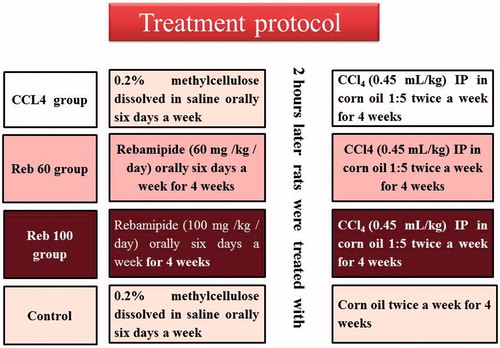
A total of 40 rats were used in the study. Rats were randomly allocated into four groups (10 rats/group) and were to be treated as follows: Group 1 (CCl4 group) rats received CCl4 by intraperitoneal (IP) injection (0.45 ml/kg) twice a week (Vassiliadis et al. Citation2011); at a density of 1.59 g/ml, this meant rats were dosed each time with 0.72 g CCl4/kg. The CCl4:corn oil solution [1:5, v/v] was prepared by adding 0.5 ml CCl4 (neat) to 2 ml corn oil. These rats were gavaged daily (6 days/week) with methylcellulose-saline vehicle for 4 weeks; on days when CCl4 was to be administered, the vehicle was given 2 h before the CCl4; Group 2 (Reb-100 group) rats received rebamipide (at 100 mg/kg/day) daily (6 days/week) by gavage for 4 weeks – on days when CCl4 was to be administered, the drug was given 2 h before the CCl4; Group 3 (Reb-60 group) rats received rebamipide (at 60 mg/kg/day) daily (6 days/week) by gavage for 4 weeks – on days when CCl4 was to be administered, the drug was given 2 h before the CCl4; and Group 4 (control group) rats were to receive corn oil in place of CCl4 – accordingly, these rats were to be gavaged daily (6 days/week) with methylcellulose-saline vehicle for 4 weeks and on days when the corn oil was to be administered, the methylcellulose-saline vehicle was given 2 h before the injection (). All rats were weighed weekly to permit adjustment of dosage volumes as needed.
Figure 1. Treatment protocols. Details are provided in the Methods section.
Preparation of blood sample and tissue homogenate
At 24 h after the final drug/vehicle gavage, all rats were euthanized by cervical dislocation and blood samples immediately obtained via cardiac puncture and collection into uncoated tubes and allowed to clot at room temperature for 60 min. The samples were then centrifuged (3000× g, 10 min, 4°C) and the resultant serum in each supernatant was recovered and stored at −20 °C until analysis.
After the blood collection, the liver of each host was carefully removed. Two 0.5-cm sections of the second largest lobe were fixed in 10% formalin (in saline) and then embedded in paraffin. For histologic examination, the embedded tissues were cut to 6-μm sections and then after affixing to slides, stained with hematoxylin-eosin. For the detection of collagen precipitates, parallel sections of the liver tissues were stained using Masson’s trichrome stain.
The remaining portions of each liver were divided into four parts and individually stored at −80 °C until analyzed. For those latter analyses, liver homogenates were prepared in 10 vol (i.e. 0.2 g tissue in 2 ml) cold phosphate-buffered saline (PBS, pH 7.4). Each resulting mixture was centrifuged (6000 × g, 20 min, 4°C) and aliquots of the derived supernatant then used for biochemical analysis and for measures of protein content using a standard kit (Wokea Medical Supplies, Changchun, China).
Indirect measures of liver function
Serum alanine aminotransferase and aspartate aminotransferase activity were each assayed using kits from Biodiagnostics (Giza) that measure the amount of pyruvate and oxaloacetate, respectively, produced from 2,4-dinitrophenylhydrazine (Reitman and Frankel Citation1957). Serum bilirubin levels were measured using kits (Biodiagnostics) that were designed based on the method of Schmidt and Eisenburg (Citation1975).
Oxidative stress
Oxidative stress was assayed via determination of liver malondialdhyde (MDA) content and superoxide dismutase (SOD) activity using kits (Biodiagnostics) designed based on assays described in Satoh et al. (Citation1978) and Nishikimi et al. (Citation1972).
Liver fibrotic markers (hyalouronic acid, pro-collagen III and TGFβ1), IL-10 and PGE2
Hyalouronic acid (HA), pro-collagen III (Procol-III), IL-10 and PGE2 levels in the liver homogenates were determined using ELISA kits (EIAab [Wuhan, China] for HA and Procol-III; Abcam [Cambridge, UK] for TGFβ1 and Wokea Medical Supplies for IL-10 and PGE2), according to manufacturer instructions. The levels of detection of the kits for HA, Procol-III, IL-10, PGE2 and TGFβ1 were 31.6, 19.5, 2.5, 10.0 and 7.8 pg/ml, respectively. To present results in terms of per-gram liver protein, protein content in the homogenate was assayed as noted above.
Statistics
All data are expressed as means ± SD. An unpaired Student’s t-test was first used to evaluate significance of differences between the control and each CCl4 group. Statistical significance between outcomes from all groups (including CCl4 only vs the drug-treated groups) was then evaluated using a one-way analysis of variance (ANOVA) with a Dunnett’s post-hoc test. Pearson’s correlation coefficients (R) were calculated to study correlations between variables. A p value < 0.05 was considered significant. All analyses were performed using Prism software (v.6.0, Graphpad, San Diego, CA).
Results
Protective effect of rebamipide on liver function
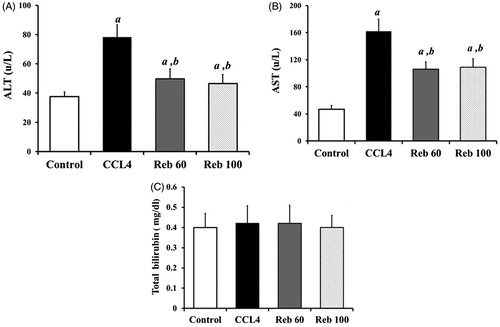
Compared to control rats, treatment with CCl4 led to significant increases in ALT (p < 0.01) and AST (p < 0.05) (). Rebamipide at both doses (60 and 100 mg/kg) significantly inhibited much of the CCl4-induced effect of ALT and AST serum level. While the drugs reduced ALT by 40.6% and AST by 32.0% vs levels from CCl4, these effects did not return host levels to control/baseline (i.e. ALT 37.5 [± 3.16] U/L and AST 47.0 [± 5.4] U/L). Furthermore, there was no dose-relatedness to these rebamipide-induced effects. With respect to bilirubin, CCl4 treatment did not cause significant changes in serum bilirubin levels (). Rebamipide also had no impact on serum bilirubin.
Figure 2. Effect of rebamipide on plasma ALT, AST and bilirubin. (A) ALT; (B) AST; (C) Bilirubin. Values shown are means ± SD (n = 10/group). Value significantly different compared to acontrol rats or bCCl4 only-treated rats (p < 0.05).
Rebamipide inhibition of hepatic oxidative stress
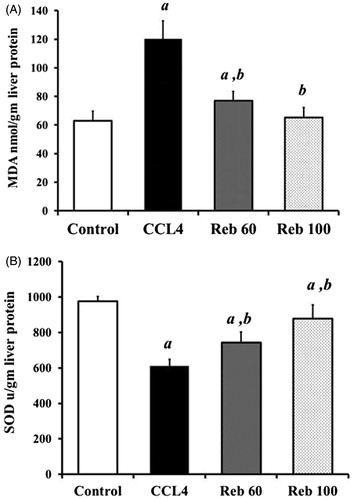
Treatment with CCl4 alone induced a significant release in levels of free radicals and resulted in induction of oxidative stress in the liver. The CCl4 induced a significant (p < 0.01) 93.5% increase in liver MDA content and a significant (p < 0.01) 37.2% decrease in SOD activity compared to values in control rat tissues (). Rebamipide at 100 mg/kg imparted a strong anti-oxidant effect; this dose resulted in a significant (p < 0.01) 45% decrease in MDA levels (to 65.4 [± 6.8] nmol/g liver protein) and 43% increase in SOD activity (to 878.8 [± 77.4] U/g protein; p < 0.01) relative to values seen in rats that received only the CCl4. In general, the drug returned host tissue levels to near control/baseline (i.e. MDA = 62.97 [± 6.6] nmol/g protein; SOD = 975.3 [± 26.9] U/g protein) levels.
Figure 3. Effect of rebamipide on hepatic oxidative stress (MDA and SOD). (A) MDA (in nmol/g liver protein). (B) SOD (in U/g liver protein). Values shown are means ± SD (n = 10/group). Value significantly different compared to a control rats or b CCl4 only-treated rats (p < 0.01).
Anti-fibrotic effect of rebamipide
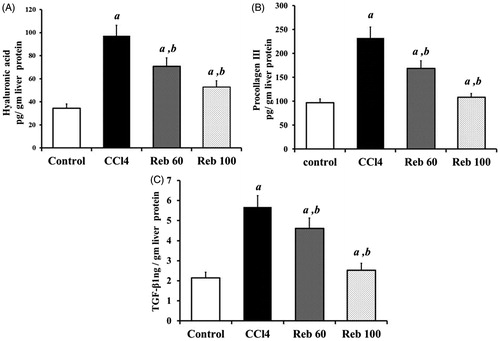
In CCl4-treated rats, liver fibrotic marker content was significantly (p < 0.01) altered compared to values in tissues from control rats. Levels of HA were increased nearly 3.0-fold 97.2 [± 9.0] pg/g liver protein) () and Procol-III nearly 2.4-fold to 232.0 [± 22.3] pg/g protein) () compared to control rat levels of, respectively, 34.5 [± 3.6] and 97.1 [± 7.4] pg/g protein). Rebamipide (100 mg/kg) treatment inhibited these CCl4-induced releases of HA and Procol-III by, respectively, 45.8% (p < 0.01) and 53.1% (p < 0.01) in liver tissues.
Moreover, CCl4 increased levels of TGFβ1 by 1.64-fold, i.e. a value of 5.7 [± 0.6] compared to control rat levels of 2.2 [± 0.3] ng/g protein (). Rebamipide at 60 or 100 mg/kg significantly inhibited TGFβ1 formation/release induced by CCl4, with levels now being measured at, respectively, 4.6 [± 0.5] and 2.5 [± 0.4] ng/g protein.
Figure 4. Effect of rebamipide on fibrotic markers (hyalouronic acid, pro-collagen-III and transforming growth factor-β1). (A) Hyalouronic acid (HA, in pg/g liver protein). (B) Pro-collagen-III (Procol-III, in pg/g liver protein). (C) Transforming growth factor (TGF)-β1 (in ng/g liver protein). Values shown are means ± SD (n = 10/group). Value significantly different compared to acontrol rats or bCCl4 only-treated rats (p < 0.01).
Impact of rebamipide on CCl4-induced hepatic fibrosis
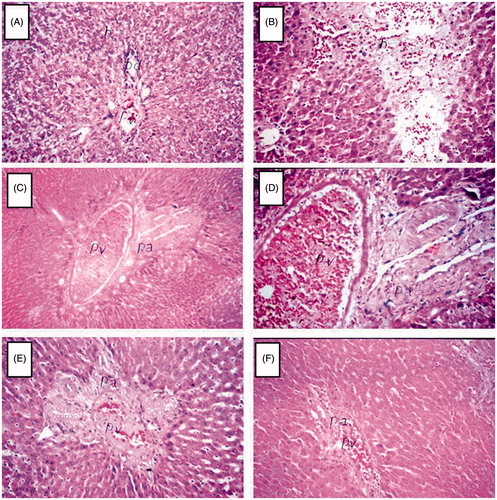
Control rats had normal histological structures in the portal area and surrounding hepatocytes (), whereas rats treated with CCl4 alone evidenced focal hemorrhaging in the hepatic parenchyma () associated with fibrosis in the portal area and severe congestion in the portal vein (). Rats treated with rebamipide (60 mg/kg) showed fibrosis in the portal area with dilatation in the portal vein (). On the other hand, rats treated with rebamipide (100 mg/kg) showed only mild congestion in their portal vein ().
Figure 5. Histology of liver. Organs were recovered at necropsy and then processed and stained with hematoxylin and eosin. (A) Control rats showed normal histological structure of the portal area and surrounding hepatocyte. (B–D) Rats treated with CCl4 alone manifested (B) focal hemorrhages in the hepatic parenchyma associated with (C and D) fibrosis in the portal area and severe congestion in the portal vein. (E) Rats treated with 60 mg rebamipide/kg evidenced fibrosis in portal area with dilatation in the portal vein. (F) Rats treated with 100 mg rebamipide/kg showed only mild congestion in the portal vein at the portal area. Images shown are representative photomicrographs. Magnification in A, B and D–F = 40×; in C = 16×.
Rebamipide suppresses collagen precipitation in liver tissues
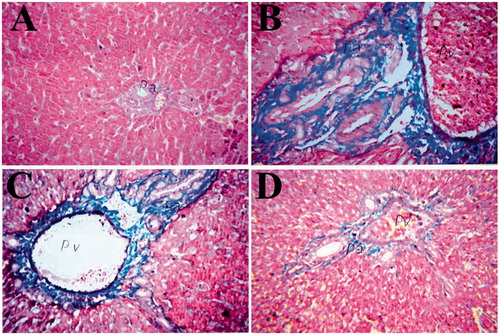
Liver tissues were stained with Masson’s trichrome stain (MTS) to examine the presence of collagen (positive reaction = blue color). Normal liver tissues from control rats had a negative reaction with MTS (i.e. no blue color) (). Liver tissues from CCL4-treated rats showed a strong positive reaction with the stain (), whereas liver tissues from rebamipide-treated rats – especially at 100 mg/kg – showed a very weak reaction ().
Figure 6. Effect of rebamipide on collagen precipitation in livers. Organs were recovered at necropsy and then processed and stained with Masson’s trichrome stain (MTS). (A) Normal liver tissues from control rats showed no collagen precipitation (no blue color). (B) Liver tissues from CCl4-treated rats show severe MTS-positive reaction (blue color). Tissues from (C) 60 and (D) 100 mg/kg/day rebamipide-treated rats show much less collagen precipitation compared to CCl4-only counterparts. Images shown are representative photomicrographs. Magnification = 40×.
Effect of rebamipide on hepatic IL-10
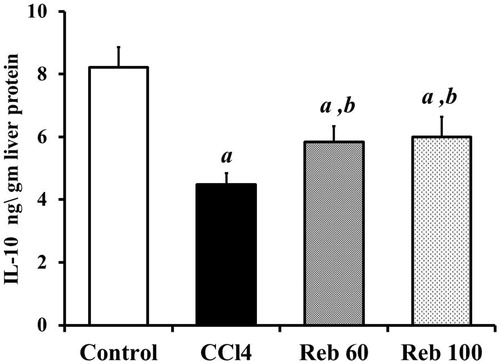
Host treatment with CCl4 alone caused a significant (p < 0.01) ≈45.5% decrease in IL-10 content in liver tissues (4.5 [± 0.4] ng/g liver protein) compared to levels in control rats (8.2 [± 0.6] ng/g protein) (). Rebamipide at both doses (60 and 100 mg/kg) significantly (p < 0.01) increased IL-10 levels by 30.5% and 34.1%, respectively, compared to CCl4 only host values (i.e. to 5.8 [± 0.5] and 6.0 [± 0.6] ng/g liver protein, respectively).
Figure 7. Effect of rebamipide on hepatic IL-10 levels. Values shown are means ± SD (n = 10/group). Value significantly different compared to a control rats or b CCl4 only-treated rats (p < 0.01).
Effect of rebamipide on hepatic PGE2
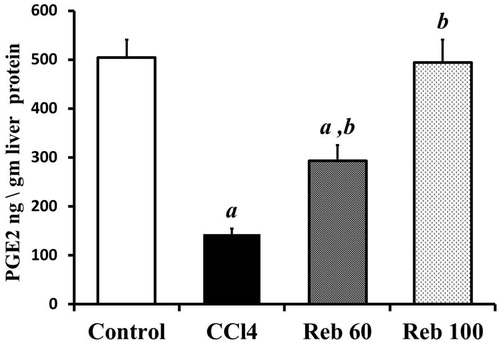
CCL4 alone treatment resulted in significant (p < 0.01) decreases in PGE2 levels in liver tissues to 143.0 [± 11.3] ng/g liver protein compared to control rat values of 504.2 [± 36.4] ng/g protein. Treatment with either rebamipide dose (60 or 100 mg/kg) led to significant (p < 0.01) increases (compared to CCl4-only host values) in PGE2 levels of 294.1 [± 31.1] and 494.6 [± 45.6] ng/g protein, respectively. The high drug dose increased PGE2 by ≈245.6% vs CCl4 only levels, with this effect being a return to near control/baseline levels to 504.2 [± 36.4] ng/g protein ().
Figure 8. Effect of rebamipide on hepatic PGE2 levels. Values shown are means ± SD (n = 10/group). Value significantly different compared to a control rats or b CCl4 only-treated rats (p < 0.01).
When analyzed in the context of changes in expression of fibrotic markers, hepatic levels of PGE2 showed a significant (p < 0.05) negative correlation with expression of both fibrotic markers HA and Procol-III (r = −0.88 and −0.70), respectively.
Discussion
Liver fibrosis is associated with significant morbidity and mortality (Sanchez-Valle et al. Citation2012). This fibrosis is characterized by an excessive accumulation of extracellular matrix proteins, including collagen, throughout the liver (Bataller Citation2005). The pathogenesis of liver fibrosis is controlled by a diversity of inflammatory cells and cytokine networks that promote activation of hepatic stellate cells (HSC), the major cell type that secretes collagen in the liver (Maher Citation2001). Cross-talk between Kupffer cells and HSC helps to regulate fibrotic progression; Kupffer cells impact both the induction and resolution of fibrosis. Specifically, Kupffer cells can promote fibrosis via secretion of fibrogenic and pro-inflammatory cytokines and by inducing oxidative stress; these same cells can retard fibrosis by secreting PGE2 and anti-inflammatory IL-10 (Wynn and Barron Citation2010).
Rebamipide is an anti-ulcer drug that acts mainly by induction of prostaglandins and free radical scavenging (Arakawa et al. Citation1998). The drug is also effective as an anti-inflammatory agent and has been useful in the treatment of inflammatory diseases such as colitis (Kishimoto et al. Citation2000) and mucositis (Kim et al. 2015). This anti-inflammatory effect is associated with an inhibition of cytokine-mediated neutrophil infiltration, inhibition of reactive oxygen species (ROS) release (Sakurai et al. 1998) and induction of IL-10 (Arakaki et al. Citation2014).
The present study investigated the potential utility of rebamipide against hepatic fibrosis induced by CCl4. Here, rebamipide showed a hepato-protective effect against damage induced by CCl4. These results were inconsistent with those of Hong et al. (Citation1998), who noted similar effects of rebamipide against liver damage induced by endotoxic shock. Further, the present results confirmed previously reported data from several studies that comment on the anti-oxidant activity imparted by rebamipide (Hahm et al. Citation1997; Kinjo et al. Citation2008; Tokuhara et al. Citation2008).
The results of the present study indicated rebamipide might also impart an anti-fibrotic effect in liver tissues. The results here showed that drug significantly inhibited increases in the presence of fibrotic markers like hyaluronic acid (HA), pro-collagen III (Procol-III) and TGFβ1 induced by host exposure to CCl4. In addition, at least the higher of two doses of rebamipide tested (100 mg/kg/day) appeared to protect the liver from portal area fibrosis and sever congestion that was associated with CCl4 exposures. These results were confirmed histologically as CCl4 induced strong positive Masson’s trichrome reactions, whereas co-treatments with rebamipide resulted in host tissues with only weak positive reactions, indicating inhibition of CCl4-induced collagen precipitation in situ.
Overall, there is a paucity of data concerning anti-fibrotic effects of rebamipide. Previous studies have reported that an increased presence of PGE2 was associated with a retarding of hepatic fibrosis in different animal models (Treffkorn et al. Citation2004; Rincón-Sánchez et al. Citation2005). Thus, in the animal model here, the observed effect of rebamipide against CCl4-induced fibrosis may be due, in part, to induction of endogenous PGE2. The results here provide support for this concept as CCl4 alone led to a significant decrease in PGE2 levels in livers, whereas treatment with rebamipide mitigated this inhibition significantly. Further proof of this linkage was obtained through the observation that showed that PGE2 level in liver tissues had a strong negative correlation with levels of the two measured fibrotic markers, HA and Procol-III. The linkage is further strengthened by earlier findings which showed that endogenous PGE2 was protective against damage associated with hepatic ischemia/re-perfusion (I/R) (Kuzumoto et al. Citation2005).
Moreover, prevention of PGE2 synthesis by cyclo-oxygenase-2 (COX-2) inhibitors like celecoxib worsened hepatic fibrosis induced by CCl4, thioacetamide (TAA) or porcine serum (Hui et al. Citation2006). Still, in contrast to the results here, Paik et al. (Citation2009) concluded that celecoxib was a good anti-fibrotic agent in that it exerted a pro-apoptotic effect on HSC in TAA-treated rats. Similarly, Wen et al. (Citation2014) reported celecoxib could ameliorate hepatic fibrosis and cirrhosis through a down-regulation of PGE2 in hepatic tissues.
To investigate possible changes in the other possible parameters that may be involved in any anti-fibrotic effects of rebamipide, the present study also investigated effects of the drug on liver IL-10 expression. The anti-fibrotic effect of this cytokine has been attributed to its ability to down-regulate expression of other pro-fibrogenic cytokines such as TGFβ1 and to inhibit HSC activation (Zhang et al. Citation2007). The present study indicated that rebamipide inhibited the expected decreases in IL-10 levels in the liver induced by CCl4. Unfortunately, there is little data available in the literature about effects of rebamipide on IL-10. Arakaki et al. (Citation2014) reported rebamipide up-regulated IL-10 expression in eye lesions. However, in contrast to the results reported here, Aihara et al. (Citation1998) noted that rebamipide suppressed production of IL-10 (as well as of TNFα and IL-1β). Clearly, further detailed studies (i.e. at molecular/gene level) are needed to clarify how rebamipide might be acting in the liver either to induce IL-10 expression OR to rebuff CCl4-related mechanisms that cause the decrease in the expression of this protein.
Conclusions
This study showed that rebamipide acts in this particular rodent model as an anti-fibrotic agent. Based on the results, it could be concluded that this effect is due, in part, to the drug causing increases in endogenous PGE2 and IL-10 in the liver. Further investigations to define more precisely the molecular targets that could be involved in the observed rebamipide anti-fibrotic effects are needed.
Acknowledgements
The authors are grateful to Dr Adel Bakeer (Professor of Pathology, Cairo University) for the kind help in performing the histopathology and interpretation of those results. The authors are also grateful to the Colleges of Pharmacy at Damanhour University and Tanta University for providing facilities to perform these studies.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Aihara M, Imagawa K, Funakoshi Y, Ohmoto Y, Kikuchi M. 1998. Effects of rebami-pide on production of several cytokines by human peripheral blood mononuclear cells. Dig Dis Sci. 43:160S–166S.
- Arakaki R, Eguchi H, Yamada A, Kudo Y, Iwasa A, Enkhmaa T. 2014. Anti-inflammatory effects of rebamipide eye drop administration on ocular lesions in a murine model of primary Sjögren’s syndrome. PLoS One. 9:e98390.
- Arakawa T, Kobayashi K, Yoshikawa T, Tarnawski A. 1998. Rebamipide: overview of mechanisms of action and efficacy in mucosal protection and ulcer healing. Dig Dis Sci. 43: 5S–13S.
- Bataller R. 2005. Liver fibrosis. J Clin Invest. 115:209–218.
- Brenner DA. 2009. Molecular pathogenesis of liver fibrosis. Trans Am Clin Climatol Assoc. 120:361–368.
- Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. 2001. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 41:661–690.
- Choe JY, Park KY, Lee SJ, Park SH, Kim SK. 2010. Rebamipide inhibits TNFα-induced IL-8 expression by suppressing the NF-κB signal pathway in human umbilical vein endothelial cells. Inflamm Res. 59:1019–1026.
- Cohen-Naftaly M, Friedman SL. 2011. Current status of novel anti-fibrotic therapies in patients with chronic liver disease. Ther Adv Gastroenterol. 4:391–417.
- de Minicis S, Brenner DA. 2007. NOX in liver fibrosis. Arch Biochem Biophys. 462:266–272.
- Diao L, Mei Q, Xu JM, Liu XC, Hu J, Jin J, Yao Q, Chen ML. 2012. Rebamipide suppresses diclofenac-induced intestinal permeability via mitochondrial protection in mice. World J Gastroenterol. 18:1059–1066.
- Dieter P, Scheibe R, Bezugla Y, Matthé E, Schuch S, Treffkorn L, Bernard B, Kamionka S, Kolada A. 2004. Regulatory role of prostaglandin E2 in liver (patho)-physiology is controlled at its site of synthesis and its action on the receptors. Comp Hepatol. 3:S35.
- Dieter P, Scheibe R, Télzer S, Kamionka S, Kolada A. 1999. Prostaglandin E2, an important regulator in (patho) physiological liver functions. In: Wisse E, Knook DL, de Zanger R, Arthur MJ, editors. Cells of the hepatic sinusoid. Leiden: Kupffer Cell Foundation. p. 6–10.
- Fischer R, Cariers A, Reinehr R, Haussinger D. 2002. Caspase 9-dependent killing of hepatic stellate cells by activated Kupffer cells. Gastroenterology. 123:845–861.
- Friedman SL. 2000. Molecular regulation of hepatic fibrosis: an integrated cellular response to tissue injury. J Biol Chem. 275:2247–2250.
- Fukuda K, Ishida W, Tanaka H, Harada Y, Fukushima A. 2014. Inhibition by reba-mipide of cytokine-induced or LPS-induced chemokine synthesis in human corneal fibroblasts. Br J Ophthalmol. 98:1751–1755.
- Funahashi Y, Yoshida M, Yamamoto T, Majima T, Takai S, Gotoh M. 2014 Intra-vesical application of rebamipide suppresses bladder inflammation in a rat cystitis model. J Urol. 191:1147–1152.
- Hahm KB, Lee KJ, Kim YS, Kim JH, Cho SW, Yim H, Joo HJ. 1998. Augmented eradication rates of Helicobacter pylori by new combination therapy with lansoprazole, amoxicillin, and rebamipide. Dig Dis Sci. 43:235–240.
- Hahm KB, Park IS, Kim YS, Kim JH, Cho SW, Lee SI, Youn JK. 1997. Role of rebamipide on induction of heat-shock proteins and protection against reactive oxygen metabolite-mediated cell damage in cultured gastric mucosal cells. Free Radic Biol Med. 22:711–716.
- Hong KW, Kim KE, Kim BY, Lee WS, Kim CD. 1998. Effect of rebamipide on liver damage and increased tumor necrosis factor in a rat model of endotoxin shock. Dig Dis Sci. 43:154S–159S.
- Hui AY, Leung WK, Chan HL, Chan FK, Go MY, Chan KK, Tang BD, Chu ES, Sung JJ. 2006. Effect of celecoxib on experimental liver fibrosis in rat. Liver Int. 26:125–136.
- Kim H, Seo JY, Kim KH. 2000. Inhibition of lipid peroxidation, NF-κB activation and IL-8 production by rebamipide in Helicobacter pylori-stimulated gastric epithelial cells. Dig Dis Sci. 45:621–628.
- Kinjo N, Kawanaka H, Akahoshi T, Yamaguchi S, Yoshida D, Anegawa G, Konishi K, Tomikawa M, Tanoue K, Tarnawski A, et al. 2008. Significance of ERK nitration in portal hypertensive gastropathy and its therapeutic implications. Am J Physiol. 295:G1016–1024.
- Kishimoto S, Haruma K, Tari A, Sakurai K, Nakano M, Nakagawa Y. 2000. Rebamipide, an anti-ulcer drug, prevents DSS-induced colitis formation in rats. Dig Dis Sci. 45: 1608–1616.
- Knolle P, Schlaak J, Uhrig A, Kempf P, Meyer zum Büschenfelde K, Gerken G. 1995. Human Kupffer cells secrete IL-10 in response to LPS challenge. J Hepatol. 22:226–229.
- Kuzumoto Y, Sho M, Ikeda N, Hamada K, Mizuno T, Akashi S, Tsurui Y, Kashizuka H, Nomi T, Kubo A, et al. 2005. Significance and therapeutic potential of prostaglandin E2 receptor in hepatic ischemia/reperfusion injury in mice. Hepatology 42:608–617.
- Li W, Zhao Y, Xu X, et al. 2015. Rebamipide suppresses TNFα-mediated inflammation in vitro and attenuates the severity of dermatitis in mice. FEBS J. 282:2317–2326.
- Liu H, Wei W, Li X. 2009. Celecoxib exacerbates hepatic fibrosis and induces hepatocellular necrosis in rats treated with porcine serum. Prostagland Lipid Mediat. 88:63–67.
- Maher JJ. 2001. Interactions between hepatic stellate cells and the immune system. Semin Liver Dis. 21:417–426.
- Muhanna N, Doron S, Wald O, Horani A, Eid A, Pappo O, Friedman S, Safadi R. 2008. Activation of hepatic stellate cells after phagocytosis of lymphocytes: a novel pathway of fibrogenesis. Hepatology 48:963–977.
- Nishikimi M, Appaji N, Yagi K. 1972. The occurrence of superoxide anion in the reaction of reduced phenazine methosulfate and molecular oxygen. Biochem Biophys Res Commun. 46:849–854.
- Otogawa K, Kinoshita K, Fujii H, Sakabe Ma, Shiga R, Nakatani K, Ikeda K, Nakajima Y, Ikura Y, Ueda M, et al. 2007. Erythrophagocytosis by liver macrophages (Kupffer cells) promotes oxidative stress, inflammation, and fibrosis in a rabbit model of steato-hepatitis: implications for pathogenesis of human non-alcoholic steato-hepatitis. Am J Pathol. 170:967–980.
- Paik YH, Kim JK, Lee JI, Kang SH, Kim DY, An SH, Lee SJ, Lee DK, Han KH, Chon CY, et al. 2009. Celecoxib induces hepatic stellate cell apoptosis through inhibition of Akt activation and suppresses hepatic fibrosis in rats. Gut 58:1517–1527.
- Poli G. 2000. Pathogenesis of liver fibrosis: role of oxidative stress. Mol Asp Med. 21:49–98.
- Ramachandran P, Iredale JP. 2012. Liver fibrosis: a bi-directional model of fibrogenesis and resolution. QJM. 105:813–817.
- Reitman S, Frankel S. 1957. A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases. Am J Clin Pathol. 28:56–63.
- Rincón-Sánchez AR, Covarrubias A, Rivas-Estilla AM, Pedraza-Chaverrí J, Cruz C, Islas-Carbajal MC, Panduro A, Estanes A, Armendáriz-Borunda J. 2005. PGE2 alleviates kidney and liver damage, decreases plasma renin activity and acute phase response in cirrhotic rats with acute liver damage. Exp Toxicol Pathol. 56: 291–303.
- Rosenbloom J, Mendoza FA, Jimenez SA. 2013. Strategies for anti-fibrotic therapies. Biochim Biophys Acta. 1832:1088–1103.
- Sakurai K, Osaka T, Yamasaki K. 2005. Rebamipide reduces recurrence of experimental gastric ulcers: role of free radicals and neutrophils. Dig Dis Sci. 50:S90–S96.
- Sánchez-Valle V, Chávez-Tapia NC, Uribe M, Méndez-Sánchez N. 2012. Role of oxidative stress and molecular changes in liver fibrosis: a review. Curr Med Chem. 19:4850–4860.
- Sangiovanni A, Prati GM, Fasani P. 2006. The natural history of compensated cirrhosis due to hepatitis C virus: a 17-year cohort study of 214 patients. Hepatology. 43:1303–1310.
- Satoh K. 1978. Serum lipid peroxide in cerebrovascular disorders determined by a new colorimetric method. Clin Chim Acta. 90:37–43.
- Schmidt M, Eisenburg J. 1975. Serum bilirubin determination in newborn infants. New micromethod. Fortschr Med. 93:1461–1466.
- Suetsugu H, Ishihara S, Moriyama N, Kazumori H, Adachi K, Fukuda R, Watanabe M, Kinoshita Y. 2000. Effect of rebamipide on prostaglandin EP4 receptor gene expression in rat gastric mucosa. J Lab Clin Med. 136:50–57.
- Tokuhara K, Hamada Y, Tanaka H, Yamada M, Ozaki T, Matsui K, Kamiyama Y, Nishizawa M, Ito S, Okumura T. 2008. Rebamipide, anti-gastric ulcer drug, up-regulates induction of iNOS in pro-inflammatory cytokine-stimulated hepatocytes. Nitric Oxide. 18:28–36.
- Treffkorn L, Scheibe R, Maruyama T, Dieter P. 2004. PGE2 exerts its effect on the LPS-induced release of TNFα, ET-1, IL-1α, IL-6, and IL-10 via the EP2 and EP4 receptor in rat liver macrophages. Prostagland Lipid Mediat. 74:113–123.
- Vassiliadis E, Larsen DV, Clausen RE, Veidal SS, Barascuk N, Larsen L, Simonsen H, Silvestre TS, Hansen C, Overgaard T, et al. 2011. Measurement of CO3-610, potential liver biomarker derived from MMP-9 degradation of collagen type III, in a rat model of reversible CCl4-induced fibrosis. Biomarker Insights. 6:49–58.
- Wang XZ, Zhang LJ, Li D, Huang YH, Chen ZX, Li B. 2003. Effects of transmitters and IL-10 on rat hepatic fibrosis induced by CCl4. World J Gastroenterol. 9:539–543.
- Wen SL, Gao JH, Yang WJ, Lu YY, Tong H, Huang ZY, Liu ZX, Tang CW. 2014. Celecoxib attenuates hepatic cirrhosis through inhibition of epithelial-to-mesenchymal transition of hepatocytes. J Gastroenterol Hepatol. 29:1932–1942.
- Wynn TA, Barron L. 2010. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 30:245–257.
- Yu J, Hui AY, Chu ES, Go MY, Cheung KF, Wu CW, Chan HL, Sung JJ. 2009. The anti–inflammatory effect of celecoxib does not prevent liver fibrosis in bile duct–ligated rats. Liver Int. 29:25–36.
- Zhang LJ, Chen YX, Chen ZX, Huang YH, Yu JP, Wang XZ. 2004. Effects of IL-10 and PDGF on expression of MMP-2 in rat and tissue inhibitor of metalloproteinase-1 in rat fibrotic liver and cultured hepatic stellate cells. World J Gastroenterol. 10:2574–2579.
- Zhang LJ, Zheng WD, Chen YX, Huang YH, Chen ZX, Zhang SJ, Shi MN, Wang XZ. 2007. Antifibrotic effects of interleukin-10 on experimental hepatic fibrosis. Hepato-gastroenterol. 54:2092–2098.