
Abstract
The potential to engineer extracellular vesicles (EV) that target specific cells and deliver a therapeutic payload has propelled a growing interest in their development as promising therapeutics. These EV are often produced from cultured cells. Very little is known about the interaction of cell culture-derived EV with cells of the immune system and their potential immunomodulatory effects. The present study evaluated potential immunotoxic effects of HEK293T-derived EV on the human monocytic cell lines THP-1 and U937. Incubation of cells with different doses of EV for 16–24 h was followed by assessment of cytotoxicity and cell function by flow cytometry. Changes in cell functionality were evaluated by the capacity of cells to phagocytize fluorescent microspheres. In addition, the internalization of labeled EV in THP-1 and U937 cells was evaluated. Exposure to EV did not affect the viability of THP-1 or U937 cells. Although lower doses of the EV increased phagocytic capacity in both cell lines, phagocytic efficiency of individual cells was not affected by EV exposure at any of the doses evaluated. This study also demonstrated that THP-1 and U937 monocytic cells are highly permissive to EV entry in a dose-response manner. These results suggest that, although HEK293T-derived EV are efficiently internalized by human monocytic cells, they do not exert a cytotoxic effect or alter phagocytic efficiency on the cell lines evaluated.
Introduction
Extracellular vesicles (EV) are naturally occurring nano-sized lipid vesicles shed from essentially all mammalian cells and are present in body fluids such as plasma, serum, breast milk, cerebrospinal fluid and serum. Based on the origin of secreted vesicles, EV can be classified into microvesicles (MV) and exosomes. MV (150–1000 nm) are formed by direct outward budding of plasma membrane, while exosomes (30–100 nm) are of endosomal origin, released from multi-vesicular bodies (MVB) fusing to plasma membrane (Johnstone et al. Citation1987; Colombo et al. Citation2014; Gyorgy et al. Citation2015). The secretion of EV has been found in both eukaryotes and prokaryotes, where it appears to be a conserved process during evolution (Raposo & Stoorvogel Citation2013). EV contain cytosolic contents such as proteins, lipids, mRNA and miRNA. They are involved in many biological processes via their internalization by recipient cells. EV protect their cargo from enzymatic degradation in the extracellular environment (Mulcahy et al. Citation2014). The ability of EV to protect their cargo while in circulation has made them attractive as disease biomarkers and drug delivery systems.
Therapeutic application for EV is promising due to their endogenous composition, their ability to attach to target cells via surface adhesion proteins, the capacity to be loaded with different cargo and the capability to engineer the EV surface to present external targeting moieties (reviewed in Batrakova and Kim (Citation2015) and in Gyorgy et al. (Citation2015)). EV produced by cultured cells are typically collected by ultracentrifugation. Following purification, small molecule drugs may be directly loaded into the EV or larger therapeutic molecules such as nucleic acid or protein are incorporated into the EV by electroporation (Alvarez-Erviti et al. Citation2011), sonication or extrusion (Haney et al. Citation2015). A variety of cell types have been developed to produce therapeutic EV, including dendritic cells (Alvarez-Erviti et al. Citation2011), mesenchymal stem cells (Chen et al. Citation2011; Yeo et al. Citation2013) and immortalized cell lines such as HEK293 (El-Andaloussi et al. Citation2012; Ohno et al. Citation2013; Yeo et al. Citation2013).
The development of therapeutic EV is rapidly moving towards the clinical trials and the need for assessment of potential risks is imminent. A critical part of the risk assessment phase for new drugs and biologicals is the investigation of potential toxicity. Evaluation of the potential adverse effects of a given drug in the immune system or immunotoxicity is a fundamental component (Galbiati et al. Citation2010). Agents that interact with cells and functions of the immune system can induce unwanted immunomodulatory effects, e.g. immunosuppression, immunogenicity, hyper-sensitivity, autoimmunity or adverse immunostimulation. Current guidelines for immunotoxicity testing largely rely on animal tests. However, regulatory bodies are actively supporting the development, characterization and validation of alternative in vitro testing methods (Gennari et al. Citation2005; ICH Citation2011; Hartung & Corsini Citation2013). Overall, in vitro immunotoxicity testing allows for early screening and prioritization for more complex immunological studies and in vivo testing.
We evaluated here in vitro immunotoxicity testing of survival and function of two human monocyte/macrophage cell lines. Monocytes are innate immunity phagocytic cells and act as environmental sensors and first responders to foreign organisms or materials. During homeostasis and inflammation, monocytes migrate into tissues and differentiate to macrophages or dendritic cells. Agents that affect monocyte survival or functional status will interfere with their activation, migration and differentiation, with consequences on the development of both innate and adaptive immune responses.
This study evaluated the impact of EV on monocytic cell viability and function by two flow cytometry-based methods previously described: the assessment of cell death by measuring the index of apoptosis and necrosis in the cell population (Roy et al. Citation2006; Li et al. Citation2011) and assessment of the internalization of EV in both monocytes and differentiated macrophages by flow cytometry and confocal microscopy (Leclerc et al. Citation2010; Berenson et al. Citation2013; Ji et al. Citation2013; Diler et al. Citation2014a,Citationb). Application of these flow cytometry-based methods for the rapid and robust screening of critical parameters of immune cells offers important advantages for the early assessment of immunotoxicity in safety studies of novel therapeutics such as EV.
Materials and methods
EV donor cell line
HEK293T cell line (ATCC CRL-11268™) was purchased from American Type Culture Collection (ATCC, Manassas, VA). HEK293T cells were cultured as adherent cells in T-flasks using Dulbecco’s Modified Eagle Medium (DMEM) (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) (Sigma, St. Louis, MO). All FBS-supplemented media used in this study was depleted of exogenous exosomes by 18 h centrifugation at 100 000 rcf at 4 °C.
For EV scale up production, HEK293T cells were adapted from adherent culture to grow in suspension using HEKPlus SFM (ATCC® ACS-4002™) as recommended by the supplier with few alterations. In brief, cells were cultured in 15-cm dishes and adapted to low serum conditions in 3:1 DMEM/10%FBS:HEKPlus for 1 week (Day 1 to Day 7). This was followed by a 1:1 ratio of media for 1 week (Day 8 to Day 14) and a 1:3 ratio for 2 weeks (Day 15 to Day 28). Cells (200 000 cells/ml) were then transferred to a 0.5 L spinner flask and cultured in 2% FBS-supplemented HEKPlus media and set at 130 rotations per minute in a 37 °C 5% CO2 incubator.
EV isolation
EV were isolated at 4 °C using a standard ultracentrifugation protocol (Thery et al. Citation2006). In brief, 0.4 L of cell culture supernatant was centrifuged at 300 rcf for 10 min, transferred to clean tubes and centrifuged at 2000 rcf for 20 min to remove cell debris. The supernatant was transferred to ultracentrifuge tubes (item 355622, Beckman Coulter, Brea, CA) and centrifuged at 10 000 rcf for 30 min in a Beckman Coulter Optima™ XE ultracentrifuge with a Type 45 Ti rotor, followed by vacuum filtration using a 0.22-μm filter (EMD Millipore, Billerica, MA). Using new ultracentrifuge tubes, filtered supernatant was centrifuged at 110 000 rcf for 75 min. The EV pellet was washed once with sterile phosphate-buffered saline (PBS, pH 7.4) and pelleted using the same conditions. Finally, the EV pellet was re-suspended in 0.3–0.5 ml phenol red free RPMI 1640 media supplemented with 10% exosome-depleted FBS. EV protein yield was measured using a Pierce™ BCA Protein Assay (Life Technologies).
EV characterization
Western blotting
Western blotting was done using manufacturer protocols and standard conditions. The EV pellet was lysed using RIPA buffer with 1× Protease and Phosphatase Inhibitor Cocktail. Protein quantification was done using the BCA protein assay. Protein lysates were mixed with 4× non-reducing lithium dodecyl sulfate sample loading buffer containing 3% 2-mercaptoethanol. An exception was for anti-CD63 antibody that was run under non-reducing conditions. Twenty micrograms of total protein was loaded per lane of a gradient 4–12% pre-cast SDS-PAGE gel (NuPAGE®, Life Technologies). Following electrophoretic separation of the proteins, the materials were electro-transferred to a nitrocellulose membrane, then blocked with 5% bovine serum albumin (BSA) solution before being incubated with primary antibodies. The primary antibodies used included: anti-Calreticulin (Cell Signaling #2891S, Danvers, MA), anti-GAPDH (SC-32233, Santa Cruz Biotechnology, Dallas, TX), anti-TSG101 antibody (SC-7964, Santa Cruz Biotechnology) and anti-CD63 (ab68418, Abcam, Cambridge, MA). The membrane was then washed using 0.05% Tween-20 in 1× PBS (PBS-T wash buffer) and then incubated in PBS-T containing 1% BSA and secondary antibody (goat anti-mouse or goat-anti-rabbit) for 1 h. Presence of proteins was visualized using an enhanced chemiluminescence (ECL) detection system (GE Healthcare Bio-Sciences, Pittsburgh, PA). GAPDH and histones were used as loading controls.
Cryo-transmission electron microscopy (Cryo-TEM)
Sample preparation for Cryo-TEM was performed at the Liquid Crystal Institute at Kent State University (Kent, OH), as described in Gao et al. (Citation2014). Briefly, the EV samples were first mildly sonicated for ∼1 min to break up any aggregates that may have formed. To prepare vitrified cryo-TEM specimens, 4 μl suspensions of the MV were applied to a grid and flash-frozen in liquid ethane within the controlled environment (22 °C and 95% relative humidity) of an automated vitrification device (FEI Vitrobot Mark IV, FEI, Hillsboro, OR). The vitrified samples were stored under liquid nitrogen before transferring to a Gatan Cryo holder (Model 626.DH) and visualized in a FEI Tecnai G2 F20 ST TEM (FEI). The microscope was operated at 200 kV and under low-dose mode to minimize radiation damage to the samples. Images were captured using a 4k × 4k Gatan Ultrascan CCD camera at magnifications of 38 000× and 43 000×.
EV staining
EV were stained with PHK67 green fluorescent dye (Sigma-MINI67) according to manufacturer protocols, with slight modifications. In brief, the EV pellet was diluted to 1 ml using Diluent C (Sigma-MINI67). In a separate tube, 4 μl PKH67 dye was added to 1 ml Diluent C, mixed, added immediately to the tube containing the diluted EV, then gently mixed three times. The tube was incubated at room temperature for 3 min with periodic mixing. PKH67 staining was inactivated by addition of 10 ml of 10% FBS-supplemented DMEM and incubated for 1 min. After staining, 25 ml PBS was added and the stained pellet was re-pelleted by ultracentrifugation at 110 000 rcf for 75 min. The EV pellet was washed twice with PBS and re-suspended in 10% FBS-supplemented RPMI without phenol red.
Human monocyte cell lines culture and differentiation
Human monocyte cells lines THP-1 (ATCC TIB-202) and U-937 (ATCC CRL-1593.2) were maintained in culture in T-75 culture flasks (Nunc, Thermo Scientific, Waltham, MA) following ATCC recommendations. U-937 cells were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated FBS (Atlanta Biologicals, Flowery Branch, GA), 10 mM HEPES, 2 mM L-glutamine and 100 U penicillin + 100 μg streptomycin/ml (Gibco, Carlsbad, CA). THP-1 cells were maintained in media similar to the one used for U-937 cells additionally supplemented with 50 μM 2-mercaptoethanol (Sigma). Cells were maintained at 37 °C in a humidified 5% CO2 atmosphere.
Differentiation of monocyte cells towards macrophage-like adherent cells was achieved by treatment with 50 ng/ml of phorbol 12-myristate 13-acetate (PMA; Sigma) for 24 h, followed by 48 h incubation in untreated cell culture media before experimental assays. Harvesting of PMA-treated cells was achieved by either gentle scraping or by treatment with Trypsin/EDTA (Gibco) for 5–10 min. For microscopic visualization of adherent cells, rounded 12-mm glass coverslips were placed in the wells of a 24-well-plate before cell plating.
Cells and EV co-incubation
Medium (900 μl) containing 500 000 cells was added to each well of a 12-well culture plate (Falcon, Corning Life Sciences, Corning, NY). After 1 h incubation at 37 °C, in a 5% CO2 humidified environment, 100 μl of EV suspension at either different concentrations, positive control treatments or cell culture media were added to each well. Plates were returned to incubate for 16–24 h according to each assay protocol. When required, cytochalasin D (CytoD, Sigma) at a final concentration of 10 μM was added to cells 1 h prior to addition of the EV. For experiments carried out in 24-well culture plates, all cell densities and treatment doses were maintained by escalating down all the volumes to half of the volume used in a 12-well plate.
Apoptosis and necrosis assay
After 24 h co-incubation of the cells with EV, the cells were harvested, washed twice with cold PBS and re-suspended in 100 μl of 1× binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2). Simultaneous staining for apoptosis and necrosis was achieved by addition of 5 μl of FITC Annexin V and 5 μl 7-Amino-Actinomycin (7-AAD) (BD Pharmingen, San Jose, CA) per sample. Incubation at room temperature in the dark for 15 min was followed by addition of 400 μl binding buffer before flow analysis. Positive controls used to induce apoptosis and necrosis were: 10 mM 5-fluorouracil (5-FU) for 24 h or 500 nM staurosporine (Sts; Sigma) for 24 h or 75% ethanol for 5 min.
Microsphere phagocytosis assay
After a 21-h co-incubation of the cells and EV, 1-μm diameter FluoSpheres Fluorescent Microspheres (Molecular Probes, Life Technologies) were added to each well at a ratio of 50 microspheres/plated cell (in 500 μl media). Following 3 h incubation at 37 °C in a 5% CO2 humidified environment, cells were harvested, washed twice with cold flow buffer (2% BSA-0.01% NaN3-PBS) and reconstituted in 200 μl flow buffer for flow cytometry analysis. Phagocytic capacity was expressed as the number of cells positive for fluorescent microspheres, while phagocytic efficiency was determined by the MFI of those positive cells. Positive controls used to stimulate phagocytosis in monocyte cell lines were 5 ng PMA/ml or 1 μg lipo-polysaccharide (LPS)/ml (Sigma) for 21 h.
Phagocytosis of labeled EV
Cells were harvested after 16 h incubation with PKH67-labeled EV. For flow cytometric analysis, cells were washed twice and re-constituted in 200 μl flow buffer. To discriminate the fluorescence signal of intracytoplasmic vesicles from vesicles attached to the surface of the cell membrane, a quenching assay (Hed et al. Citation1987; Feng et al. Citation2010; Lehmann et al. Citation2000; Parker et al. Citation2010) was performed. After the final wash with flow buffer, cell suspensions were re-constituted in 0.05% trypan blue (Invitrogen, Life Technologies), followed by flow cytometry analysis. Permeabilization of cells for intracellular quenching was achieved by treatment with BD Cytofix/Cytoperm solution kit (BD Biosciences, San Jose, CA) following manufacturer instructions. In brief, cells were incubated for 20 min in 200 μl Fixation/Permeabilization solution at 4 °C in the dark, washed twice with 1× BD Perm/Wash buffer, re-suspended in 200 μl of 0.05% trypan blue solution prepared in 1× BD Perm/Wash buffer and incubated for 20 min at 4 °C in the dark before flow acquisition. LPS and PMA were used as internal positive controls for phagocytosis, given that they stimulate activation and/or maturation of myelo-monocytic cells.
Confocal microscopy
For confocal microscopy analysis, non-differentiated cells were harvested, washed twice in cold 2% FBS-PBS and counted by trypan blue exclusion. Samples of 50 000 cells in 200 μl were prepared on cover slides using a Cytospin centrifuge (Fisher Scientific, Pittsburgh, PA) for staining. PMA-differentiated cells attached to glass coverslips were washed twice with 2% FBS-PBS before staining. Cell membrane and nuclear staining of cells was performed following manufacturer recommendations. In brief, 150 μl of a 3 μg/ml solution of Wheat Germ Agglutinin (WGA; AlexaFluor conjugate; Invitrogen) was added to each sample and the mixture was incubated at 37 °C in the dark for 10 min followed by two gentle washes with Hank’s Balanced Salt Solution (HBSS) (Invitrogen). The coverslip was then mounted onto a glass slide using ProLong® Diamond Antifade with DAPI (Life Technologies).
Flow cytometry and confocal microscopy
Flow cytometry analysis was performed in a BD Accuri C6 flow cytometer (BD Biosciences), acquiring a minimum of 10 000 events/sample. Data was analyzed using BD CFlow Plus software. Confocal microscopy images were captured using a FV1000-Spectral Confocal System (Olympus, Pittsburgh) under a 40× oil objective, at the Campus Microscopy and Imaging Facility (CMIF) at the Ohio State University.
Statistical analyses
Data shown are the result of 3–4 replicate experiments for each assay. Data were analyzed by analysis of variance with repeated measures, incorporating observational dependencies across different doses within the same day of experiments. Holm’s procedures were applied to adjust the multiplicity to control the familywise error rate at 0.05. Data analysis was performed by using SAS 9.3 (SAS, Inc., Cary, NC).
Results
Characterization of HEK293T-derived EV
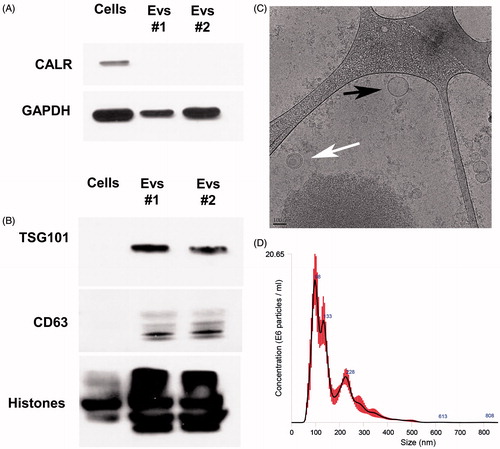
To evaluate the purity of the EV, calreticulin (CALR) was blotted in both cells and EV lysates. Lack of CALR detection in EV lysates confirmed the absence of endoplasmic reticulum and apoptotic body contaminants () (van Deun et al. Citation2014) . Enrichment of exosomal markers, TSG101 and CD63 (Thery et al. Citation2006) was present in the EV pellets but not the cell lysates (). Cryo-TEM and Nanoparticle Tracking Analysis were performed to study the shape, number and size distribution of the isolated EV. shows EV with either a single membrane or bi-layer membrane. The EV had a mean size distribution of 166.5 nm (mean) and 97.9 nm (mode) from the Nanoparticle Tracking Analysis ().
Figure 1. Extracellular vesicles (EV) characterization. (A) Purity of isolated EV pellets is shown by the absence of calreticulin (CALR) in EV lysates by Western blot (20 μg protein loaded/lane). (B) EV were enriched in multi-vesicular bodies (MVB) and exosomal markers TSG101 and CD63 as detected by Western blot (10 μg protein loaded/lane). (C) Cryo-TEM images for HEK293T-isolated EV. Bar size = 100 nm. EV with double (white arrow) and single (black arrows) membranes were detected. (D) Particle concentration and size distribution of EV as evaluated using Nanoparticle Tracking Analysis. EV collected from two different batches (EVs #1 and EVs #2) of the same cell line are shown to demonstrate reproducibility.
Lack of cytotoxicity in EV-treated THP-1 and U937 cells
Given the critical role of myelo-monocytic cells in innate and adaptive immune response, a reduction in cell number due to increased necrosis or apoptosis from EV-based therapies could have a negative impact on immune and overall health. Flow cytometry detection of apoptosis in cells was achieved by Annexin V staining of exposed phosphatidylserine (PS), a marker of cell apoptotic process, while staining of nucleic acid with the vital dye 7-AAD revealed cytoplasmic membrane damage indicative of necrosis. Determination of type and stage of cell death was determined as follows: Annexin V+/7-AAD- cells indicated early apoptosis, Annexin V+/7-AAD+ cells indicated late apoptosis and Annexin V−/7-AAD+ cells indicated necrosis.
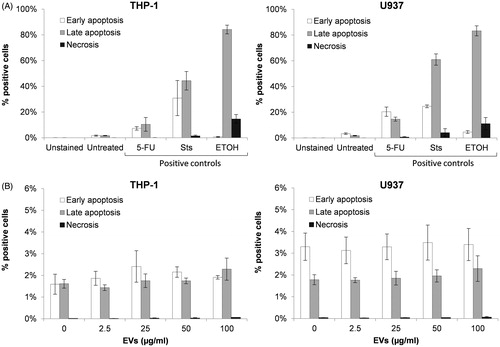
A low fraction of untreated non-differentiated THP-1 and U937 cells exhibited an apoptotic phenotype (), while the percentage of necrotic cells was negligible (). Although both cell lines were of the myelo-monocytic lineage, there were differences between both the percentage and proportion of early vs late apoptosis and in response to chemical agents known to induce early/late apoptosis and necrosis (). Specifically, while both THP-1 and U937 cells had markedly elevated levels of late apoptosis following ethanol treatment, U937 cells had a decrease in the ratio of cells in early vs late apoptosis following exposure to staurosporine (Sts). Exposure to HEK293T-derived EV at doses of 2.5, 25, 50 and 100 μg/ml for 24 h did not induce apoptotic or necrotic changes in THP-1 or U937 (). The EV failed to increase any of the parameters of necrosis, early apoptosis or late apoptosis, regardless of the dosage, although U937 cells had a slightly overall higher percentage of cells in early vs late apoptosis (3.30% in early apoptosis and 1.78% in late apoptosis) vs the similar percentage of early vs late apoptosis (1.59% in early apoptosis, 1.61% in late apoptosis) seen in THP-1 cells. In summary, incubation of THP-1 or U937 cells with HEK293T-derived EV at concentrations between 2.5–100 μg/ml for 24 h did not affect the percentage of cells experiencing apoptosis or necrosis or the proportion of cells undergoing early vs late apoptosis as compared to untreated cells.
Figure 2. Assessment of EV effects on apoptosis and necrosis in THP-1 and U937 cells. Apoptosis and necrosis of untreated or 24 h-treated cells was evaluated by flow cytometry based on the detection of exposed phosphatidylserine (PS) and DNA labeling with the viability stain 7-AAD. (A) Induction of early apoptosis (white bar), late apoptosis (grey bar) and necrosis (black bar) in THP-1 and U937 cells by 5-fluorouracil (5-FU), staurosporine (Sts) and ethanol (ETOH) compared to untreated cells. (B) Detection of early apoptosis, late apoptosis and necrosis in cells exposed to different concentrations of EV compared to untreated cells. Error bars show SEM. Data representative of at least three experiments.
Maintenance of phagocytic efficiency of individual EV-treated THP-1 and U937 cells
The capacity of the non-differentiated monocyte THP-1 and U937 cell lines to phagocytize fluorescently-labeled 1-μm polystyrene microspheres (FluoSpheres) was assessed by flow cytometry (). Cell phagocytic activity of untreated THP-1 and U937 cells was below 12% and significantly increased after stimulation with LPS or PMA (). A cell line-specific phagocytosis capacity was observed, with a higher percentage of untreated U937 cells positive for FluoSpheres (11.46% positive cells) compared to untreated THP-1 cells (8.74% positive cells). Likewise, efficiency of phagocytosis, determined by the mean fluorescence intensity (MFI) of FluoSphere+ cells, was larger in U937 cells (MFI = 14 259) than in THP-1 cells (MFI = 13 138). Interestingly, U937 cells responded less robustly to stimulation with LPS and PMA than THP-1 cells.
Figure 3. Assessment of the capacity of THP-1 and U937 cells to phagocytize fluorescent microspheres. Untreated cells or 21 h-treated cells were exposed to 1 μm FluoSpheres for 3 h. Percentage of positive cells and fluorescence intensity was detected by flow cytometry. (A) Phagocytosis of FluoSpheres by untreated cells or cells stimulated with either lipopolysaccharide (LPS) or phorbol 12-myristate 13-acetate (PMA). (B) Percentage of cells positive for FluoSpheres in the absence of treatment or after exposure to different concentrations of EV. (C) Mean fluorescence intensity (MFI) of cells positive to FluoSpheres in presence or absence of EV at different concentrations. Error bars show SEM. Data representative of at least three experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
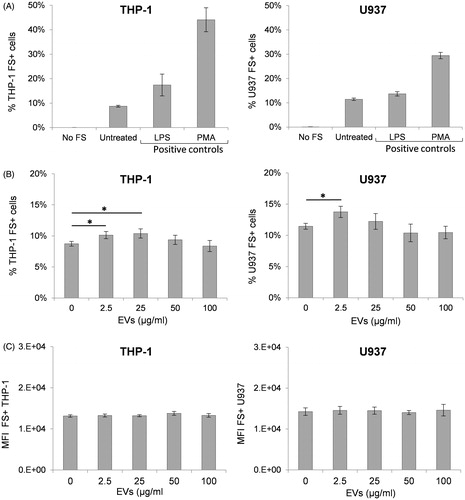
A decreased overall ability to phagocytize foreign particles or a reduction in efficacy of phagocytosis could negatively impact effective immune responses. The effect of the EV on the phagocytic function of THP-1 and U937 cells was evaluated after a 21 h co-incubation of cells with either 2.5, 25, 50 or 100 μg EV/ml. The phagocytic capacity of both THP-1 and U937 cells was not altered following exposure to either 50 or 100 μg/ml of EV (). Interestingly, a small but significant increase in the number of cells positive for FluoSpheres was detected after incubation with lower EV concentrations, i.e. 25 μg/ml in THP-1 cells and 2.5 μg/ml in both THP-1 and U937 cells. However, the MFI of FluoSphere+ cells remained unchanged, regardless of EV dosage (), indicating that EV exposure did not impact the efficiency of FluoSphere phagocytosis in these cell lines.
Internalization of EV by non-differentiated THP-1 and U937 cells was dose-dependent
To further evaluate the results observed in the apoptosis/necrosis and phagocytosis assays, cellular uptake of labeled EV was evaluated by flow cytometry and confocal microscopy techniques. Detection of fluorescently-labeled EV signals by flow cytometry in non-differentiated THP-1 and U937 cells followed a dose-response effect (). The MFI of cells exposed to PKH67-labeled EV increased proportionally to the EV dose (10, 20, 50 and 100 μg/ml) in a linear fashion (R2 = 0.9981 in THP-1 and R2 = 0.9972 in U937 cells) ().
Figure 4. Phagocytosis of labeled EV by THP-1 and U937 cells. Cells were incubated for 16 h with or without PKH67-labeled EV at different concentrations; PKH67 fluorescence was then detected in individual cells by flow cytometry. (A) Overlapped histograms representing the fluorescence signal of cells exposed to different concentrations of EV compared to untreated cells. (B) MFI of cells in (A) (NoTB, white bars) compared to same cell suspension after addition of 0.05% trypan blue (TB, grey bars) or after permeabilization treatment followed by 0.05% trypan blue (Perm + TB, black bars). Error bars show SEM. Data representative of at least three experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
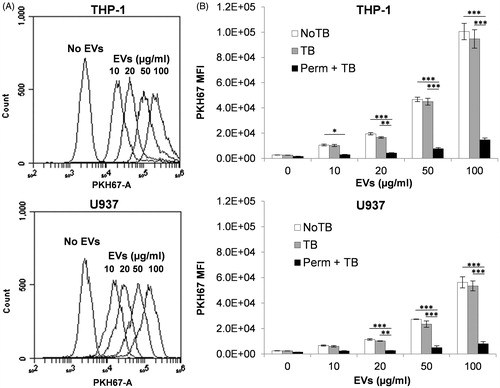
Addition of 0.05% trypan blue (TB) to cell suspensions previous to flow cytometry induced a small but non-significant reduction in PKH67 MFI at all EV doses evaluated (), suggesting the detected fluorescent signal came from internalized EV rather than from EV attached to the outer leaf of the cell membrane. The quenching efficacy of trypan blue on PKH67 fluorescence was demonstrated by induction of a dramatic reduction of intracellular PKH67 fluorescence after cell membrane permeabilization (). Exposure of THP-1 and U937 cells to unlabeled EV neither altered cell morphology, as evaluated by forward and side scatter plots, nor exhibited any detectable fluorescence compared to cells without EV, demonstrating absence of confounding morphological or auto-fluorescence effects (data not shown).
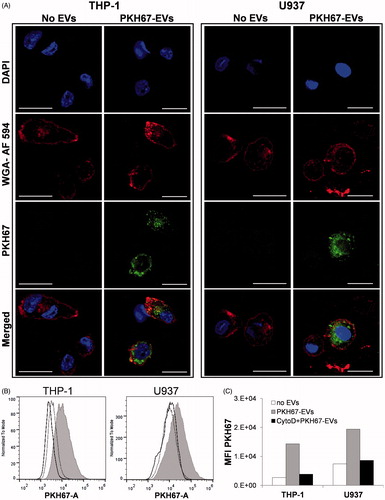
PMA-differentiated THP-1 and U937 cells internalize EV
With the purpose of visualizing intracellular fluorescently-labeled EV by confocal microscopy, THP-1 and U937 cells were differentiated with PMA towards an adherent macrophage-like phenotype, characterized by expanded cytoplasm and increased cytoplasm-to-nucleus ratio. shows confocal microscopy images of intra-cytoplasmic labeled-EV in both cell lines after 16 h incubation with a low dose of 7 μg EV/ml. Cells containing few internalized labeled EV were difficult to capture by confocal microscopy due to fast quenching of the PKH67 signal. Detection of the PKH67 signal in PMA differentiated cells incubated with labeled EV was also assessed by flow cytometry. A shift of the entire cell population suggests an efficient uptake of EV by all the cells in both cell lines (). Treatment of PMA-differentiated THP-1 and U937 cells with CytoD (inhibitor of actin polymerization) dramatically reduced EV internalization, as shown by the fall in PKH67 fluorescence detection to background levels (). Similarly, imaging of CytoD-treated cells by confocal microscopy showed very few labeled EV that were internalized (data not shown).
Figure 5. Phagocytosis of labeled EV by differentiated THP-1 and U937 cells. Adherent, PMA-differentiated cells were incubated for 16 h in the absence or presence of 7 μg PKH67-labeled EV/ml. Uptake of EV was detected by confocal microscopy and flow cytometry. (A) Confocal microscopic images of cells with or without PKH67-EV (green) labeled with the nuclear stain DAPI (blue) and the cell membrane stain WGA-Alexa Fluor 594 (red) (Scale bar = 20 μm). (B) Overlapped histograms comparing the PKH67 signal from cells exposed to 7 μg/ml labeled EV (filled grey), cells without EV (solid line) and cells treated with cytochalasin D (CytoD) prior to incubation with EV (dotted line). (C) MFI of cells represented in (B). PKH67-labeled staining (green) localizes to the cyoplasmic region while the cellular membrane of a mid-cellular section is demonstrated by a linear (red) band of WGA-Alexa Fluor 594 staining.
Discussion
The primary focus of this study was to develop a fast, reliable, reproducible screening assay for evaluating immunotoxicity of EV on non-activated macrophage-precursor cells that can be readily used across laboratories. Studies investigating the immunomodulatory effects of EV have been restricted to endogenous EV produced by immune and non-immune cells in normal homeostasis and in disease (e.g. DC, placental EV, cancer cells, CNS) (Saenz-Cuesta et al. Citation2014; Zhou et al. Citation2014; de Rivero Vaccari et al. Citation2016). In addition, EV were studied from treated, stimulated or tolerized immune cells (e.g. DC, macrophages) or using EV as immunotherapy (Romagnoli et al. Citation2014; Zhang et al. Citation2014; Greening et al. Citation2015). Thus, the present study sought to identify potential effects of human non-immune cell culture-derived EV, developed as potential delivery vehicles of exogenous therapeutic biomolecules, on human monocytic cell lines using flow-cytometry-based in vitro techniques. THP1 and U937 cells, rather than primary monocytes, were selected for the in vitro assay since cell lines are more reproducible than primary cells, have a homogeneous genetic background, lack infectious viruses or toxic products and may be cultured for many passages. The proposed assays are limited to the screening of effects of EV on monocyte/macrophage survival and phagocytic function. Changes in these variables could be indicative of immunotoxic effects associated with inflammation, immunosuppression or immunostimulation. In vitro tests involving other immune cell populations (e.g. T-cells, NK cells, granulocytes) and in vivo studies in appropriate animal models would be required to address other critical immunotoxic effects including hypersensitivity and autoimmunity.
EV from a variety of human cell lines have been investigated as potential delivery vehicles of exogenous therapeutic biomolecules. For the present study, EV were isolated from the HEK293T human embryonic kidney cell line. This line has been used for pharmaceutical and biomedical research purposes for their high transfection efficiency, stable transduction and production of potentially therapeutic EV (Dovrat et al. Citation2014; Lai et al. Citation2014a; Valiunas et al. Citation2015). In addition, HEK cells shed a high number of EV (Yeo et al. Citation2013). Biochemical stimulation of vesicular exocytosis (Dolovcak et al. Citation2009), EV isolation and purification (Jeppesen et al. Citation2014), miRNA content enrichment (Guduric-Fuchs et al. Citation2012) and generation of targeted EV (Ruiss et al. Citation2011; Ohno et al. Citation2013) have been described in HEK293 cells.
Herein, we describe a novel method to reproducibly generate suspension cultures of HEK293T cells in low-serum media that reduced the time, cost and labor needed to isolate sufficient quantities of EV. Characterization of the EV derived from adapted HEK293T cells demonstrated purity and a presence of 50–150 nm EV, as reported in previous studies (Tickner et al. Citation2014). This further supports the use of adapted HEK293T to scale-up EV production and is in agreement with previous studies using adapted HEK293 cells in efficient recombinant protein production (Durocher et al. Citation2002).
Due to their size and membrane composition, EV have a high bioavailability and can interact with multiple cell types. They also have the potential to modulate functions of the immune system including influencing the onset and maintenance of an immunological response (Li et al. Citation2006; Lai et al. Citation2014b). Several studies have demonstrated the potent immunomodulatory effects of EV and related exosomes (reviewed in Robbins and Morelli (Citation2014), Lai et al. (Citation2014b) and Greening et al. (Citation2015)). The route of EV administration can determine the immune cells that will be primarily exposed to EV. We demonstrated here the effect of EV on two human myelo-monocytic (i.e. myeloid leukemia) cell lines, THP-1 and U937, which exhibit a monocyte phenotype and have been widely used and evaluated in cytotoxicity studies (Ferrara et al. Citation2005; Lessig et al. Citation2011; Arimilli et al. Citation2012; Prach et al. Citation2013; Diler et al. Citation2014b; Ramery and O’Brien Citation2014).
Several mechanisms can lead to immunotoxicity generating diverse outcomes: direct cytotoxicity or interference with basic immune cell functionality (Hartung and Corsini Citation2013). Direct cell toxicity by chemicals and biological materials is often identified by assessing the occurrence of the two major models of cell death: apoptosis and necrosis. Identification of the precise mechanisms and stages of cell death are essential to understand further consequences for the immune system, driven by the particular set of molecules exposed and secreted during those specific processes. It has been shown that the recognition and internalization of dying cells by phagocytes can result in an anti- or pro-inflammatory response depending on the type of dying cell (early apoptotic vs late apoptotic/necrotic cell, respectively) (Poon et al. Citation2010). A low percentage of necrotic cells are usually expected under experimental conditions due to the physical stress generated by cell manipulation. Our findings demonstrated the maintenance of homeostatic levels of apoptosis and necrosis in THP-1 and U937 cells after exposure to increasing doses of EV. Verification of internalization of labeled EV by monocyte recipient cells by both flow cytometry and confocal microscopy further supports the lack of direct cytotoxic effects of internalized EV on the monocyte recipient cells evaluated. The flow cytometry method used in this study is a straightforward assay that can be used as an effective early screening tool to determine the display of PS vs cell permeability (i.e. Annexin V vs 7-AAD, respectively).
The major function of classical monocytes is the removal of microorganisms, lipids and apoptotic cells via phagocytosis (Yang et al. Citation2014). Phagocytosis initiates the innate immune response by facilitating the removal and killing of pathogens while priming the adaptive immune response. For that reason, alterations in the phagocytic capacity of monocytes can interfere with the development of innate and adaptive immunity. Interestingly, we found that the exposure to higher levels of EV had no effect on the percentage of synthetic particles (FluoSpheres) phagocytized by the myelo-monocytic cells, while exposure to low EV dosages did result in an increase in the percentage of cells able to phagocytize FluoSpheres – suggesting that EV could increase the functional ability of these myelo-monocytic cells. It is worth noting that the observed differences did not correlate with changes in the phagocytic capacity of individual monocytic cells to internalize labeled particles. Further studies exploring the effect of EV on monocyte functional parameters (e.g. proliferation, maturation, induction of adaptive immune responses, etc.) could unveil subtle immunomodulatory effects that are beyond the scope of these initial immunotoxicity studies.
Several studies investigating the uptake and internalization of EV by recipient cells have suggested the presence of cell-type specific and non-specific uptake. Multiple mechanisms have been described: clathrin-mediated endocytosis, caveolin-dependent endocytosis, phagocytosis, lipid raft-mediated uptake and micropinocytosis (reviewed in Mulcahy et al. Citation2014). The contribution of each of these mechanisms may depend on the molecules exposed on the surface of both the vesicle and the target cell, as well as on the size and heterogeneity of EV. Human erythroleukemia or T-cell leukemia derived exosomes are internalized more efficiently by phagocytic cells that non-phagocytic cells (Feng et al. Citation2010). In another study, bovine milk exosomes were incorporated by differentiated THP-1 cells, but not by undifferentiated THP-1 cells (Izumi et al. Citation2015). Detection of intracellular EV in a permissive cell is dose- and time-dependent, with studies showing a saturation level between 12–14 h of incubation (Feng et al. Citation2010; Tian et al. Citation2014). The current results demonstrated that, after 16 h incubation, HEK293T-derived EV were efficiently internalized by undifferentiated THP-1 and U937 cells in a dose-dependent manner, reaching 100% in cells exposed to doses as low as 20 μg EV/ml. Interestingly, this clear dose-dependent EV internalization did not correlate with any changes in cell survival or in the phagocytic efficiency of labeled microspheres, suggesting a lack of cell reactivity to the presence of intracellular EV. The present study also demonstrated that EV were internalized by PMA-differentiated cells, primarily by an endocytic pathway. This finding supported those from previous studies with EV derived from diverse cell origins (DeFife et al. Citation1999; Morelli et al. Citation2004; Feng et al. Citation2010; Escrevente et al. Citation2011).
A concern of the immunological safety of EV-associated therapy is the potential of using EV from allogeneic or xenogeneic origins. Mammalian cell-derived EV carry a variety of allogeneic proteins, including MHC. To reduce the risk of an immunological reaction, it may be necessary to derive therapeutic EV from autologous sources, i.e. primary cells, or multipotent or induced pluripotent stem cells derived from the patient. Although there is evidence that allogeneic or xenogeneic EV are tolerated and functional in immune-competent animals, the response to repeated injections of non-syngeneic EV has not been investigated (reviewed by Gyorgy et al. Citation2015). Addressing these questions is beyond the scope of the present study.
To our knowledge, this is the first study to evaluate the intrinsic immunomodulatory effects of non-immune, cell culture-derived EV intended to be used as vehicles for therapeutic molecules. Taken together, this study demonstrated that, HEK293T-derived EV are internalized efficiently by the myelomonocytic THP-1 and U937 cell lines in a dose-dependent manner, these EV do not alter the capacity of individual cells to phagocytize synthetic particles and they do not exhibit a cytotoxic effect as assessed by induction of apoptosis and necrosis. The distinctive and consistent response of THP-1 and U937 cells to different treatments and stimuli during our study, supports the use of these well characterized human cell lines for early in vitro screening tests. The flow cytometry-based methods described herein allowed for the evaluation of potential deleterious effects in an unbiased, quantitative and reproducible manner and will contribute to the standardization of EV toxicological studies. Moreover, this systematic approach can be used to evaluate immunotoxicity of other substances and therapeutics on other types of cells at earlier stages of drug development.
Conclusions
This work evaluated the potential toxic effects of HEK293T-derived extracellular vesicles (EV) on the human monomyelocytic cell lines THP-1 and U937. These cells represent a population essential in innate and immune responses. We have demonstrated that, although EV were efficiently internalized by these cells, and that there was no evidence of cytotoxicity or functional alterations due to the EV exposure. This paper expands our understanding of the interactions between EV and cells of the immune system, offers an in vitro method to systematically evaluate immunotoxicity of wild type and engineered EV and, most importantly, presents new evidence that supports the lack of immunotoxicity of EV. Therefore, the current findings support the development of EV for pharmacological and genetic therapies.
Acknowledgments
The authors thank Dr Min Gao at Kent State University TEM lab for capturing TEM images and Brian Kemmenoe at the Campus Microscopy and Imaging Facility (CMIF) at the Ohio State University for technical assistance in confocal microscopy. This work was supported by the National Institutes of Health [3UH2TR000914 to TDS and MAP].
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. 2011. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 29:341–345.
- Arimilli S, Damratoski BE, Bombick B, Borgerding MF, Prasad GL. 2012. Evaluation of cytotoxicity of different tobacco product preparations. Regul Toxicol Pharmacol. 64:350–360.
- Batrakova EV, Kim MS. 2015. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J Contr Rel. 219:396–405.
- Berenson CS, Kruzel RL, Eberhardt E, Sethi S. 2013. Phagocytic dysfunction of human alveolar macrophages and severity of chronic obstructive pulmonary disease. J Infect Dis. 208: 2036–2045.
- Chen TS, Arslan F, Yin Y, Tan SS, Lai RC, Choo AB, Padmanabhan J, Lee CN, de Kleijn DP, Lim SK. 2011. Enabling a robust scalable manufacturing process for therapeutic exosomes through oncogenic immortalization of human ESC-derived MSCs. J Transl Med. 9:47.
- Colombo M, Raposo G, Thery C. 2014. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 30:255–289.
- de Rivero Vaccari JP, Brand F, Adamczak S, Lee SW, Barcena JP, Wang MY, Bullock MR, Dietrich WD, Keane RW. 2016. Exosome-mediated inflammasome signaling after central nervous system injury. J Neurochem. 136(Suppl 1):39–48.
- DeFife KM, Jenney CR, Colton E, Anderson JM. 1999. Disruption of filamentous actin inhibits human macrophage fusion. FASEB J. 13:823–832.
- Diler E, Schicht M, Rabung A, Tschernig T, Meier C, Rausch F, Garreis F, Brauer L, Paulsen F. 2014a. The novel surfactant protein SP-H enhances the phagocytosis efficiency of macrophage-like cell lines U937 and MH-S. BMC Res Notes. 7:851.
- Diler E, Schwarz M, Nickels R, Menger MD, Beisswenger C, Meier C, Tschernig T. 2014b. Influence of external calcium and thapsigargin on the uptake of polystyrene beads by the macrophage-like cell lines U937 and MH-S. BMC Pharmacol Toxicol. 15:16.
- Dolovcak S, Waldrop SL, Fitz JG, Kilic G. 2009. 5-Nitro-2-(3-phenylpropylamino)-benzoic acid (NPPB) stimulates cellular ATP release through exocytosis of ATP-enriched vesicles. J Biol Chem. 284:33894–33903.
- Dovrat S, Caspi M, Zilberberg A, Lahav L, Firsow A, Gur H, Rosin-Arbesfeld R. 2014. 14-3-3 and β-catenin are secreted on extracellular vesicles to activate the oncogenic Wnt pathway. Mol Oncol. 8:894–911.
- Durocher Y, Perret S, Kamen A. 2002. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucl Acids Res. 30:E9.
- El-Andaloussi S, Lee Y, Lakhal-Littleton S, Li J, Seow Y, Gardiner C, Alvarez-Erviti L, Sargent IL, Wood MJ. 2012. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat Protoc. 7:2112–2126.
- Escrevente C, Keller S, Altevogt P, Costa J. 2011. Interaction and uptake of exosomes by ovarian cancer cells. BMC Cancer. 11:108.
- Feng D, Zhao WL, Ye YY, Bai XC, Liu RQ, Chang LF, Zhou Q, Sui SF. 2010. Cellular internalization of exosomes occurs through phagocytosis. Traffic. 11:675–687.
- Ferrara F, Bertelli A, Falchi M. 2005. Evaluation of carnitine, acetylcarnitine and isovaleryl-carnitine on immune function and apoptosis. Drugs Exp Clin Res. 31:109–114.
- Galbiati V, Mitjans M, Corsini E. 2010. Present and future of in vitro immunotoxicology in drug development. J Immunotoxicol. 7:255–267.
- Gao M, Kim YK, Zhang C, Borshch V, Zhou S, Park HS, Jakli A, Lavrentovich OD, Tamba MG, Kohlmeier A, et al. 2014. Direct observation of liquid crystals using cryo-TEM: specimen preparation and low-dose imaging. Microsc Res Tech. 77:754–772.
- Gennari A, Ban M, Braun A, Casati S, Corsini E, Dastych J, Descotes J, Hartung T, Hooghe-Peters R, House R, et al. 2005. The use of in vitro systems for evaluating immunotoxicity: Report and recommendations of an ECVAM Workshop. J Immunotoxicol. 2:61–83.
- Greening DW, Gopal SK, Xu R, Simpson RJ, Chen W. 2015. Exosomes and their roles in immune regulation and cancer. Semin Cell Dev Biol. 40:72–81.
- Guduric-Fuchs J, O'Connor A, Camp B, O'Neill CL, Medina RJ, Simpson DA. 2012. Selective extracellular vesicle-mediated export of an overlapping set of microRNAs from multiple cell types. BMC Genomics. 13:357.
- Gyorgy B, Hung ME, Breakefield XO, Leonard JN. 2015. Therapeutic applications of extracellular vesicles: Clinical promise and open questions. Annu Rev Pharmacol Toxicol. 55:439–464.
- Haney MJ, Klyachko NL, Zhao Y, Gupta R, Plotnikova EG, He Z, Patel T, Piroyan A, Sokolsky M, Kabanov AV, et al. 2015. Exosomes as drug delivery vehicles for Parkinson's disease therapy. J Contr Rel. 207:18–30.
- Hartung T, Corsini E. 2013. Immunotoxicology: Challenges in the 21st Century and in vitro opportunities. ALTEX. 30:411–426.
- Hed J, Hallden G, Johansson SG, Larsson P. 1987. The use of fluorescence quenching in flow cyto-fluorometry to measure the attachment and ingestion phases in phagocytosis in peripheral blood without prior cell separation. J Immunol Meth. 101:119–125.
- ICH (I.C.O.H). 2011. [Internet]. Pre-clinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals. Bethesda, MD: U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER), July 1997. Available from: http://www.ich.org/products/guidelines/safety/safety-single/article/preclinical-safety-evaluation-of-biotechnology-derived-pharmaceuticals.html.
- Izumi H, Tsuda M, Sato Y, Kosaka N, Ochiya T, Iwamoto H, Namba K, Takeda Y. 2015. Bovine milk exosomes contain microRNA and mRNA and are taken up by human macrophages. J Dairy Sci. 5:2920–2933.
- Jeppesen DK, Hvam ML, Primdahl-Bengtson B, Boysen AT, Whitehead B, Dyrskjot L, Orntoft TF, Howard KA, Ostenfeld MS. 2014. Comparative analysis of discrete exosome fractions obtained by differential centrifugation. J Extracell Ves. 3:25011.
- Ji W, Chen X, Ma Y, Hu D, Lu R, Zhou X, Wei L. 2013. Effect of Kruppel-like factor 4 gene silencing on apoptosis and phagocytosis of murine RAW264.7 macrophages. Xi Bao Yu. 29:341–344.
- Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. 1987. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J Biol Chem. 262:9412–9420.
- Lai CP, Mardini O, Ericsson M, Prabhakar S, Maguire CA, Chen JW, Tannous BA, Breakefield XO. 2014a. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano. 8:483–494.
- Lai FW, Lichty BD, Bowdish DM. 2014b. Microvesicles: Ubiquitous contributors to infection and immunity. J Leukocyte Biol. 2:237–245.
- Leclerc L, Boudard D, Pourchez J, Forest V, Sabido O, Bi V, Palle S, Grosseau P, Bernache D, Cottier M. 2010. Quantification of microsized fluorescent particles phagocytosis to a better knowledge of toxicity mechanisms. Inhal Toxicol. 22:1091–1100.
- Lehmann AK, Sornes S, Halstensen A. 2000. Phagocytosis: Measurement by flow cytometry. J Immunol Meth. 243:229–242.
- Lessig J, Neu B, Glander HJ, Arnhold J, Reibetanz U. 2011. Phagocytotic competence of differentiated U937 cells for colloidal drug delivery systems in immune cells. Inflammation. 34:99–110.
- Li, Q, Kobayashi, M, Kawada, T. 2011. Ziram induces apoptosis and necrosis in human immune cells. Arch Toxicol. 85:355–361.
- Li, XB, Zhang ZR, Schluesener HJ, Xu SQ. 2006. Role of exosomes in immune regulation. J Cell Mol Med. 10:364–375.
- Morelli AE, Larregina AT, Shufesky WJ, Sullivan ML, Stolz DB, Papworth GD, Zahorchak AF, Logar AJ, Wang Z, Watkins SC, et al. 2004. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood. 104:3257–3266.
- Mulcahy LA, Pink RC, Carter DR. 2014. Routes and mechanisms of extracellular vesicle uptake. J Extracell Ves. 3:24641–24654.
- Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, Fujita K, Mizutani T, Ohgi T, Ochiya T, et al. 2013. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 21:185–191.
- Parker H, Chitcholtan K, Hampton MB, Keenan JI. 2010. Uptake of Helicobacter pylori outer membrane vesicles by gastric epithelial cells. Infect Immun. 78:5054–5061.
- Poon IK, Hulett MD, Parish CR. 2010. Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ. 17:381–397.
- Prach M, Stone V, Proudfoot L. 2013. Zinc oxide nanoparticles and monocytes: Impact of size, charge and solubility on activation status. Toxicol Appl Pharmacol. 266:19–26.
- Ramery E, O'Brien PJ. 2014. Evaluation of cytotoxicity of organic dust components on THP1 monocytes-derived macrophages using high content analysis. Environ Toxicol. 29:310–319.
- Raposo G, Stoorvogel W. 2013. Extracellular vesicles: Exosomes, microvesicles, and friends. J Cell Biol. 200:373–383.
- Robbins PD, Morelli AE. 2014. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. 14:195–208.
- Romagnoli GG, Zelante BB, Toniolo PA, Migliori IK, Barbuto JA. 2014. Dendritic cell-derived exosomes may be a tool for cancer immunotherapy by converting tumor cells into immunogenic targets. Front Immunol. 5:692.
- Roy MK, Takenaka M, Kobori M, Nakahara K, Isobe S, Tsushida T. 2006. Apoptosis, necrosis. and cell proliferation inhibition by cyclosporine A in U937 cells (a human mono-cytic cell line). Pharmacol Res. 53:293–302.
- Ruiss R, Jochum S, Mocikat R, Hammerschmidt W, Zeidler R. 2011. EBV-gp350 confers B-cell tropism to tailored exosomes and is a neo-antigen in normal and malignant B-cells - a new option for treatment of B-CLL. PLoS One. 6:e25294.
- Saenz-Cuesta M, Osorio-Querejeta I, Otaegui D. 2014. Extracellular vesicles in multiple sclerosis: What are they telling us? Front Cell Neurosci. 8:100.
- Thery C, Amigorena S, Raposo G, Clayton A. 2006. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protocol Cell Biol. Chapter 3:Unit 3.22. DOI: 10.1002/0471143030.cb0322s30.
- Tian T, Zhu YL, Zhou YY, Liang GF, Wang YY, Hu FH, Xiao ZD. 2014. Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery. J Biol Chem. 289:22258–22267.
- Tickner JA, Urquhart AJ, Stephenson SA, Richard DJ, O'Byrne KJ. 2014. Functions and therapeutic roles of exosomes in cancer. Front Oncol. 4:127.
- Valiunas V, Wang HZ, Li L, Gordon C, Valiuniene L, Cohen IS, Brink PR. 2015. A comparison of two cellular delivery mechanisms for small interfering RNA. Physiol Rep. 3:e12286.
- van Deun J, Mestdagh P, Sormunen R, Cocquyt V, Vermaelen K, Vandesompele J, Bracke M, de Wever O, Hendrix A. 2014. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J Extracell Ves. 3:24858–24871.
- Yang J, Zhang L, Yu C, Yang X, Wang H. 2014. Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomarker Res. 2:1.
- Yeo RW, Lai RC, Zhang B, Tan SS, Yin Y, Teh BJ, Lim SK. 2013. Mesenchymal stem cell: An efficient mass producer of exosomes for drug delivery. Adv Drug Deliv Rev. 65:336–341.
- Zhang B, Yin Y, Lai RC, Lim SK. 2014. Immunotherapeutic potential of extracellular vesicles. Front Immunol. 5:518.
- Zhou M, Chen J, Zhou L, Chen W, Ding G, Cao L. 2014. Pancreatic cancer-derived exosomes regulate expression of TLR4 in dendritic cells via miR-203. Cell Immunol. 292:65–69.