
Abstract
Blinded readers examined peripheral smears of 108 children with steady sickle cell (SC) disease and controls by counting ten 100× microscope fields and calculating percent of irreversible and reversible SC from total red cell population SC index (SCI). SCI was correlated to disease severity, and transfusion, hydroxyurea, or neither. Controls had a mean of 0.28% SC (range 0–0.64). Children with hemoglobin SS had a mean SCI of 5.12% ± 5.37 (range 0–30). SCI increased 0.33% with each increasing year (p <0.0001). Patients with SCI > 0.64 were 3.32 times as likely to experience clinical complications (p = 0.0124). Although blood transfusions and hydroxyurea decreased percent of SC, 72% treated patients had SCI >0.64, correlating with persistent sickling. This standardized method quantifies SC in peripheral smears. Percent of SC increased with age and correlated with disease severity, especially hemolytic complications, providing readily available information with minimal or no extra cost.
INTRODUCTION
Although the peripheral blood smear is readily available in the clinic or clinical laboratory, the hematologist or technician does not routinely quantify the number of sickle cells because the utility of such quantification has not been established. Peripheral smears are occasionally reviewed to assess for the presence of sickled forms and macrocytosis in the context of hydroxyurea therapy; it is expected that there will be less sickled forms and red cells will become macrocytic [Citation1].
Our aim was to standardize the count of sickle cell forms in the peripheral smear and to reassess whether such quantification will provide any prognostic benefit. Other methods have been previously used to quantify sickle cells either by morphology [Citation2] or based on density [Citation3].
METHODS
The study was reviewed by the University of Miami Institutional Review Board and approved with waiver of informed consent. The patient population was well characterized and was followed by a single clinician (OA). All consecutive nonselected peripheral smears were obtained at the time of a complete blood count during routine outpatient visits at Jackson Memorial Hospital Pediatric Sickle Cell Clinic during a 4-month period. Peripheral smears were evaluated by a hematopathologist (NM) and a medical technologist with expertise in bone marrow and peripheral blood cell morphology (MM). The readers were blinded to the subjects’ diagnosis (i.e. SS, SC and normal).
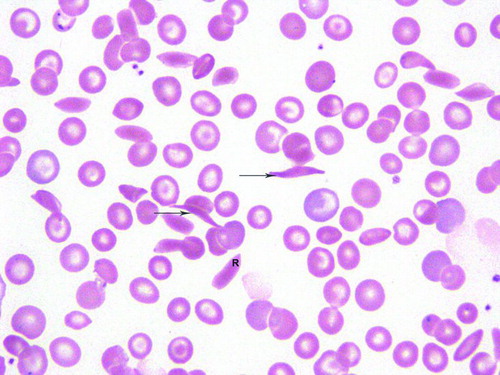
Sickle cell cells (reversible and irreversible) were quantified in ten 100× microscopic fields. The total number of red cells was also recorded per field. Sickled forms were classified as reversible if they were normochromic and the abnormal shape had a holly leaf, splinter, boat, envelope or ellipsoid configuration. Irreversible sickled cells have deformed membranes that have lost the capacity to return to a normal shape even on exposure to oxygen [Citation4]. These hyperchromic red cells have their length twice than the width, and the erythrocytes have sharp angular contours [Citation5], in the shape of a crescent with two elongated and pointed ends. Figure shows the morphology of reversible and irreversible sickle cells. Sickle cell index (SCI) was defined as the mean percentage of the number of reversible and irreversible sickle cells in ten 100× microscopic fields.
Figure 1. Peripheral blood smear depicts a reversible (R) sickled cell and two irreversible cells (arrows). There is anisocytosis and target cells.
In addition, patient demographic information (age, gender, sickle cell genotype, history of splenectomy and sickle cell-related complications) and therapeutic information (use of hydroxyurea, chronic transfusions or no treatment) were recorded. Information and the peripheral smears from three subjects with Hb SS who were on both transfusion and hydroxyurea were excluded from analysis.
Statistical Analysis
Frequencies, means and standard deviations were calculated for the demographic data and SCI. SCI calculations were distributed into quartiles, in general and for specific complications. t tests were performed to detect differences in SCI depending on the history of clinical complications yes/no, age <2 years and ≥2 years, SS, vs. other genotypes, gender and treatments (hydroxyurea, transfusions or neither). SCI was also analyzed with age in years as a continuous variable. Differences were considered statistically significance at an alpha = 0.05.
RESULTS
Eighty one subjects had one peripheral smear reviewed; 25 had two smears examined, 4 had three smears examined and 1 subject had four smears examined during the study period. The total number of slides reviewed was 147 in 111 subjects. The mean age of the subjects was 9.91 ± 6.44 years, range 0.24–21.37 years. Eighty eight (79.3%) had hemoglobin SS, 16 (14.4%) had hemoglobin SC, 4 (3.6%) had sickle beta thalassemia plus, and 3 (2.7%) had sickle beta thalassemia zero. There were 57 (51.4%) males and 54 (48.6%) females.
Sickle Cell Index
Control smears had a mean of 0.28% red cells (range 0–0.64) that were identified as sickle cells. The mean SCI of all subjects was 4.59% ± 5.56 (0–30). The mean SCI for SS group was 5.12% ± 5.37(0–30), all ages included. The subjects with non-SS genotypes had significantly lower SCI by 6.38% compared to subjects with the SS genotype (p < 0.001). Females had 1.6% lower SCI than males, but the difference was not statistically significant (p = 0.0814).
Table presents the data for SCI, age, sickle cell genotypes and treatment received. We observed significant differences between those children less than 2 years of age and older than 2 years of age. Interestingly, SCI increased 0.33% with each increasing year in age (p < 0.0001). Figure presents the linear correlation between age and SCI in the patients with SS who were not receiving transfusions or hydroxyurea. For patients with more than 1 smear, there were no statistically significant differences in the sickle cell indices between the peripheral smears. (Mean difference = –1.02; p = 0.2383.)
Table 1. Sickle Index per Age Category, Genotype, and Treatment.
Figure 2. The plot demonstrates a gradual increase in sickle cell index (SCI) as patient's age progresses.
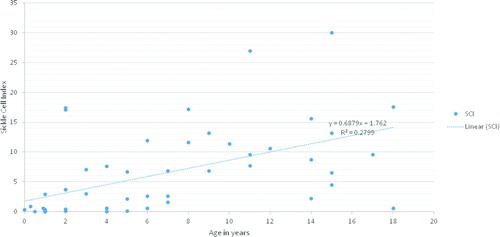
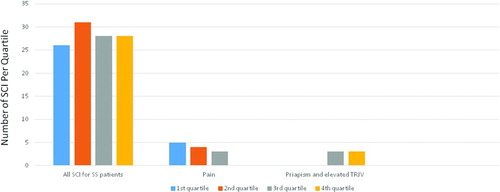
Figure 3. Quartile distributions for SCI for all SS patients 2 years and older. TRJV: tricuspid valve jet velocity.
There were differences in SCI among the patients who were on transfusions, on hydroxyurea, or on neither therapy. In the presence of sex, age and genotype (SS vs. non-SS), the transfusion only group (32 subjects) had a SCI that was 5.25% lower than the untreated (61 subjects) who were not receiving transfusion or hydroxyurea, p < 0.0001. The hydroxyurea group (15 subjects) had a SCI 4.68% lower than the untreated group (p = 0.0026). Although blood transfusions and hydroxyurea decreased the percent of sickle cells, 72% patients on those treatments had SCI higher than controls (>0.64), correlating with persistent sickling. There were no statistically significant inverse correlations between the SCI and hemoglobin A and hemoglobin F within the transfusion and the hydroxyurea group respectively or in general, probably due to sample size.
The significant complications for this patient population were: stroke (N = 10 patients), abnormal transcranial Doppler (CitationN = 12), cerebral vasculopathy or abnormal MRA [Citation5], recurrent pain [Citation12], albuminuria, proteinuria, or chronic renal failure [Citation6], asthma or history of acute chest syndrome [Citation6], history of increased tricuspid regurgitation velocity [Citation3], history of priapism [Citation3], splenic sequestration [Citation2], other significant history [Citation4] and no significant sickle cell complications (51 patients). Of the 78 patients who had SCI > 0.64, 59% had a complication. Additionally, patients with a SCI in excess of 0.64 were 3.32 times as likely to experience clinical complications (p = 0.0124; 95% CI for the Odds Ratio 1.26–9.36). We did not detect differences between the mean SCI from patients with Hb SS 2 years and older with or without complications. However, none of the patients without history of complications were on hydroxyurea or chronic transfusions while 80% of patients with past history of complications were on those treatments at the time of the assessments, potentially confounding the values. When we looked at specific complications, we observed that the patients with elevated tricuspid regurgitation velocity and history of priapism had SCI > 3.8 (third and fourth quartiles), while none of the patients with recurrent pain were in the upper quartile (Figure ).
DISCUSSION
The expected hematological parameters such as hemoglobin, reticulocyte count, white cell counts for children with sickle cell anemia have been previously described [Citation6]. Sickle cells may constitute about 5–50% of the total number of red cells in the peripheral smear [Citation2]. The literature suggested that there is a decrease in the number of sickle cells during a painful episode [Citation7–9]. Other investigators did not find an association between the number of sickle cells and clinical characteristics or differences between the steady-state and during the acute pain episode [Citation10–12]. We decided to rexamine this finding in the current work for patients at steady state. Contrary to most of the previous reports, we counted both reversible and irreversible sickled forms.
We found that a standard method for the quantification of sickle cells was feasible in the clinical practice as performed by a hematopathologist and technologist. The mean number of sickle cells, both reversible and irreversible present in ten 100× microscope field as the numerator over the total number of red cells × 100 was calculated (SCI). Children younger than 2 years of age and non-SS children had SCI usually less than 1 probably related to less hemoglobin S polymerization. Patients with Hb SS less than 2 years of age had low number of sickle cells comparable to controls without sickle cell disease. SCI increased by 0.33% with each year of age. Possible reasons for the increased sickling could be erythroid in origin (i.e. lower Hb F, increasingly defective membrane), vascular in origin (progressive vasculopathy), or decreased clearance from circulation due to impaired spleen function. We do not have data on pitted cells or Howell Jolly body quantification. However, data from the Pediatric Hydroxyurea in Sickle Cell Anemia BABY HUG Trial showed that the mean number of Howell Jolly bodies and pitted cells were higher at age 3 compared with age 1 even on patients who were receiving hydroxyurea [Citation13].
We found an incomplete treatment effect from hydroxyurea and blood transfusions because sickle cell forms were still increased in number despite these treatments. However, we do not have data comparing pre- or posttreatment smears; therefore, specific changes due to hydroxyurea or transfusions could not be fully examined. Similarly, we evaluated the smears at steady state; thus, changes during an acute pain episode could not be ascertained. For those few patients who had several smears, the number of sickle cell forms per patient was consistent. Therefore, a single patient assessment may be informative.
This study was limited because we did not know the pretreatment SCI in many patients with history of complications who were already receiving therapy (either transfusions or hydroxyurea). Certainly, the validity of SCI should be confirmed with a larger sample size, preferably in a prospective manner. However, the quantification of sickle cells is simple and may be informative to the treating physician to assess treatment response and to detect those patients with high rate of hemolysis. In particular, an SCI > 3.8 was seen in those patients with the hemolytic phenotype [Citation14] associated with elevated tricuspid regurgitation jet velocity and priapism. Furthermore, even children with SCI > 0.64 were 3.32 times more likely to experience sickle-cell related complications. The use of automated quantification of red cell morphology may be helpful in the future.
Declaration of Interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
REFERENCES
- Charache S, Barton FB, Moore RD, et al. Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive “switching” agent. The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine (Baltimore) 1996;75:300–302.
- Rodgers GP, Noguchi CT, Schechter AN. Irreversible sickled erythrocytes in sickle cell anemia: a quantitative reappraisal. Am J Hematol 1985;20:17–23.
- Clark MR, Mohandas N, Embury SH, Lubin BH. A simple laboratory alterative to irreversible sickled cells (ISC) counts. Blood 1982;60:659–662.
- Serjeant GR, Serjeant BE. Sickle Cell Disease. 3rd edn. Oxford University Press, 2001.
- Jensen WN, Rucknage DL, Taylor WJ. In vivo study of the sickle cell phenomenon. J Clin Med 1960;56:854–865.
- Brown AK, Sleeper LA, Miller ST, et al. Reference values and hematologic changes from birth to 5 years in patients with sickle cell disease. Cooperative Study of Sickle Cell Disease. Arch Pediatr Adolesc Med 1994;148:796–804.
- Barreras L, Diggs LW. Bicarbonates, pH, and percentage of sickle cells in venous blood in patients with sickle cell crisis. Am J Med Sci 1964;247:710–718.
- Zipursky A, Chachula DM, Brown EJ. The reversibly sickled cell. Am J Pediatr Hematol Oncol 1993;15:219–225.
- Asakura T, Asakura K, Obata K, et al. Blood samples collected under venous pressure from patients with sickle cell disease contain a significant number of a new type of reversibly sickled cells: Constancy of the percentage of sickled cells in individual patients during steady state. Am J Hematol 2005;80:249–256.
- Smith CM 2nd, Krivit W, White JG. The irreversibly sickled cell. Am J Pediatr Hematol Oncol 1982;4:307–315.
- Steinberg MH, Dreiling BJ, Lovell WJ. Sickle cell anemia: Erythrokinetics, blood volumes, and a study of possible determinants of severity. Am J Hematol 1977;2:17–23.
- Hebbel RB, Boogarerts MAB, Eaton JW, Steinberg MH. Erythrocyte adherence to endothelium in sickle cell anemia. N Engl J Med 1980;302:992–995.
- Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 2011;377:1663–1672.
- Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev 2007;21:37–47.