
Abstract
Congenital nephrotic syndrome (CNS) caused by a mutation in the Wilms tumor 1 suppressor gene (WT1) is part of Denys Drash Syndrome or Frasier syndrome. In the framework of genetic counseling, the diagnosis of CNS can be refined with gene mutation studies on long-term stored formalin-fixed paraffin-embedded tissue from postmortem examination. We report a case of diffuse mesangial sclerosis with perinatal death caused by a de novo mutation in the WT1 gene in a girl with an XY-genotype. This is the first case of Denys Drash Syndrome with the uncommon missense c.1097G>A [p.(Arg366His)] mutation in the WT1 gene which has been diagnosed on long-term stored formalin-fixed paraffin-embedded tissue in 1993. This emphasizes the importance of retained and adequately stored tissue as a resource in the ongoing medical care and counseling.
Introduction
Congenital nephrotic syndrome (CNS) has been arbitrarily defined as the occurrence of nephrotic syndrome in patients less than 3 months of age. Later onset, in between 3 months to 1 year, is defined as infantile nephrotic syndrome. In the past two decades, the molecular genetic bases of several conditions that may cause CNS have been identified. Most often CNS is caused by recessive genes (NPHS1, NPHS2, LAMB2, and others) and is associated with early onset of disease, often beginning during fetal development (; [Citation1–3]). In contrast, dominant gene mutations usually lead to later onset, with the exception of mutations in the Wilms tumor 1 suppressor gene (WT1; ). Mutations in the NPHS1 gene, coding for nephrin (), lead to the Finnish-type of CNS; a recessively inherited disorder characterized by massive proteinuria detectable at birth, a large placenta, marked edema, and characteristic radial dilatations of the proximal tubules. The NPHS2 gene codes for podocin () and causes familial focal segmental glomerulosclerosis [Citation1, 2, Citation4]. In addition, the LAMB2 gene, coding for laminin beta 2 (), is reported as the cause for Pierson syndrome [Citation5, 6]. Less than 3% of cases with CNS are caused by mutations in the WT1 gene (; [Citation1, Citation7]), which is responsible for Denys Drash Syndrome (DDS) or Frasier syndrome. Frasier syndrome, first described in 1964, presents at birth with male pseudohermaphroditism, streak gonads, a 46 XY-genotype, progressive glomerulopathy and high risk of gonadoblastoma development [Citation8]. DDS, first described in 1970, is a heterogeneous disorder of the urogenital system with the triad of CNS, structural urogenital anomalies with pseudohermaphroditism and a risk of Wilms tumor development [Citation9]. These components are not uniformly present in all patients. The WT1 gene has been recognized in 1990 and is located at chromosome 11p13 [Citation10, 11]. It spans 10 exons and encodes a zinc-finger-transcription factor presumed to regulate the expression of numerous target genes through DNA binding (). The so called hot spot mutation causing DDS, with 40% of cases, replaces the amino acid arginine with the amino acid tryptophan at protein position 394 (p.(Arg394Trp); [Citation12]).
Figure 1. The majority of CNS in the first three month of life is caused by the following genes: The NPHS1 gene codes for nephrin and is responsible for the Finnish type CNS; the NPHS2 gene codes for podocin and leads to familial focal segmental sclerosis. Less common is a WT1 gene mutation causing DDS or Frasier syndrome. The LAMB2 gene is also associated with CNS and leads to Pierson syndrome. Other infrequent mutations in PLCE1, LMX1B (responsible for Nail–Patella syndrome) and LAMB3 have also been described. A small part is attributed to a nongenetic etiology with CNS due to congenital syphilis, congenital toxoplasmosis, congenital CMV infection or neonatal autoantibodies against endopeptidase.
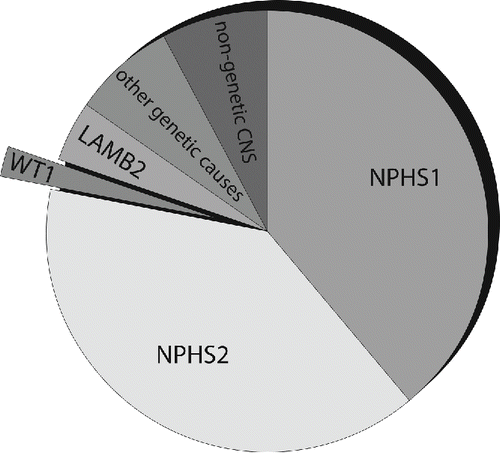
Figure 2. Location of the proteins responsible for the majority of CNS: Nephrin (NPHS1 gene) is a transmembrane protein of the immunoglobulin superfamily and interacts through its C-terminal part with podocin (NPHS2 gene), a harpin-like scaffolding protein both localized in the slit diaphragm (SD) in between the podocytic processes. WT1 encodes a zinc-finger transcription factor that plays a key role during kidney and genital development. Laminin beta 2 (LAMB2 gene) is a component of the glomerular basement membrane (GBM).
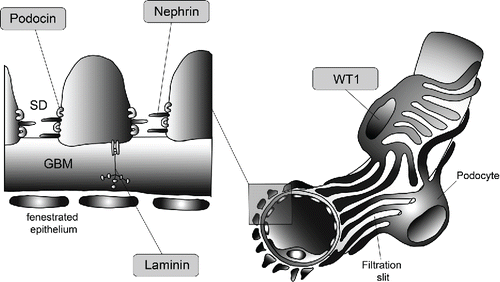
We report about a patient with an uncommon missense mutation c.1097G>A (p.(Arg366His); OMIM 607102.0004) in the WT1 gene, which could be detected on 21-year-old paraffin embedded tissue.
Clinical report
The patient was the second child of healthy, nonconsanguineous parents, and was born in 1993. Cardiotocograph decelerations and fetal distress required an emergency Caesarean section at 36 + 5/7 weeks of gestation. The child presented with severe cardiorespiratory distress leading to postpartum death. Laboratory investigations indicated an umbilical cord pH 6.66 and serum hypoalbuminemia (<1.0 gm/dL). Birth weight was 2570 g (90th centile), crown-heel length was 46 cm (90th centile), foot length 7 cm (90th centile) and head circumference 33.0 cm (>95th centile). The cranial sutures were widely separated. The child had phenotypic normal external female genitalia and cyanotic acra. There were no external dysmorphic features.
Postmortem X-ray imaging showed a left pneumothorax, fracture of the 6th left rib and signs of previous cardiac puncture (status after cardiopulmonary resuscitation and adrenaline injection). At autopsy, symmetrically enlarged hyperlobulated kidneys with macroscopically small cystic change at the corticomedullary transition were observed (38.6 g, normal combined weight for age 25.4 g, a). There was an uterus didelphys with duplicated cervix and a single vagina (b). The brain was enlarged (380 g, normal weight for age 315 g) with a regular development. There were subarachnoidal bleedings and congestive leptomeningeal veins. The lungs showed sporadic pleural point bleedings and signs of meconium aspiration. Ureteric dilatation, signs of hydronephrosis or signs pointing to oligohydramnios were not reported, thereby making obstructive changes unlikely. Macroscopic abnormalities of gonads, heart or diaphragm were not reported. A histological work up from the gonads was not performed.
Figure 3. Histological, macroscopic, and electron microscopy observations from postmortem examination. a: Enlarged hyperlobulated kidney with macroscopically small cystic change at the corticomedullar transition zone. b: Uterus didelphys (**) with duplicated cervix and a single vagina. Normal development of rectum (*). c: Histology of the kidneys with glomeruli showing diffuse mesangial sclerosis and hypercellularity. There is marked interstitial fibrosis and tubular atrophy with formation of microcystic tubule dilatations and a lymphocytic inflammation (H.E. staining). d: Electron microscopy of the kidney shows extensive effacement of podocytes with fatty vacuoles.
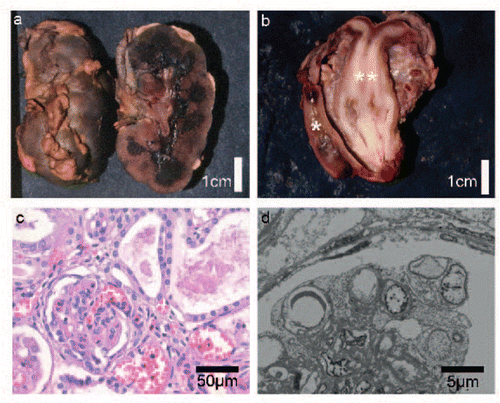
Microscopic examination of the kidneys showed glomeruli of varying size and of different developmental stage. Some glomeruli were markedly shrunken while others were enlarged. The glomeruli showed diffuse mesangial proliferation with capillary collapse and focal segmental glomerulosclerosis (FSGS), with <5% of glomeruli with global sclerosis (c). Silver stain preparations revealed mesangial interposition and double basement membranes. Marked interstitial fibrosis and tubular atrophy was present with formation of microcystic tubule dilatations and mononuclear lymphocytic inflammation. Extensive effacement of podocytes with fatty vacuoles was evident on electron microscopy (d). The placenta was enlarged (615 g, >90th centile) with hydropic edematous change. Microscopic examination showed discrete intervillous thrombosis and signs of chorionitis; erythroblastosis was not noted.
In line with the premature birth, the fetal distress, the enlarged kidneys, the hypoalbuminemia, the enlarged hydropic placenta, and the microscopically seen diffuse mesangial glomerular sclerosis with extensive podocytic effacement, the diagnosis of CNS was made. Genetic analysis was not performed at that moment.
Twenty years later, in 2013, the older sister (born in 1987) presented at our clinic for genetic counseling to be informed about a possible recurrence risk. The parents and the older sister showed no mutations in the major causative genes responsible for CNS. In the following, it was possible to isolate DNA of sufficient quality for ongoing genetic study from the 21-year-old formalin-fixed paraffin-embedded tissue that had been preserved from the autopsy. Cell-rich tissue from lymph node and thymus was selected for genomic DNA extraction. Fluorescent in situ hybridization (FISH) analysis demonstrated an XY-karyotype revealing an intersex-child, which is a typical finding in patients with DDS. With sequencing analysis a heterozygous missense mutation in exon 8 of the WT1 gene a 1097G > A mutation was detected, which had resulted in replacement of arginine at amino acid 366 (p.(Arg366His)) in the WT1 protein (for overview of reported cases see [Citation13-Citation23]). The WT1 mutation was absent in both parents and in the healthy sister of the patient. Taken together, the CNS was caused by a de novo missense mutation in the WT1 gene. The older sister could be reassured about the recurrence risk, which is not increased. The diagnosis from 1993 was refined to CNS caused by DDS with an unusual de novo WT1 mutation as the underlying cause.
Table 1. Cases reported with c.1097G>A (p.(Arg366His)) mutation in the WT1 gene.
Discussion
We report a deceased newborn with female phenotype, hypoalbuminemia, a duplicated uterus and cervix, an enlarged placenta and diffuse mesangial glomerular sclerosis at postmortem examination leading to the diagnosis of CNS. A de novo WT1 mutation and a XY-genotype could be documented on the long term stored formalin-fixed paraffin embedded tissue. So, the WT1 mutation confirmed the diagnosis of DDS.
From a clinical point of view, there are several situations to consider DDS in a patient. In the prenatal setting it should be considered in the ultrasound diagnosis of a diaphragmatic hernia or signs of lung hypoplasia, especially if there is a 46 XY karyotype in a female phenotype at ultrasound. However, our case showed neither a diaphragm hernia [Citation13–15] nor signs of lung hypoplasia [Citation16], which has earlier been suggested to be associated with this uncommon mutation in the WT1 gene (see ).
WT1 mutations causing DDS or Frasier syndrome cover a broad age range with varying onset of symptoms and wide variability in clinical expression. The spectrum of early onset nephrotic syndrome and a possible association with distinct WT1 mutations has been investigated in earlier studies, but to date a clear-cut relation between the type of mutation and the clinical phenotype of DDS has not been found [Citation7, Citation12].
In our patient the infrequent c.1097G>A (p.(Arg366His)) WT1 mutation was documented. Of the cases which have—as far as we are aware of—been reported in the literature, 12 showed ante- or perinatal onset of CNS (see ). In the remaining two cases, onset of symptoms was not reported [Citation12, Citation17]. In 8 cases reported, death occurred at birth or up to 1 year of age. Follow up was reported in 1 case until the age of 26 months [Citation14]. In 1 patient, a gonadoblastoma was found [Citation17] and in 2 other individuals a unilateral Wilms tumor was documented [Citation12, Citation18]. Median age of Wilms tumor onset has been reported as 12.5 months and without WT1 gene alteration as 36 months [Citation12]. The fact that over half of the reported patients carrying the c.1097G>A(p.(Arg366His)) WT1 mutation died within the first year explains the relatively rare association of this mutation with Wilms tumor.
In 11 of the 14 patients, abnormal internal genital organs were reported. In two cases, there was no report about abnormalities in genital organs [Citation12, Citation19]. The remaining case showed a 46 XX karyotype and normal gonadal development [Citation20]. Our patient showed abnormal internal genital development with a uterus didelphys and a duplicated cervix. Macroscopic abnormalities of the gonads were not reported. However, further histological work up was not performed in 1993 and therefore the presence of gonadal dysgenesis, hypoplasia, or streak gonads remains indistinct in our case.
Although formalin-fixed paraffin-embedded tissue has great diagnostic morphological value, most often its use is limited for ongoing molecular study. Cross-linking and fragmentation of nucleic acids, loss of enzymatic activity, and insufficient nucleic acid concentrations represent major problems. We show that even after long-term storage for 21 years, it was technically possible to preserve and retrieve DNA of sufficient quality for molecular studies from formalin-fixed paraffin embedded tissue.
In conclusion, this case underlines the importance of adequate long-term tissue storage after initial diagnosis in a surgical pathology department as part of the patient's clinical record [Citation21]. Especially, in the field of fetal and pediatric pathology preservation and storage of tissue may benefit other family members in the future in ways not conceived at the time that the tissues were originally obtained. Although economic and spatial factors of pathology archives remain a persistently discussed issue, they are an ultimate necessity by providing a unique source for applying new diagnostic possibilities for descendants. Last but not least, pathology archives constitute a unique biobank for research purpose and retrospective studies.
Acknowledgments
We thank the parents and the older sister of the reported deceased patient for their consent.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
References
- Hinkes BG, et al. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics 2007;119(4):e907–e919.
- Hildebrandt F, Heeringa SF. Specific podocin mutations determine age of onset of nephrotic syndrome all the way into adult life. Kidney Int 2009;75(7):669–671.
- Trautmann A, et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol 2015;10(4):592–600.
- Boute N, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 2000;24(4):349–354.
- Hasselbacher K, et al. Recessive missense mutations in LAMB2 expand the clinical spectrum of LAMB2-associated disorders. Kidney Int 2006;70(6):1008–1012.
- Zenker M, et al. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet 2004;13(21):2625–2632.
- Schumacher V, et al. Spectrum of early onset nephrotic syndrome associated with WT1 missense mutations. Kidney Int 1998;53(6):1594–1600.
- Frasier SD, Bashore RA, Mosier HD. Gonadoblastoma associated with pure gonadal dysgenesis in monozygous twins. J Pediatr 1964;64:740–745.
- Drash A, et al. A syndrome of pseudohermaphroditism, Wilms’ tumor, hypertension, and degenerative renal disease. J Pediatr 1970;76(4):585–893.
- Call KM, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 1990;60(3):509–520.
- Gessler M, et al. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature 1990;343(6260):774–778.
- Royer-Pokora B, et al. Twenty-four new cases of WT1 germline mutations and review of the literature: genotype/phenotype correlations for Wilms tumor development. Am J Med Genet A 2004;127A(3):249–257.
- Devriendt K, et al. Diaphragmatic hernia in Denys-Drash syndrome. Am J Med Genet 1995;57(1):97–101.
- Cho HY, et al. Hydrothorax in a patient with Denys–Drash syndrome associated with a diaphragmatic defect. Pediatr Nephrol 2006;21(12):1909–1912.
- Antonius T, et al. Denys–Drash syndrome and congenital diaphragmatic hernia: another case with the 1097G > A(Arg366His) mutation. Am J Med Genet A, 2008;146A(4):496–499.
- Dharnidharka VR, et al. Pulmonary dysplasia, Denys–Drash syndrome and Wilms tumor 1 gene mutation in twins. Pediatr Nephrol 2001;16(3):227–231.
- Pelletier J, et al. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys–Drash syndrome. Cell 1991;67(2):437–447.
- Baird PN, et al. Constitutional mutations in the WT1 gene in patients with Denys–Drash syndrome. Hum Mol Genet 1992;1(5):301–305.
- Lin HC, et al. Denys–Drash syndrome. J Formos Med Assoc 2004;103(1):71–74.
- Hahn H, et al. Two cases of isolated diffuse mesangial sclerosis with WT1 mutations. J Korean Med Sci 2006;21(1):160–164.
- Bevilacqua G, et al. The role of the pathologist in tissue banking: European Consensus Expert Group Report. Virchows Arch 2010;456(4):449–454.
- Jeanpierre C, et al. Identification of constitutional WT1 mutations, in patients with isolated diffuse mesangial sclerosis, and analysis of genotype/phenotype correlations by use of a computerized mutation database. Am J Hum Genet 1998;62(4):824–833.
- Suri M, et al. WT1 mutations in Meacham syndrome suggest a coelomic mesothelial origin of the cardiac and diaphragmatic malformations. Am J Med Genet A 2007;143A(19):2312–2320.