Abstract
Background Multiple epiphyseal dysplasia (MED) is a common genetically and clinically heterogeneous skeletal dysplasia characterized by early-onset osteoarthritis, mainly in the hip and knee, and mild-to-moderate short stature. Here we report on a 6-generation MED family with 17 affected members.
Method The clinical and radiographic data on the 12 affected members still living were scrutinized. A structured inquiry comprising state of health and MED-related symptoms since birth up to the present time and the osteoarthritis outcome (KOOS) questionnaire were sent to all living family members with MED. The 5 known gene loci for autosomal dominant MED were analyzed for linkage, using fluorescence-labeled microsatellite markers. Linkage was ascertained with markers close to the COL9A2 gene, which was analyzed for mutations by sequencing.
Results We identified an exon 3 donor splice mutation in the COL9A2 gene in all affected family members. Clinical, radiographic, and questionnaire data from affected family members suggested that MED caused by COL9A2 mutations starts in early childhood with knee pain accompanied by delayed ossification of femoral epiphyses. The disease then either stabilizes during puberty or progresses with additional joints becoming affected; joint surgery might be necessary. The progression of the disease also affects muscles, with increasing atrophy, resulting in muscle fatigue and pain. Muscular atrophy has not been reported earlier in cases with COL9A2 mutations.
Interpretation In a patient with clinically suspected or verified MED, it is important to perform DNA-based analysis to identify a possible disease-causing mutation. This information can be used to carry out genetic risk assessment of other family members and to achieve an early and correct diagnosis in the children.
Multiple epiphyseal dysplasia (MED) is an autosomal dominant skeletal dysplasia that affects approximately 1 in 10,000 individuals. It was first described in 1937, by the Swedish radiologist Ribbing. The predominant features of the disease are delayed and irregular ossification of epiphyses and early onset of osteoarthritis. The MED symptoms appear during early childhood, usually with pain in the knees after exercise. Children who are affected often have difficulty in getting up from the floor and have a waddling gait. They frequently complain of fatigue during long walking. Adult height is usually in the lower range of normal. The limbs are relatively short in comparison to the trunk. Pain and joint deformities progress with age, resulting in early-onset osteoarthritis, particularly of the large weight-bearing joints. The spine is usually normal (Briggs and Chapman Citation2002, Jakkula et al. Citation2005, Zanki et al. Citation2007). The diagnosis of the dominant form of MED is based on physical examination and radiographic findings in the proband and other family members (Lachman et al. Citation2005).
In many cases, the disease-causing gene mutation is known; mutations in 5 different genes have been identified to cause dominant MED. These genes are COMP, which encodes cartilage oligomeric matrix protein; COL9A1, COL9A2, and COL9A3, which code for the 3 alpha chains of cartilage-specific type IX collagen; and MATN3, which encodes matrilin-3, a cartilage extracellular matrix protein. COMP is mutated in 80% of all MED samples analyzed. In approximately 10–20% of all samples tested, a mutation cannot be identified in any of the 5 known genes mentioned above, suggesting that mutations in other hitherto unidentified genes are also involved in the pathogenesis of dominant MED (Briggs and Chapman Citation2002, Zanki et al. Citation2007). There is also a rare autosomal recessive form of MED that is caused by mutations in a sulfate transporter gene (DTDST). This form of MED is characterized by malformations of the hands, feet, and knees, with a double-layered patella and scoliosis (Ballhausen et al. Citation2003).
We present a clinical and genetic study of a 6-generation family with autosomal dominant MED. The individuals affected were of different ages and some of them had had MED symptoms for almost 90 years. The aim of the study was to investigate the symptoms of the affected family members, to analyze the development of the disease over time, and to identify the mutation that causes the disease in this family.
Material and methods
Family material and clinical investigations
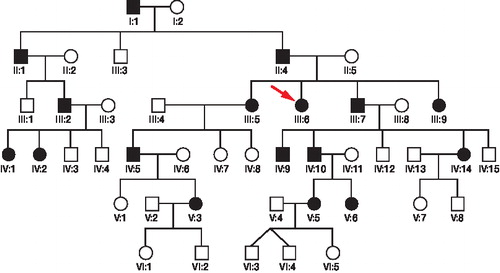
In the present 6-generation MED family, 17 members were or had been affected (). The hospital records with clinical and radiographic data concerning the 12 affected members who were still living were scrutinized. 12 cases in 3 generations of the family have been examined earlier clinically and radiographically (Ödman Citation1959). A structured inquiry covering the state of health and MED-related symptoms from birth up to the present time and also the Swedish version of knee injury and osteoarthritis outcome (KOOS) questionnaire (CitationRoos et al. 2003) were sent to all living family members with MED.
Figure 1. Pedigree of the 6-generation family showing the 17 members affected by multiple epiphyseal dysplasia. Black symbols represent affected individuals. An arrow indicates the proband.
Genotyping of candidate genes and mutation analysis
Blood samples were collected from 24 family members (12 affected, 9 healthy, and 3 whose disease state was unknown) after obtaining informed consent. Genomic DNA was extracted from peripheral lymphocytes according to standard procedures. The 5 known gene loci (COMP, MATN3, COL9A1, COL9A2, COL9A3) for autosomal dominant MED were genotyped using fluorescence-labeled microsatellite markers and 25 ng of DNA per PCR reaction, as described previously (Klar et al. Citation2004). Genotypes were analyzed with PeakScanner v.1.0 (Applied Biosystems), haplotypes were drawn with Cyrillic v.2.1.3, and 2-point logarithms of odds (LOD) scores were calculated using MLINK (from the LINKAGE package v.5.1) (Lathrop and Laouel Citation1984). The COL9A2 gene was analyzed for mutations by DNA sequencing of exon 3 and intron 3, using the BigDye Terminator v3.1 Cycle Sequencing Kit (Invitrogen) and an ABI PRISM 3730 DNA analyser (Applied Biosystems). The DNA sequences obtained were analyzed with Sequencher v.4.1.2 (Gene Codes Corporation, Ann Arbor, MI).
Results
Clinical findings
The presenting symptoms of the 12 living family members with MED were pain in the knees and fatigue during long walks at 2–7 years of age (). The main radiographic findings were delayed ossification of the epiphyses of the hips and knees, irregular bone structures, and irregular jagged contours of the distal femoral epiphyses (). Most of the individuals affected had relatively mild manifestations during early adulthood (), but at 35–40 years of age many suffered from increasing pain in the knees and hips, resulting in functional disability. Restricted movements of the elbows and knees were common. Degenerative osteoarthritis and sometimes osteochondritis dissecans () was seen on radiographs. A few cases had undergone surgical removal of loose cartilaginous fragments from the knees. In a few cases, total hip replacement was required. After the age of 40 many had developed a scoliosis. All the affected family members had had slim muscles since birth, and after the age of 35–40 slight-to-moderate muscular atrophy was common. In one of the patients, severe muscular atrophy after the age of 80 was seen. The adult heights of the MED patients were in the lower normal range or slightly shorter. The hands and feet were small.
Clinical findings in the MED patients.
Figure 2. Subject IV:10. Left knee at age 12. The figure shows typical findings with irregular ossification of the epiphyses and delayed ossification of the patella.
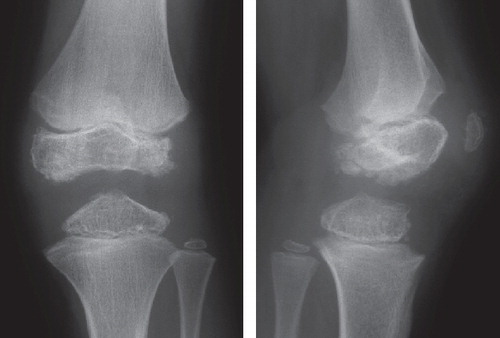
Figure 3. Subject IV:2. Left knee at age 15. The femoral epiphyses are flattened, especially the medial one.
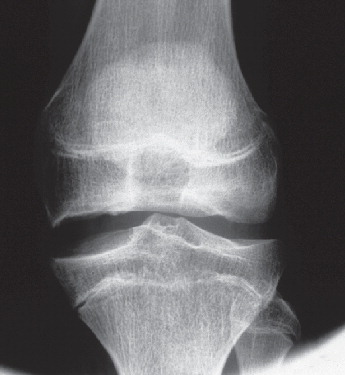
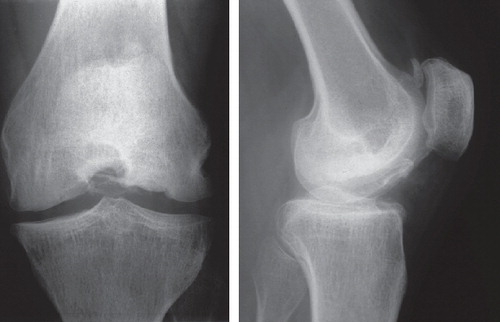
Figure 4. Subject III:6. Left knee at age 40. The femoral condyles are flattened and there is obvious osteoarthritis. A separate bone fragment dorsal to the patella is seen in the lateral view.
The disorder varied strikingly within the family. For example, a 95-year-old male patient (III:2) had a relatively mild form of the disease with only slight pain in the knees during early childhood. He was then free of symptoms until the age of 27, after which he had a few episodes of pain in the knees caused by osteochondritis dissecans. No surgery was needed. By the age of 60 he suffered from slight pain in the hips, and this pain began to increase at the age of 88, requiring bilateral hip replacement when he was 93. In contrast, a 93-year-old female patient (III:6) had a severe form of MED. From the age of 4, she had pain in the knees when weight bearing and difficulty in walking. At the age of 14 she began to suffer from dull ache in the knees. Knee surgery with removal of loose cartilaginous fragments was performed when she was 19 years old, and again at the age of 85. After the age of 35 she had increasing, disabling pain in several joints and in muscles. She had total hip replacements at the age of 65 and at 76. At the age of 85 severe muscular atrophy developed, with motor disability, and she became confined to a wheelchair.
The 8 females affected had a more severe form of MED than the 4 males, and together they had undergone 11 orthopedic operations. Only 1 of the 4 males needed orthopedic surgery. All the adult family members affected had a university education and full-time employment.
Linkage and mutation analysis
Haplotype construction and linkage analysis of the 5 gene loci for autosomal dominant MED was performed with DNA from the 24 family members. Linkage was excluded for the COMP, MATN3, COL9A1, and COL9A3 loci. A positive linkage to the disease was obtained with marker D1S1598 close to the COL9A2 gene (maximum LOD score of 2.7). The COL9A2 gene was analyzed for mutations by sequencing of exon 3 and intron 3—the part of the gene where mutations have been identified in previous MED patients (Muragaki et al. Citation1996, Holden et al. Citation1999, Fiedler et al. Citation2002). A single nucleotide alteration, a T to C transition (IVS3DS [+2] T>C), was identified in the exon 3/intron 3 donor splice site in a heterozygous state in all 12 affected individuals from whom DNA was available. Analysis of the 9 healthy family members and of 3 individuals whose disease state was unknown confirmed that they did not carry the mutation.
Discussion
Our study of a 6-generation MED family is unique in that the affected family members were followed clinically from infancy to old age—the oldest up to the age of 95. Our findings show that MED caused by this particular COL9A2 mutation generally starts in early childhood with knee pain, which is accompanied by delayed ossification of femoral epiphyses. The disease then either stabilizes during puberty, with occasional knee pain, or progresses. Frequently, additional joints such as the elbows, hands, and hips then become affected and joint surgery may be necessary. Progression of the disease also affects muscles, with increasing atrophy, resulting in muscle fatigue and pain.
All of the affected family members in our study were found to carry an identical mutation at the donor splice site of exon 3/intron 3 of the COL9A2 gene, which was dominantly inherited in this 6-generation family. There is a 1 in 2 (or 50%) chance that each child of an affected parent will inherit the mutation and thus be affected with the disorder. The exons of the COL9A2 gene are translated into protein (a subunit of collagen IX), while the introns of the gene are removed and not translated. When the mutation is found at the donor splice site, it means that it is located right at the site of the cutoff between the exon and intron. Muragaki et al. (Citation1996) previously identified the mutation (IVS3DS [+2] T>C) in one family, and concluded that its effect is that exon 3 is not translated at all, resulting in a loss of 12 amino acids in the protein product. Less than 5% of the mutations that are known to cause dominant MED are located in the COL9A2gene; this type of MED is defined as EDM2 (Briggs and Chapman Citation2002, Jakkula et al. Citation2005). The EDM2 mutations are all located at different positions within the donor splice site of intron 3 and result in an in-frame deletion of 12 amino acids from the COL3 domain of the collagen chain (Holden et al. Citation1999, Spayde et al. Citation2000, Fiedler et al. Citation2002, Nakashima et al. Citation2005, Takahashi et al. Citation2006). The COL3 domain has proven to be essential for the binding between collagen IX and MATN3, an extracellular matrix protein in cartilage (Fresquet et al. Citation2007). Together with COL9A1 and COL9A3, COL9A2 forms the collagen IX heterotrimer. Previous studies suggest that collagen IX functions as a bridge between collagen fibrils and matrix macromolecules in the joint cartilage (Olsen Citation1997).
In addition to the genetic heterogeneity in MED, there is a marked intrafamilial variability in the clinical phenotype, suggesting that environmental and/or modifying genetic factors also play a role (Chapman et al. Citation2003). Despite intrafamilial differences in severity of the phenotype, all reported cases of EDM2 in addition to our cases have shared the features of more severe involvement of the knees and relative sparing of the hips (Barrie et al.Citation1958, van Mourik et al. Citation1998, Holden et al. Citation1999, Fiedler et al. Citation2002). A few cases with rare additional symptoms, such as double-layered patellae (Nakashima et al. Citation2005) and ulnar club hands (Takahashi et al. Citation2006), have been reported. Muscular atrophy, which was common in our family, has not been reported earlier in cases of EDM2. Muscular atrophy has been reported in families with mutated COMP and COL9A3 genes (BÖnnemann at al. Citation2000, Jakkula et al. Citation2003).
In a patient with MED, it is important to carry out a DNA-based analysis to identify a possible disease-causing mutation. This information can be used in genetic risk assessment of other family members and to achieve an early and correct diagnosis in children in the family with suspected MED. The risk for each child of being an affected person is 1 in 2, whereas the risk for the offspring of an unaffected person is insignificant. There is as yet no curative treatment for the disease, but much can be done to alleviate the symptoms, counteract deformities, and compensate for functional limitations. Families that have children with MED need to be in early contact with a pediatric orthopedic surgeon; operations may be necessary to minimize joint deformation and to preserve mobility. The families should also be connected to a center for pediatric and youth rehabilitation at an early stage. The occupational therapist can work out techniques to help the child to manage everyday activities at school and at home. The physiotherapist is responsible for evaluation, treatment, and programs for motor training. The various therapeutic components should be co-ordinated to achieve the best possible results. Adults with MED need to continue to interact with an orthopedic surgeon, an occupational therapist, and a physiotherapist, and sometimes with a rehabilitation center for adults. Joint replacement may become unavoidable with age.
KHG was the initiator of the study, scrutinized the hospital records, compiled family, clinical, and radiographic data, and wrote the results and the discussion of the clinical observations. JD and HÖ performed genotyping of candidate genes, mutation analysis, and wrote the results of the linkage and mutation analysis. HM performed genotyping of candidate genes and revised the manuscript. ND contributed with genetic analysis and manuscript revision. TL examined and evaluated the radiographs.
We thank the patients and their families for their excellent cooperation. The study was supported by the Swedish Research Council, the Torsten and Ragnar SÖderberg Foundations, Uppsala University, and Uppsala University Hospital.
References
- Ballhausen D, Bonafe L, Terhal P, Unger SL, Bellus G, Classen M, . Recessive multiple epiphyseal dysplasia (rMED): phenotype delineation in eighteen homozygotes for DTDST mutation R279W. J Med Genet 2003; 40:65-71.
- Barrie H, Carter C, Sutcliffe J. Multiple epiphyseal dysplasia. Br Med J 1958; 2:133-7.
- Briggs MD, Chapman KL. Pseudoachondroplasia and multiple epiphyseal dysplasia. Mutation review, molecular interactions and genotype to phenotype correlations. Human Mutation 2002; 19:465-78.
- Bönnemann C, Cox G, Shapiro F, Wu J-J, Feener C, Thompson T, Anthony D, Eyre D, Darras B, Kunkel L. A mutation in the alpha 3 chain of type IX collagen causes autosomal dominant multiple epiphyseal dysplasia with mild myopathy. PNAS 2000; 97(3):1212-7.
- Chapman KL, Briggs MD, Mortier GR. Review: clinical variability and genetic heterogeneity in multiple epiphyseal dysplasia. Ped Pathol Mol Med 2003; 22:53-75.
- Fiedler J, Stöve J, Heber F, Brenner RE. Clinical phenotype and molecular diagnosis of multiple epiphyseal dysplasia with relative hip sparing during childhood (EDM2). Am J Med Gen 2002; 112:144-53.
- Fresquet M, Jowitt T, Ylöstalo J, Coffey P, Meadows R, Ala-Kokko L, Thornton D, Briggs M. Structural and functional characterization of recombinant Matrilin-3 A-domain and implications for human genetic bone diseases. J Biol Chem 2007; 282(48):34634-43.
- Holden P, Canty E, Mortier G, Zabel B, Spranger J, Carr A, Grant M, Loughlin J, Briggs M. Identification of novel pro-2(IX) collagen gene mutations in two families with distinctive oligo-epiphyseal forms of multiple epiphyseal dysplasia. Am J Hum Genet 1999; 65:31-8.
- Jakkula E, Lohiniva J, Capone A, Bonafe L, Marti M, Schuster V, Giedion A, Eich G, Boltshauser E, Ala-Kokko L, Superti-Furga A. A recurrent R718W mutation in COMP results in multiple epiphyseal dysplasia with mild myopathy: clinical and pathogenic overlap with collagen IX mtuations. J Med Genet 2003; 40:942-8.
- Jakkula E, Makitie O, Czarny-Ratacjak M, Jackson GC, Damignani R, Susic M, . Mutations in the known genes are not the major cause of MED; distinctive phenotypic entities among patients with no identified mutations. Euro J Hum Genet 2005; 13:292-301.
- Klar J, Gedde-Dahl Jr T, Larsson M, Pigg M, Carlsson B, Tentler D, Vahlquist A, Dahl N. Assignment of the locus for ichthyosis prematurity syndrome to chromosome 9q33.3-34.13. J Med Genet 2004; 41:208-12.
- Lachman RS, Krakow D, Cohn D, Rimoin DL. MED, COMP, multilayered and NEIN: an overview of multiple epiphyseal dysplasia. Pediatr Radiol 2005; 35:116-23.
- Lathrop GM, Laouel JM. Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet 1984; 36(2):460-5.
- Muragaki Y, Mariman EC, van Beersum SE, Perälä M, van Mourik JB, Warman ML, Olsen BR, Hamel BC. A mutation in the gene encoding the alpha 2 chain of the fibril-associated collagen IX, COL9A2, causes multiple epiphyseal dysplasia (EDM2). Nat Genet 1996; 12;103-5.
- Nakashima E, Ohashi H, Kimizuka M, Nishimura G. Double-layered patella in multiple epiphyseal dysplasia is not exclusive to DTDST mutation. Am J Med Genet 2005; 133A:106-7.
- Olsen B. Molecules in focus: Collagen IX. Int J Biochem Cell Biol 1997; 29:555-8.
- Ribbing S. Stüdien über hereditäre, multiple Epiphysenstörungen. Acta Radiol (Suppl 34) 1937: 7-107.
- Roos EM, Roos HP, Ekdahl C and Lohmander LS. Knee injury and Osteoarthritis Outcome Score (KOOS) - validation of a Swedish version. Scand J Med Sci Sports 1998,8:439-48 and health City: WHO 2001, 2003: 31.
- Spayde E, Joshi A, Wilcox W, Briggs M, Cohn D, Olsen B. Exon skipping mutation in the COL9A2 gene in a family with multiple epiphyseal dysplasia. Matrix Biol 2000; 19:121-8.
- Takahashi M, Matsui Y, Goto T, Nishimura G, Ikegawa S, Ohashi H, Yasui N. Intrafamilial phenotypic diversity in multiple epiphyseal dysplasia associated with a COL9A2 mutation (EDM2). Clin Rheumatol 2006; 25:591-5.
- van Mourik J, Hamel B, Mariman E. A large family with multiple epiphyseal dysplasia linked to COL9A2 gene. Am J Med Genet 1998; 77:234-40.
- Zanki A, Jackson G, Mittaz Crettol L, Taylor J, Elles R, Mortier G, Spranger J, Zabel B, Unger S, Le Merrer M, Cormier-Daire V, Hall C, Wright M, Bonafe L, Superti-Furga A, Briggs M. Preselection of cases through expert clinical and radiological review significantly increases mutation detection rate in multiple epiphyseal dyslasia. Eur J Hum Genet 2007; 15:150-4.
- Ödman P. Hereditary enchondral dysostosis: twelve cases in three generations mainly with peripheral location. Acta Radiol 1959; 52:97-113.
Database references.
- OMIM (Online Mendelian Inheritance in Man) Internet: http://www.ncbi.nlm.nih.gov/omim. Search: epiphyseal dysplasia.
- GeneReviews (University of Washington) Internet: http://www.genetests.org (select GeneReviews). Search: multiple epiphyseal.