Abstract
Characterization of DNA methylation is one assessment of chromatin remodeling in early embryos. Unfortunately, evaluation at specific loci is hindered by their small cell numbers. Our objective was to determine if bisulfite sequencing could be optimized for preimplantation embryos, comparing conversion times, primer design, and DNA amplification methods. Methylation at three loci, SATI, OCT4, and IGF2, was investigated in bovine in vitro produced (IVP) embryos, somatic cells, and no template controls. Bisulfite treatment for 15–16 h gave higher quality DNA than treatment for 18 h. Three step primer design improved bisulfite primer specificity, yielding more PCR product than primers previously reported. Following optimization, methylation data were obtained from as few as 4 cell equivalents. Finally, DNA amplification efficiencies were evaluated using miniprep, TempliPhi, or 96-well glycerol stocks with automated TempliPhi. While TempliPhi was better than standard minipreps, the 96-well format proved most efficient. Preliminary methylation profiles of bovine IVP 2-cell, 8-cell, blastocyst stage embryos and somatic cells were 25, 10, 22, and 74% for SATI and 88, 88, 79, and 88% for OCT4, respectively, suggesting that SATI is demethylated during early embryonic reprogramming, while OCT4 remains hypermethylated. IGF2 methylation was 84, 28, and 84% for bovine IVP 8-cell, blastocyst stage embryos and somatic cells; blastocyst stage embryos exhibited more variability, ranging from 0 to 80%. This new assay will enhance assessment of chromatin remodeling in embryos, and be especially useful for evaluating those produced by assisted reproductive technologies.
| Abbreviations | ||
| IVP: | = | in vitro produced |
| CpG: | = | cytosine-guanine |
| MSRE-PCR: | = | methylation-sensitive restriction enzyme PCR |
| 5MeC: | = | methylated cytosines |
| TP: | = | TempliPhi™ |
| MP: | = | miniprep |
| LP: | = | large scale prep |
| LB/Amp: | = | Luria Broth with ampicillin |
INTRODUCTION
Assisted reproductive technologies, such as somatic cell nuclear transfer and in vitro production of embryos, are increasingly used for the production of genetically superior livestock, and for the production of genetically modified animal models for the study of human and animal diseases. Unfortunately, the efficiencies of producing live offspring using these technologies are reduced when compared to in vivo produced counterparts. There are numerous reports of early embryonic losses, placental abnormalities, organ and skeletal malformations, and fetal overgrowth [Cibelli et al. Citation1998; Young et al. Citation1998; Wells et al. Citation1999; Hill et al. Citation2000]. After nearly two decades of research, the cause(s) have still not been determined, but epigenetic alterations are capturing major attention.
Epigenetic modifications have key roles in chromatin remodeling impacting the regulation of gene expression, as well as maintaining genome stability. Methylation of cytosine residues in cytosine-guanine (CpG) dinucleotides and CpG islands is one such modification that alters transcription factor binding in promoter regions, as well as attracting other proteins that help to modify chromatin structure and lead to gene silencing [Bird Citation1986]. DNA methylation affects a broad range of biological processes, including embryonic reprogramming, stem cell pluripotency, genetic imprinting, and X-chromosome inactivation [Farthing et al. Citation2008; Lopez-Serra and Esteller Citation2008]. Assessment of DNA methylation is valuable for understanding the regulation of these processes, as well as for developing new modalities for modulating embryonic differentiation and development [Waggoner Citation2007].
Current methods for assessing DNA methylation include techniques such as methylation-sensitive restriction enzyme PCR (MSRE-PCR) and other PCR based assays [Singer-Sam et al. Citation1990], as well as immunocyto-chemistry [Dean et al. Citation2001]. These methods are used to obtain a genome-wide view of methylation, but have many limitations. Most importantly they are not capable of quantitative analysis at particular loci [Millar et al. Citation2002]. For analysis at specific loci, the method of choice has been bisulfite sequencing, allowing the determination of methylation status of all cytosines within the given sequence [Frommer et al. Citation1992]. Bisulfite treatment of single-stranded DNA deaminates unmethylated cytosine residues to uracil, while leaving methylated cytosines (5MeC) unchanged [Shapiro et al. Citation1974]. PCR amplification with specific primers for loci of interest replaces uracil with thymine, and sequencing allows for the detection and quantitation of methylated (C) versus unmethylated (T) cytosines. While this method gives a detailed methylation profile, it is time consuming and optimally requires large amounts of high quality DNA.
Unfortunately, obtaining DNA methylation profiles is often hindered when analyzing small quantities of DNA. Bisulfite sequencing can be especially difficult when evaluating early preimplantation embryos [Warnecke et al. Citation1998a, Citation1998b]. Therefore, a more efficient method for evaluating locus-specific methylation in the early preimplantation embryo is needed and would allow further assessment of chromatin remodeling ultimately leading to improved in vitro embryo production systems and increased production of live offspring. Our objective was to determine if current bisulfite sequencing methods could be enhanced to analyze early bovine embryos, through optimizing primer design and bisulfite conversion methods, along with comparing the use of illustra™ TempliPhi (GE Healthcare, Buckinghamshire, UK) rolling circle DNA amplification and sequencing to standard methods. We hypothesized that these modifications would improve DNA yield, quality, product specificity, and reduce overall time and expense. These new adaptations should lead to decreased costs and improved high throughput analyses of embryos, making this more valuable for assessing early embryonic reprogramming, especially those produced using assisted reproductive technologies.
RESULTS
Locus-Specific PCR of Bisulfite Treated DNA
Initial experiments using previously published bisulfite primers for IGF2 (Bisulfite-10.1 and -10.2; [Gebert et al. Citation2006]) yielded no PCR products () or inconsistent amplification results when using published conditions for evaluating small samples. This included adjusting temperature, [Mg + 2], and adding DMSO, with no improvements. Upon in silico analysis using MacVector and Mfold, these primers displayed self duplexing or formed hairpin structures at the annealing temperature to be used. Redesign and evaluation of primers using our three step process produced bisulfite primer sets that had closer melting temperatures, a reduced chance of self-duplexing or primer-dimer formation, and an increased likelihood of binding to target DNA, yielding more consistent PCR results for small samples (, ).
FIGURE 1 Bisulfite primer design impacts PCR amplification. (A) IGF2 PCR products using previously published primers and conditions (bisulfite-10.1 and 10.2 F and R primer pairs; [Gebert et al. Citation2006]); Lane 1–2: 1,000 cells, Lane 3: IVP blastocyst-stage embryo, Lane 4: no template control ; Expected product size should have been 419 bp; (B) IGF2 PCR product from primers designed using 3 step method; Lane 1: IVP blastocyst-stage embryo; Lane 2: no template control. M: 100 bp ladder; Expected product size was obtained, 404 bp.
![FIGURE 1 Bisulfite primer design impacts PCR amplification. (A) IGF2 PCR products using previously published primers and conditions (bisulfite-10.1 and 10.2 F and R primer pairs; [Gebert et al. Citation2006]); Lane 1–2: 1,000 cells, Lane 3: IVP blastocyst-stage embryo, Lane 4: no template control ; Expected product size should have been 419 bp; (B) IGF2 PCR product from primers designed using 3 step method; Lane 1: IVP blastocyst-stage embryo; Lane 2: no template control. M: 100 bp ladder; Expected product size was obtained, 404 bp.](/cms/asset/7d861ff4-1a18-45b6-bcb1-11ebc655041c/iaan_a_433292_f0001_b.jpg)
TABLE 1 Primer Pairs Utilized in Bisulfite Sequencing.
PCR optimization results for bisulfite treated DNA from embryos or small samples of cells is shown in . A large pool of 100–200 cells [Han et al. Citation2006] was used for the initial optimization, so that the first PCR product produced a faint band at the optimal temperature and would be used in a gradient of the second nested PCR reaction. Detectable amplicons for Satellite I were seen for all sample concentrations tested (bisulfite reaction starting material of 1000, 50, and 25 cell equivalents), while no template bisulfite treated samples and PCR negative controls did not amplify. Single copy genes were more difficult to amplify from small amounts of DNA, and required hemi-nested PCR (). The final optimal number of cells in an initial bisulfite-treated sample was 80 cells, allowing assessment of DNA in duplicate, across several loci from 8 cell equivalents per reaction.
FIGURE 2 Optimization of PCR for bisulfite treated DNA from small sample sizes. (A) Satellite I ; Lane 1: 1,000 cells; lane 2: 50 cells; lane 3: 25 cells; lane 4: 1 blastocyst-stage embryo; lane 5: bisulfite negative control; lane 6: PCR negative control; lane 7: positive control. (B) Satellite I for bovine embryos; Lane 1: 50 × 2-cell embryos; lane 2: 10 × 8-cell embryos; Lane 3: 1 blastocyst-stage embryo; Lane 4:1 blastocyst-stage embryo; Lane 5: no template control. (C) Oct4 for embryos; lanes 1 and 2: 1 blastocyst-stage embryo; lane 3 and 4: 50 × 2-cell embryos; Lane 5 and 6:10 × 8-cell embryos, lane 7: no template control; M: 100 bp ladder.
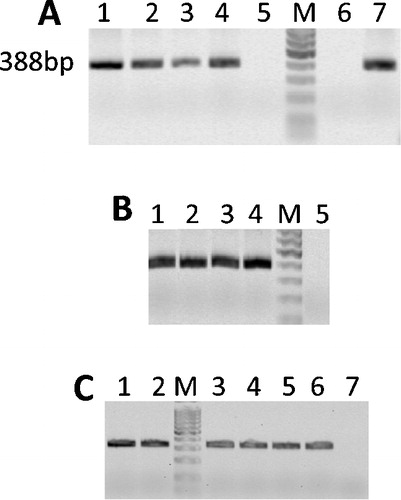
Bisulfite treatment of DNA for 15 to 16 h was of better quality (improved DNA integrity) than DNA treated for 18 h, based on the quality of PCR amplification, the appearance of sharp bands on electrophoresis, and longer reads following sequencing. Bisulfite-treated DNA samples were successfully used for locus-specific PCR for about 6 months from treatment, whereas samples used after 6 months led to inconsistent amplification results, yielding incomplete sequences with many gaps, and improper base calling.
Bisulfite Treated DNA Amplification
Locus specific PCR products from bisulfite treated DNAs were cloned into plasmids for further analysis of template methylation within the pool. Following bacterial transformation, DNA within colonies were amplified using three different methods prior to sequencing. TempliPhi™ (TP) alone was better than miniprep (MP) for small scale experiments, reducing the time to acquiring results by approximately 12 h, and increasing the maximum number of samples (bacterial colonies) processed within each run, from 20 colonies using minipreps to a maximum of 46 colonies using PCR tube strips with TempliPhi™. However, it was the 96-well glycerol stocks with automated TempliPhi™ amplification and sequencing (large prep; LP) that proved to be the most cost-effective, high-throughput method for bisulfite sequencing of small samples. Using this approach, final data was acquired in 48 h on 95 samples and 1 no template control per plate.
Methylation Analysis
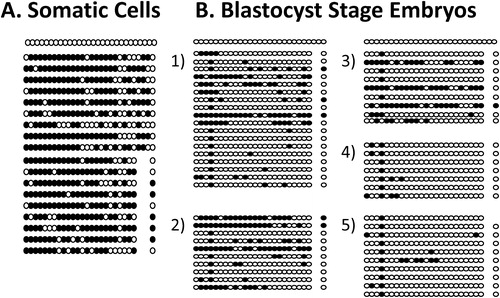
Preliminary results are presented here for somatic cells, and preimplantation bovine embryos to show how the revised protocol can be of benefit for analyzing small samples. A representative methylation plot for SAT I is presented in . Somatic cell methylation was 74% for Satellite I (). However, overall percent methylation of bovine IVP 2-cell, 8-cell, and blastocyst stage embryos for SATI was 25, 10, and 22%, respectively. The percent methylation for OCT4 was 88, 88, and 79 % for bovine IVP 2-cell, 8-cell, and blastocyst stage embryos, respectively, similar to somatic cells that had 88% methylation at OCT4. The percentage methylation for IGF2 was 84% and 28% for bovine IVP 8-cell and blastocyst stage embryos, whereas somatic cells had 84% methylation. The IGF2 gene had the most variability at the blastocyst stage, with individual embryos ranging from 0 to 80% methylation. Methylation of IGF2 was not evaluated for 2-cell embryos. All bisulfite conversion reactions had less than 5% unconverted cytosines present outside CpGs.
FIGURE 3 Satellite I methylation profile. Circles indicate individual CpGs within the locus, with black and white indicating methylated and unmethylated cytosine residues, respectively. (A) Methylation plot for somatic cells (74% methylation). (B) Methylation plot for five blastocyst stage embryos: embryo 1 had 22% methylated cytosines, embryo 2 had 35% methylated cytosines, embryo 3 had 29% methylated cytosines, embryo 4 and 5 had 8% methylated cytosines.
DISCUSSION
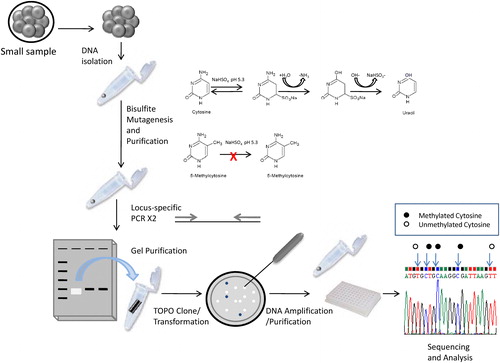
Bisulfite sequencing is a powerful method for assessing DNA methylation profiles at specific loci, but ideally requires large amounts of DNA and is very time consuming. Here we show that through modifications in primer design, optimizing bisulfite conversion time, as well as through the use of automated TempliPhi™ amplification and sequencing, these disadvantages can be overcome, allowing for more cost-effective high-throughput analysis of DNA methylation in bovine preimplantation embryos. gives an overview of this improved bisulfite sequencing protocol.
FIGURE 4 Overview of locus specific DNA methylation analysis of bovine IVP embryos.
When designing a bisulfite sequencing assay to assess methylation at a specific locus it is important to first determine the region of interest within the particular gene. Gene regulation through methylation may occur within CpG islands of the promoter region; however, for imprinted genes, regulatory CpG islands or differentially methylated regions (DMRs) are often located within introns or in regions upstream or downstream from the gene of interest. Therefore, CpG islands must be identified that possess regulatory significance before attempting to design primers.
Bisulfite primer design is crucial for optimal priming, especially with small samples. It is clear that knowing the necessary properties of bisulfite primers, such as avoiding CpGs or long stretches of the same base, is a basic step towards creating effective primer pairs [Warnecke et al. Citation2002; Han et al. Citation2006]. Moreover, selection of primer pairs that anneal within 1–2˚C of each other can further improve results, especially when working with limiting samples. As we have shown, these conditions alone are not sufficient for optimal bisulfite priming reactions and can lead to no or very low amplification if dealing with low quantities of starting template DNA.
Our three step bisulfite primer design resolves these problems. First, Methyl Primer Express® reduces the challenges of non-specific primer amplification and mispriming. The second step using MacVector™ evaluates primers for hairpin or dimer structures, which should be eliminated from the pool of possibilities. As a final step, Mfold software evaluates the template DNA for secondary or tertiary structure under PCR conditions to be used. An example is shown in . We demonstrate that production of primers using this three step approach significantly enhances product yields, and should be strongly considered when analyzing small samples, such as embryos.
FIGURE 5 Analysis of template DNAs using Mfold software [Mathews et al. Citation1999; Zuker Citation2003]. Bisulfite converted sequences for loci of interest were evaluated for predicted secondary or tertiary structure under the proposed PCR conditions, and the regions of primer binding analyzed. Folding of template DNA sequences with (A) excessive secondary and tertiary structure or (B) lacking secondary and tertiary structure. Primer binding sites are indicated by arrows. Arrows in panel (A) are crossed out, indicating the primers would not bind in these regions of higher order structure.
![FIGURE 5 Analysis of template DNAs using Mfold software [Mathews et al. Citation1999; Zuker Citation2003]. Bisulfite converted sequences for loci of interest were evaluated for predicted secondary or tertiary structure under the proposed PCR conditions, and the regions of primer binding analyzed. Folding of template DNA sequences with (A) excessive secondary and tertiary structure or (B) lacking secondary and tertiary structure. Primer binding sites are indicated by arrows. Arrows in panel (A) are crossed out, indicating the primers would not bind in these regions of higher order structure.](/cms/asset/9be670f2-d2a6-40d2-a9be-6ebe2a2dc4dc/iaan_a_433292_f0005_b.jpg)
Complete bisulfite conversion is another key factor for successful assessment of DNA methylation profiles in embryos. It is essential to balance the issue of achieving complete bisulfite conversion while avoiding DNA degradation [Clark et al. Citation1994; Grunau et al. Citation2001; Lucifero et al. Citation2002]. We determined that sample treatment with higher concentrations of bisulfite for 15–16 h gave the highest quality DNA, when compared to longer conversion times. Other reports have also shown that increasing bisulfite concentrations allow for reduced treatment times and improved DNA quality [Raizis et al. Citation1995; Millar et al. Citation2002]. Additionally, since the starting template is low, there is no need for prior restriction enzyme digestion or to shear the DNA, since bisulfite treatment alone is sufficient to knick the DNA during the incubation period. Noteworthy, results from bisulfite treated samples older than 6 months post-treatment were unpredictable, due to DNA degradation, often leading to fragmented sequences, and miscalled bases. Similar findings of bisulfite-treated DNA stability were reported by Millar et al. [Citation2002].
One of the biggest challenges of using bisulfite sequencing with embryos is the limited amount of DNA available for experiments. Optimizing for small cell numbers, first using abundant samples, such as a somatic cell line, allows for proper experimental design, taking into consideration the entire scope of the experiment and all necessary repetitions. Multiple PCR repetitions are also necessary in order to avoid amplification bias, as was previously demonstrated [Millar et al. Citation2002]. Previous reports analyzing porcine or bovine embryos reported using from 50 to 80 cell equivalents/PCR reaction [Kang et al. Citation2001, Citation2002]. While bisulfite PCR products could be obtained from as few as 4 cell equivalents in this study, we determined that use of 8 cell equivalents was more robust and gave more consistent results. This is a substantial improvement, making this protocol more applicable for analyzing embryos.
Dealing with small quantities of template DNA leads to further challenges with PCR, and can impact amplicon detection, purification, ligation, and amplification. To overcome low copy number, we and others [Lucifero et al. Citation2002; Warnecke et al. Citation2002; Gebert et al. Citation2006] have shown that use of hemi-nested or nested PCR can substantially increase PCR products from single copy genes, allowing detection and purification of sufficient quantities for cloning into plasmid vectors. Moreover, we found that increasing the ligation time to 30 min further increased efficiencies, as evidenced by increased colony numbers after bacterial transformation and overnight culture.
Amplification of plasmid DNAs and purification to produce adequate amounts of DNA for bisulfite sequencing is traditionally accomplished by plasmid miniprep. Minipreps require overnight growth of bacteria, adding an additional day to this already time consuming procedure, so we evaluated other options. Previous research has shown that rolling circle replication using bacteriophage Φ29 DNA polymerase improves the quality and quantity of DNA generated due to the enzyme's high processivity, proofreading, and strand displacement activity [Nelson et al. Citation2002; Reagin et al. Citation2003]. Microgram quantities of DNA can be produced in as few as 4 h from picogram quantities of starting template [Nelson et al. Citation2002; Reagin et al. Citation2003]. Furthermore, we demonstrate here that using this approach with the TempliPhi™ Amplification kit, enabled preparation of templates for sequencing without the need for plasmid minipreps, making this a more time and cost efficient approach. One important consideration when using this method however, is that Luria broth (LB) agar can inhibit the amplification reaction completely, so careful handling and transfer of bacterial colonies from agar plates are necessary to ensure success.
Automation of TempliPhi™ amplification and sequencing in this experiment proved to be an even more efficient way to obtain sequences of clones from large numbers of small samples. This is in agreement with a previous report that used large quantities of template DNA [Kobayashi et al. Citation2006]. Here we demonstrate that this method allows duplicate PCR reactions on 8 samples to be performed, 12 colonies per sample are grown in a 96-well culture plate overnight and glycerol stocks are processed by automated TempliPhi™ and sequencing. The cost of this method is about 50% of the small scale TempliPhi™ procedure making it the most time- and cost-efficient and reliable method tested.
These adaptations to the bisulfite sequencing protocol were successfully used here to evaluate and compare DNA methylation profiles for three loci in a fibroblast cell line and in early preimplantation embryos. Our preliminary data indicate that the heterochromatin marker, SAT I, is demethylated in the early embryo when compared to adult somatic cells. However, the euchromatic OCT4 locus remains highly methylated and is unchanged between bovine IVP embryos and adult somatic cells. Interestingly, IGF2, an imprinted gene, showed the most variability in methylation at the blastocyst stage, with 4 of 6 embryos having very low or no methylation and 2 embryos exhibiting methylation patterns similar to somatic cells. Further research of locus specific methylation patterns may help us elucidate problems found in early embryonic reprogramming produced by assisted reproductive technologies.
In conclusion, by adapting elements of other molecular protocols, we have optimized the popular bisulfite sequencing protocol, further simplifying the process and providing a more efficient and cost-effective method for dealing with large numbers of samples with small starting amounts of DNA. These improvements should prove fruitful for the analysis of locus specific methylation in embryos, and will be useful for assessing assisted reproduction systems and their effects on the early embryo epigenome.
MATERIALS AND METHODS
DNA Isolation from Cultured Cells and Embryos
A primary bovine cell line (bull skin fibroblast) was established according to procedures described by Moore et al. [Citation2007]. Cells were grown to passage 4, trypsinized, counted in a hemocytometer, and frozen at −80˚C in 50µL aliquots [68,000 cells/mL in Dulbecco's Modified Eagles Medium (low glucose, GlutaMAX™; Invitrogen, Carlsbad, CA, USA) supplemented with fetal bovine serum (FBS, 15%; Mediatech Inc, Herndon, VA, USA), non-essential amino acids (10%; Invitrogen), and 2-mercaptoethanol (0.1%; Sigma-Aldrich, St. Louis, MO, USA).
Bovine embryos were produced in vitro as previously described [Moore et al. Citation2007]. Two-cell, 8-cell, and blastocyst stage embryos were collected on days 2, 4, and 7 following fertilization (day 0), washed in PBS/BSA (0.4%; Sigma-Aldrich) and zona pellucidas were removed with pronase (0.5% solution; Sigma-Aldrich) by incubating in drops at 37˚C for 1 to 2 min, followed by extensive washing in PBS/BSA. Single embryos (blastocysts) or pools of embryos at the 2-cell stage (groups of 40) or 8-cell stage (groups of 10) were transferred in 2–5 µL of PBS/BSA to 1.5 mL microfuge tubes and stored at −80˚C.
DNA was isolated from different amounts of cell lysate (25–2,500 cell equivalents) or from embryos or embryo pools (80–150 cells) using a modified Proteinase K/SDS method [Millar et al. Citation2002]. Briefly, lysis buffer (Proteinase K, SDS, and tRNA; Sigma-Aldrich) was added to each sample and incubated at 37˚C for 24 h. The reaction was stopped by incubation of samples at 95˚C for 10 min, immediately prior to bisulfite treatment.
Bisulfite Treatment of DNA
Bisulfite treatment of DNA was adapted from methods previously described [Clark et al. Citation1994; Millar et al. Citation2002; Kang et al. Citation2003]. Briefly, DNA samples were denatured by the addition of freshly prepared 3 M NaOH (Sigma-Aldrich) and incubation at 37˚C for 30 min. Bisulfite reactions were performed by adding a freshly prepared solution of metabisulfite/hydroquinone into the denatured samples to a final concentration of 2 M sodium metabisulfite/10 mM hydroquinone (Sigma-Aldrich) and incubating in the dark at 55˚C for 15–16 h. Additionally, to achieve full bisulfite conversion, samples were ramped to 95˚C for 1 min every 2 h during the incubation period to ensure DNA remained denatured. Nuclease-free water (Ambion, Austin, TX, USA) was treated (no template) alongside samples as a bisulfite negative control.
Bisulfite-treated samples were desalted using the Wizards DNA Clean-up System (Promega, Madison, WI, USA) according to manufacturer's instructions. Samples were then desulfonated with freshly prepared 3 M NaOH, at 37˚C for 15 min, neutralized with 7.5 M ammonium acetate (Fisher Scientific, Pittsburgh, PA, USA) and precipitated overnight at −80˚C with 100% ethanol (Aaper, Shelbyville, KY, USA). Precipitated bisulfite-treated DNA was recovered by centrifugation, washed with 70% ethanol, air dried, resuspended in nuclease-free water, and stored in −80˚C for up to 6 months.
Bisulfite Primer Design
A three step process was utilized for bisulfite primer design. First, the genomic sequences of interest were acquired on Genbank. Methyl Primer Express Software (AB Applied Biosystems, Foster City, CA, USA) was used to evaluate each DNA region of interest for CpG islands, bisulfite sequence conversion in silico, and primer design. Secondly, primer pairs were tested using MacVector Software™ (Oxford Molecular Ltd., Campbell, CA, USA) to remove primers that formed hairpins, self-dimers and/or primer-dimers [Rastogi Citation2000]. Finally, the bisulfite converted sequence for the locus of interest was investigated using Mfold software, version 3.2 [Mathews et al. Citation1999; Zuker Citation2003] to predict any secondary or tertiary structures at the proposed PCR annealing temperature and buffer sodium and magnesium concentrations (50 mM and 2 mM, respectively), in order to avoid selecting primers located in these regions (). For single copy genes, where the expected PCR product yield was low, hemi-nested primers were also designed using the same approach ().
Locus-Specific PCR of Bisulfite Treated DNA
PCR reactions with bisulfite treated DNA were first optimized using large DNA pools from cell lines to obtain visible amplification products. Methods were then scaled down for low cell numbers, and finally tested on embryos for validation. Loci of interest were amplified by PCR using bisulfite primers for SAT I, OCT4, and IGF2 (). Each 25 µL PCR reaction contained 2 µL of bisulfite-treated template DNA (10% of the bisulfite reaction), 0.625 U HotStar Taqs Plus DNA polymerase (Qiagen, Valencia, CA, USA), 0.6 µM primers, 80 µM dNTPs (Roche Diagnostics GmbH, Mannheim, Germany) and the manufacturer's provided buffer. Bisulfite-treated DNAs, positive controls (somatic cells), and negative controls (bisulfite blank and PCR water blank) were denatured at 95˚C for 15 min, followed by 35 cycles of 95˚C for 30 s, 50–60˚C for 60 s (see for specific primer annealing temperature), 72˚C for 90 s, and the final extension at 72˚C for 10 min. For hemi-nested PCR, a 1 µL aliquot of PCR products from the first reaction was transferred to a second reaction containing the forward and the hemi-nested reverse primer, and amplified for up to an additional 35 cycles. Optimized conditions for each loci were used for all further amplification reactions. A minimum of two PCR reactions were performed for each bisulfite-treated sample and combined prior to ligation [Warnecke et al. Citation1998a, Citation1998b; Zhang et al. Citation2005].
Locus-specific PCR products were evaluated using gel electrophoresis (1.5% low melting agarose gel, 0.5X TA no EDTA buffer at 90 V) and photographed using a Typhoon Variable Mode Scanner (GE Healthcare, Buckinghamshire, UK). Amplified PCR products were excised and purified from agarose gel using Wizard® SV Gel Clean-up System (Promega) according to the manufacturer's protocol, and ligated into TOPO® TA II dual promoter vector (Invitrogen) for 30 min. TOP 10 One Shot® competent cells (Invitrogen) were transformed with ligation reactions or control DNA (pUC19) and plated on LB/Amp (100 µg/mL)/X-gal (80 mg/L) plates for overnight growth and blue/white screening as instructed by the manufacturer.
Plasmid DNA Amplification
Three methods were tested for amplification of the plasmid DNAs containing PCR products from bisulfite DNAs to improve efficiency. Ten to 12 bacterial colonies per bisulfite-treated DNA sample were evaluated. In the first method, miniprep (MP), individual white bacterial colonies were picked from plates, transferred into Luria Broth with ampicillin (LB/Amp; 100 µg/mL; Sigma-Aldrich) and grown overnight. Plasmid DNA was harvested using the QIAprep® Spin Miniprep Kit (Qiagen) according to the manufacturer's instructions. In the second small scale method, TempliPhi™ (TP; GE Healthcare, Buckinghamshire, UK), individual white colonies were picked from plates with a straight wire avoiding LB/agar contaminants. Single colonies were transferred into 50 µL of 10 mM Tris, pH 8.0, buffer, vortexed for 30 s and 0.5 µL of this bacterial solution was transferred into DNA amplification reactions using the TempliPhi™ Amplification Kit according to manufacturer's instructions. Finally, in the third method, large scale prep (LP), individual white colonies were picked from LB/Amp agar plates with sterile pipet tips and used to inoculate 175 µL LB/Amp solution previously aliquoted into wells of 96-well plates. Bacteria were grown overnight at 37˚C, 75 µL of glycerol (50% v/v; Sigma) was added to each well and plates were frozen at −80˚C for later use. The plasmid DNAs from 96-well plates containing frozen glycerol stocks were amplified by the TempliPhi™ Amplification Kit according to manufacturer's instructions.
Sequencing
DNAs produced by the three methods (TP, MP, LP) were utilized as templates in BIGDYE™ PCR amplification reactions (Applied Biosystems, University Park, IL, USA) with M13 Forward and Reverse primers using ABI Prism BigDye Terminator cycle sequencing protocols (Applied Biosystems, Perkin-Elmer Corp., Foster City, CA, USA). Excess dye-labeled terminators were removed by ethanol precipitation. Purified extension products were resuspended in Hi-Di formamide (Applied Biosystems) and were loaded onto POP-7 sieving matrix on 50-cm capillaries in an ABI Prisms® 3730 Genetic Analyzer (Applied Biosystems) at the University of Florida DNA Sequencing Core or on an ABI Prisms® 377XL Automated DNA Sequencer (Applied Biosystems) at the University of Florida Center for Epigenetics Core Laboratory. Sequencing data were analyzed by ABI Sequencing Analysis software (v. 5.2) and KB Basecaller (Applied Biosystems).
Methylation Analysis
Sequencing data were analyzed from each bisulfite treated sample and were represented by a minimum of 8 bacterial clones per sample. Sequences from clones were aligned with known bisulfite converted sequences for each locus using ChromasPro software (Technelysium Pty Ltd, Helensvale, Australia), methylated cytosines (C) and non-methylated cytosines (T) were identified, and the percentage of methylated CpGs over total CpGs in the locus was calculated [Warnecke et al. Citation1998a, Citation1998b]. Complete bisulfite conversion was validated when less than 5% unconverted cytosines were present outside CpGs [Warnecke et al. Citation2002; Lucifero et al. Citation2006].
ACKNOWLEDGMENT
This project was supported by National Research Initiative Competitive Grant no. 2006-35203-16620 from the USDA Cooperative State Research, Education, and Extension Service. We thank Dr. Jingda Shi, at the University of Florida Center for Epigenetics Core Laboratory, for his assistance and advice with DNA sequencing.
Declaration of Interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Bird, A. P. (1986) CpG-rich islands and the function of DNA methylation. Nature 321:209–213.
- Clark, S. J., Harrison, J., Paul, C. L. and Frommer, M. (1994) High sensitivity mapping of methylated cytosines. Nucleic Acids Res 22:2990–2997.
- Cibelli, J. B., Stice, S. L., Golueke, P. J., Kane, J. J., Jerry, J., Blackwell, C., Ponce de Leon, F. A. and Robl, J. M. (1998) Cloned transgenic calves produced from nonquiescent fetal fibroblasts. Science 280:1256–1258.
- Dean, W., Santos, F., Stojkovic, M., Zakhartchenko, V., Walter, J., Wolf, E. and Reik, W. (2001) Conservation of methylation reprogramming in mammalian development: Aberrant reprogramming in cloned embryos. Proc Natl Acad Sci USA 98:13734–13738.
- Farthing, C. R., Ficz, G., Ng, R. K., Chan, C. F., Andrews, S., Dean, W., Hemberger, M. and Reik, W. (2008) Global mapping of DNA methylation in mouse promoters reveals epigenetic reprogramming of pluripotency genes. PLoS Genet 4:e1000116.
- Frommer, M., McDonald, L. E., Millar, D. S., Collis, C. M., Watt, F., Grigg, G. W., Molloy, P. L. and Paul, C. L. (1992)Agenomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA 89:1827–1831.
- Gebert, C., Wrenzycki, C., Herrmann, D., Groger, D., Reinhardt, R., Hajkova, P., Lucas-Hahn, A., Carnwath, J., Lehrach, H. and Niemann, H. (2006) The bovine IGF2 gene is differentially methylated in oocyte and sperm DNA. Genomics 88:222–229.
- Grunau, C., Clark, S. J. and Rosenthal, A. (2001) Bisulfite genomic sequencing: Systematic investigation of critical experimental parameters. Nucleic Acids Res 29:E65–E65.
- Han, Y. M., Kim, S. H. and Kang, Y. K. (2006) Analysis of DNA methylation profiles in preimplantation embryos using bisulfite mutagenesis. Methods Mol Biol 325:251–260.
- Hill, J. R., Burghardt, R. C., Jones, K., Long, C. R., Looney, C. R., Shin, T., Spencer, T. E., Thompson, J. A., Winger, Q. A. and Westhusin, M. E. (2000) Evidence for placental abnormality as the major cause of mortality in first-trimester somatic cell cloned bovine fetuses. Biol Reprod 63:1787–1794.
- Kang, S. H., Kiefer, C. M. and Yang, T. P. (2003) Role of the promoter in maintaining transcriptionally active chromatin structure and DNA methylation patterns in vivo. Mol Cell Biol 23:4150–4161.
- Kang, Y. K., Koo, D. B., Park, J. S., Choi, Y H., Kim, H. N., Chang, W. K., Lee, K. K. and Han, Y M. (2001) Typical demethylation events in cloned pig embryos. Clues on species-specific differences in epigenetic reprogramming of a cloned donor genome. J Biol Chem 276:39980–39984.
- Kang, Y. K., Park, J. S., Koo, D. B., Choi, Y H., Kim, S. U., Lee, K. K. and Han, Y M. (2002) Limited demethylation leaves mosaic-type methylation states in cloned bovine pre-implantation embryos. Embo J 21:1092–1100.
- Kobayashi, H., Suda, C., Abe, T., Kohara, Y., Ikemura, T. and Sasaki, H. (2006) Bisulfite sequencing and dinucleotide content analysis of 15 imprinted mouse differentially methylated regions (DMRs): Paternally methylated DMRs contain less CpGs than maternally methylated DMRs. Cytogenet Genome Res 113:130–137.
- Lopez-Serra, L. and Esteller, M. (2008) Proteins that bind methylated DNA and human cancer: Reading the wrong words. Br J Cancer 98:1881–1885.
- Lucifero, D., Mertineit, C., Clarke, H. J., Bestor, T. H. and Trasler, J. M. (2002) Methylation dynamics of imprinted genes in mouse germ cells. Genomics 79:530–538.
- Lucifero, D., Suzuki, J., Bordignon, V., Martel, J., Vigneault, C., Therrien, J., Filion, F., Smith, L. C. and Trasler, J. M. (2006) Bovine SNRPN methylation imprint in oocytes and day 17 in vitro-produced and somatic cell nuclear transfer embryos. Biol Reprod 75:531–538.
- Mathews, D. H., Sabina, J., Zucker, M. and Turner, D. H. (1999) Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol 288:911–940.
- Millar, D. S., Warnecke, P. M., Melki, J. R. and Clark, S. J. (2002) Methylation sequencing from limiting DNA: Embryonic, fixed, and microdissected cells. Methods 27:108–113.
- Moore, K., Rodriguez-Sallaberry, C. J., Kramer, J. M., Johnson, S., Wroclawska, E., Goicoa, S. and Niasari-Naslaji, A. (2007) In vitro production of bovine embryos in medium supplemented with a serum replacer: Effects on blastocyst development, cryotolerance and survival to term. Theriogenology 68:1316–1325.
- Nelson, J. R., Cai, Y C, Giesler, T. L., Farchaus, J. W., Sundaram, S. T., Ortiz-Rivera, M., Hosta, L. P., Hewitt, P. L., Mamone, J. A., Palaniappan, C., et al. (2002) TempliPhi, phi29 DNA polymerase based rolling circle amplification of templates for DNA sequencing. Biotechniques (Suppl):44–47.
- Raizis, A. M., Schmitt, F. and Jost, J. P. (1995) A bisulfite method of 5-methylcytosine mapping that minimizes template degradation. Anal Biochem 226:161–166.
- Rastogi, P. A. (2000) MacVecor. Integrated sequence analysis for the Macintosh. Methods in Molecular Biology 132:47–69.
- Reagin, M. J., Giesler, T. L., Merla, A. L., Resetar-Gerke, J. M., Kapolka, K. M. and Mamone, J. A. (2003) TempliPhi: A sequencing template preparation procedure that eliminates overnight cultures and DNA purification. J BiomolTech 14:143–148.
- Shapiro, R., DiFate, V. and Welcher, M. (1974) Deamination of cytosine derivatives by bisulfite. Mechanism of the reaction. J Am Chem Soc 96:906–912.
- Singer-Sam, J., LeBon, J. M., Tanguay, R. L. and Riggs, A. D. (1990) A quantitative HpaII-PCR assay to measure methylation of DNA from a small number of cells. Nucleic Acids Res 18:687.
- Waggoner, D. (2007) Mechanisms of disease: Epigenesis. Semin Pediatr Neurol 14:7–14.
- Warnecke, P. M., Biniszkiewicz, D., Jaenisch, R., Frommer, M. and Clark, S. J. (1998) Sequence-specific methylation of the mouse H19 gene in embryonic cells deficient in the Dnmt-1 gene. Dev Genet 22:111–121.
- Warnecke, P. M., Mann, J. R., Frommer, M. and Clark, S. J. (1998) Bisulfite sequencing in preimplantation embryos: DNA methylation profile of the upstream region of the mouse imprinted H19 gene. Genomics 51:182–190.
- Warnecke, P. M., Stirzaker, C., Song, J., Grunau, C., Melki, J. R. and Clark, S. J. (2002) Identification and resolution of artifacts in bisulfite sequencing. Methods 27:101–107.
- Wells, D. N., Misica, P. M. and Tervit, H. R. (1999) Production of cloned calves following nuclear transfer with cultured adult mural granulose cells. Biol Reprod 60:996–1005.
- Young, L. E., Sinclair, K. D. and Wilmut, I. (1998) Large offspring syndrome in cattle and sheep. Rev Reprod 3:155–163.
- Zhang, Y L, Chen, T., Jiang, Y., Zhong, Z. S., Liu, S. Z., Hou, Y., Schatten, H., Chen, D. Y and Sun, Q. Y (2005) Active demethylation of individual genes in intracytoplasmic sperm injection rabbit embryos. Mol Reprod Dev 72:530–533.
- Zuker, M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Research 31:3406–3415.