Abstract
Severe male factor infertility may have a presently identifiable genetic basis. Y chromosomal microdeletions (e.g., an AZFc microdeletion), karyotypic anomalies (e.g., Klinefelter Syndrome), and mutations in both alleles of the cystic fibrosis transmembrane conductance regulator (CFTR) gene may be found, depending upon the etiology of the reproductive compromise. Which patients should be tested, what are the tests that can be performed, when should those tests be ordered, and what might a positive outcome mean, are all critically valuable clinical questions for the couple that help guide evaluation and management. It is imperative that they be asked and results discussed prior to any intervention such as testis tissue extraction, microsurgical epididymal sperm aspiration, or intracytoplasmic sperm injection so that the couple can incorporate them in their decisions moving forward vis-à-vis their reproductive choices and options. The role of reproductive medicine clinicians should not be limited to just helping couples establish a pregnancy, but instead be expanded to educating them about the reasons and causes of their reproductive failure that affect not only them as individuals but also may have implications for their offspring.
Introduction
When couples experience infertility, reproductive deficiency in the male is contributory in approximately 50% of cases. Therefore, an evaluation of the male partner should always be performed and include a detailed history, a comprehensive physical examination, semen analyses, and any other appropriate tests that those findings suggest [Sigman et al. Citation2009]. There are several known genetic anomalies and aberrations that result in severe male factor infertility and many, many more that we are not aware of yet [Matzuk and Lamb Citation2008; Nuti and Krausz Citation2008]. In which patients do we look for genetic anomalies and aberrations, what are the variety of tests that we can presently perform, when should we perform those tests, what does a positive result of those tests mean to the patient and his partner, and what might be the outcomes vis-à-vis the choices couples might make as a response to those positive results. These are all critically important clinical questions that guide the evaluation and management of each couple in terms of the elucidation of a potential genetic cause.
In whom do we look?
For the severely oligospermic (<5 million/cc) or azoospermic male, the immediate question arises as to what genetic tests to order. The answer lies not in a shotgun diagnostic approach where every test outlined below is obtained but rather by a more targeted approach that follows a detailed history and comprehensive physical examination [Oates and Lamb Citation2009]. For example, if the patient has three children easily conceived prior to intensive spermatotoxic chemotherapy for lymphoma and is presently azoospermic, there is no need for a Y chromosomal microdeletion assay (YCMD), a karyotype, or a cystic fibrosis mutation analysis. The etiology of his azoospermia is the residual of his cytotoxic therapy and is not secondary to a genetic anomaly. If physical examination demonstrates that the vasa are not palpable and the testes are normal in size and the azoospermic semen analysis shows low volume, and acidic pH, the diagnosis is congenital bilateral absence of the vas deferens (CBAVD) and a YCMD and karyotype do not need to be obtained while a cystic fibrosis mutation assay does. Finally, if the azoospermic semen volume is normal, the testes are small and soft, and the vasa are palpable, the clinical diagnosis is non-obstructive azoospermia (NOA) and a YCMD and karyotype are appropriate but a cystic fibrosis mutation analysis is not necessary. To summarize, a combination of history, physical examination, and semen analysis is used to broadly classify the patient's diagnosis either non-obstructive azoospermia (or severe oligospermia) or congenital bilateral absence of the vas deferens.
What tests should be done and what are they?
If the history, physical examination, and semen analysis demonstrate NOA or severe oligospermia, both a Y chromosomal microdeletion assay and karyotype need to be obtained [Oates and Lamb Citation2009]. The molecular geography of the Y chromosome is unusual and distinctive (). On the long arm there are eight palindromic stretches [Kuroda-Kawaguchi et al. Citation2001; Skaletsky et al. Citation2003]. In each, there is a central point from which arms with nearly identical base pair sequences radiate out in opposite directions. Each arm itself is comprised of subdivisions termed amplicons. These repetitive stretches allow for non-allelic homologous recombination (NAHR), a process of intra-chromosomal recombination between homologous repetitive sequences leading to the deletion of variably sized pieces [Reijo et al. Citation1995; Repping et al. Citation2002]. There are many genes and gene families that reside in these palindromes that are testis specific and probably involved in the spermatogenic process (DAZ, BPY, CDY1, etc) [Skaletsky et al. Citation2003]. When NAHR results in a microdeletion of a few specific chromosomal regions, spermatogenesis can be severely or completely abolished secondary to loss of the gene(s) that resided within these stretches. There are three clinically relevant microdeletions termed: AZFa, AZFb, and AZFc [Krausz and Degl'Innocenti Citation2006; Vogt Citation2005]. The length of a complete AZFa microdeletion is 0.8 Mb [Kamp et al. Citation2000; Sun et al. Citation2000]. The AZFa region is not within the eight palindromic areas on Yq but it is still the process of NAHR which is the causal mechanism for microdeletion [Sun et al. Citation2000]. AZFb (also termed P5/proximal P1), P4/distal P1, AZFc (also termed b2/b4), and AZFb/c combined microdeletion (also termed P5/distal P1) are overlapping microdeletion intervals resulting from different NAHR events, all within an expanse of Yq ranging from the distal aspect of the P5 palindrome all the way to the distal aspect of the P1 palindrome. Significant numbers of base pairs are lost: AZFb 6.2 Mb, P4/distal P1 7.0 Mb, AZFb/c 7.7 Mb, and AZFc 3.5 Mb [Repping et al. Citation2002]. The YCMD assay is a PCR-based blood test that detects the presence or the absence of defined sequence tagged sites (STSs) and therefore defines by the pattern of presence or absence any clinically relevant microdeletion region [Simoni et al. Citation2004]. Depending on the Y haplogroup and other unknown factors, the frequency of the various microdeletions found in an NOA male is, approximately: AZFa 1%, AZFb 1%, AZFb/c 1%, and AZFc 10%. AZFc microdeletions are also found in approximately 5% of the severely oligospermic male population [Simoni et al. Citation2008].
Figure 1. Clinically significant microdeletion intervals along the long arm of the Y chromosome (Tq).

The human male karyotype demonstrates 46 chromosomes (44 autosomes in 22 pairs and XY sex chromosomes) but anomalies of structure or number are often found in men with severe spermatogenic compromise. For example, 47, XXY Klinefelter syndrome occurs in 5 to 10% of NOA and in 1:600 live male births [Bojesen and Gravholt Citation2007]. Klinefelter men demonstrate deficits in both principal functions of the adult testis, spermatogenesis and testosterone production [Oates Citation2003]. There is a wide phenotypic spectrum and not all 47, XXY males are hypogonadal with a eunichoid body habitus as some may be first diagnosed at presentation for infertility [Yoshida et al. Citation1997]. If the androgenic output is adequate enough during teenage years, virilization and puberty may occur on schedule. Many of these men will have testosterone values in the range of 200 300 ng/dl. Therefore, it is quite simply the level of Leydig cell function during puberty that determines the non-testicular phenotype. All 47, XXY men, will have very small, atrophic testes [Paduch et al. Citation2008]. Karyotypic analysis in the NOA male may reveal 46, XX testicular disorder of sex development (46, XX male syndrome) which occurs with an incidence of 1:20,000 male newborns. The external phenotype is male, the internal phenotype is male, and the gonads are testes. The genetic etiology is typically a translocation of distal Yp containing SRY (a gene responsible for initiating the genetic cascade driving gonadal differentiation along testicular lines) [Vorona et al. Citation2007; Wang et al. Citation2009]. Truncated Y chromosomes, ring Y chromosomes, and isodicentric Y chromosomes, as well as autosomal translocations, may be rarely discovered in the azoospermic male with spermatogenic failure [Lange et al. Citation2009]. In summary, for an NOA male, Y chromosomal microdeletions are detected in approximately 13%, Klinefelter syndrome in up to 10%, and translocations and Y chromosomal structural anomalies in approximately 1%.
If the history, physical examination, and semen analyses demonstrate CBAVD, cystic fibrosis mutation analysis needs to be obtained. In CBAVD, the seminal vesicles are usually dysplastic, atrophic, and/or aplastic [Samli et al. Citation2006]. Since they contribute 70% of the fluid to the ejaculate and all of the alkalinity, the CBAVD male will have a low volume (approximately 0.6 mL) and acidic (approximate pH 6.5) azoospermic ejaculate [Turner Citation2009]. The cystic fibrosis gene is located on 7q, and encodes the cystic fibrosis transmembrane conductance regulator (CFTR) which is responsible for maintaining proper fluidity of epithelial secretions in the respiratory and exocrine pancreatic systems (Database: http://www.genet.sickkids.on.ca/cftr). When both maternally and paternally inherited CFTR alleles are mutated and the total pool of CFTR is severely dysfunctional (either quantitatively, qualitatively, or both), these secretions become thick and tenacious leading to obstructive pulmonary disease and exocrine pancreatic failure [Wilschanski et al. Citation2006]. Males will also have bilateral vasal agenesis. While clinical cystic fibrosis (CF) is at one end of the CFTR dysfunction spectrum, CBAVD is at the other as pulmonary and pancreatic function are adequate with vasal absence being the only phenotypic evidence. Where one falls between these two extremes depends on the combination of mutations inherited. If both mutations are ‘severe’ CF will result but if at least one mutation is ‘mild’, perhaps only CBAVD will be recognized clinically [Oates and Amos Citation1994; Uzun et al. Citation2005]. Of course a given individual may fall in between the two extremes with CBAVD and some mild respiratory or pancreatic disease. Cystic fibrosis mutation analysis can be carried out in many different ways, depending upon the level of intensity of the search for CFTR gene aberrations. A limited panel looking for 30 to 40 of the most common mutations, an expanded search panel searching a broader range of 100 or so abnormalities, or full gene sequencing with multiplex ligation-dependent probe amplification may be performed [Bareil et al. Citation2007; Database]. The most common mutation combination found in CBAVD men is ΔF508/5T. A recent study by Ratbi et al. [Citation2007] showed that 81% of their CBAVD men had both mutations identified with ΔF508 found in 30% of alleles and 5T on 27% of alleles.
Why order these genetic tests, why not just perform therapeutic interventions?
If the genetic basis for NOA or CBAVD had no clinical consequences for either the patient himself or for the couple, but simply provided an answer to an intellectual question, there would be no reason for the patient or the couple to pursue testing. Quite the opposite is true. In the NOA male, the detection of an AZFa, AZFb, or AZFb/c microdeletion on a YCMD assay predicts a complete absence of spermatogenesis [Brandell et al. Citation1998; Krausz et al. Citation2000]. In the NOA male, detection of 46, XX male syndrome (the entire Yq is absent) and many other severe Y chromosome karyotypic structural aberrations predicts a complete absence of spermatogenesis [Hopps et al. Citation2003]. Therefore, no testis sperm extraction (TESE) or testis biopsy for histological analysis is warranted. It is neither necessary nor helpful. In these circumstances, the YCMD and/or karyotype is prognostic and saves the male unnecessary surgery and cost and his female partner unnecessary stimulation and cost if a simultaneous TESE and intracytoplasmic sperm injection (ICSI) cycle would otherwise be the planned approach. How could it be fair to the patient or the couple not to have this predictive information before intensive, invasive, and expensive procedures are performed all without any hope of success?
For an NOA male with an AZFc microdeletion as the proximate cause for his spermatogenic impairment, the chances he will have sperm found on TESE are quite good (approximately 70%) and those sperm can effect fertilization, embryo development, and term pregnancy [Oates et al. Citation2002; Patrat et al. Citation2010]. This is encouraging information for the patient and his partner. Such is the case when 47, XXY Klinefelter syndrome is diagnosed as approximately 50% of those men have spermatozoa recovered with microsurgical TESE and, when combined with ICSI, fertilization, embryo development, and term pregnancy are all possible [Schiff et al. Citation2005]. It appears, as well, that the fears of 47, XXY or 47, XXX offspring have not come true with over 100 babies being reported, all either 46, XY or 46, XX [Fullerton et al. Citation2010; Sciurano et al. Citation2009]. There is, however, a slightly higher incidence of embryo aneuploidy involving autosomes 18 and 21, suggesting a possible role for preimplantation genetic screening [Staessen et al. Citation2003]. It is also important to diagnose the Klinefelter male so that the other clinical aspects/consequences of this condition can be managed appropriately. It is not just about his fertility [Swerdlow et al. Citation2005]. Those associations include low testosterone output (possible decreased libido, erectile capability, bone density, and an increased risk of metabolic syndrome); a possible increased incidence of Leydig cell, intracranial germ cell, and mediastinal germ cell tumors; a possible increased risk of breast carcinoma; and social and intellectual dysfunction.
For the male with CBAVD, it is important to pursue CF mutation analysis because he most likely has siblings, typically of childbearing age. He may be the first in his family to present with CF mutation related disease. Therefore, each one of his siblings may carry one or both of the mutations of the patient. Family counseling becomes very important to each and every brother and sister to refine and define their risk of passing along cystic fibrosis mutation related disease if their partner is a carrier for a CF mutation. It is also important to define CFTR status to allow for proper classification of the patient's prior and present medical history. For example, if the patient has had a lifelong history of sinusitis or bronchitis, perhaps the knowledge that he has cystic fibrosis mutation related disease changes the management of those two conditions for him. Therefore, it is not appropriate just to test the female partner in cases of CBAVD.
Might the patient's offspring benefit from these genetic tests?
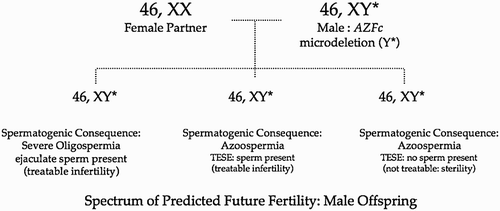
When we consider why we should order these tests, we should also view the offspring and their health as equally important, whether the offspring is male or female. We should consider the offspring our patients as well and feel a responsibility to them. For example, if the male patient carries an AZFc microdeletion, then all male offspring will inherit that microdeleted Y chromosome [Oates et al. Citation2002] (). It would be predicted, therefore, that the quantitative aspects of spermatogenesis in that male offspring will be severely reduced, the mildest expression being severe oligospermia and the most severe being azoospermia with sperm absent from the testis tissue. All male offspring will experience infertility or sterility. If it has been defined that an AZFc microdeletion is present prior to the initiation of advanced reproductive technologies (such as TESE and ICSI) then the couple can make choices [Stouffs et al. Citation2005]. Their choice may be to not use the sperm but to use donor spermatozoa instead. The couple may choose to use the male's sperm for ICSI with the realization that any daughter born will be normal both somatically and gonadally, and that every boy will experience spermatogenic compromise to a severe degree. However, that is their choice to make. The couple may also choose to employ preimplantation genetic diagnosis (PGD) so that only female embryos will be transferred. This is an appropriate use of PGD and couples should be encouraged to consider this option. Once the couple has been fully educated in regards to all of these options, then the choice they make is, by definition, the correct one for that couple because they made it with a strong educational foundation. If the couple elects to have an AZFc microdeleted son, they will need to be aware of accumulating knowledge/interventions that may help their son preserve or optimize any future fertility. In the next several years we may find out more in regards to the biology and pathophysiology of an AZFc microdeletion, perhaps even discovering therapies that may need to be instituted early in life, at puberty, or only as an adult. The awareness of their son's AZFc microdeletion may allow the couple to employ these possible therapies to help their son become a biological parent.
Figure 2. AZFc microdeletion: vertical transmission to all male offspring.
Certainly CF mutation analysis should always be ordered in both partners to define the risk for CF mutation related disease in their offspring. Depending upon the mutational status of the female partner, disease in an offspring could be quite severe (). Once again, the couple may elect to perform PGD and transfer only those embryos that will not be afflicted with cystic fibrosis mutation related disease in the future, by transferring only simple heterozygotes or completely wild type embryos.
Figure 3. Offspring phenotypes/genotypes: CBAVD male (DF508/5T) and carrier female (W1282X/+).

What about future genetic knowledge?
We must expand our thought processes to include consideration of the genetic basis of other conditions that may afflict our patients and may also lead to infertility. For example, there are many pediatric cancers which are now being cured, allowing those males to possibly achieve pregnancy, either naturally or through assisted means. If there is a genetic basis underlying that pediatric malignancy, we may be unwittingly passing that along to the next generation. In the field of male reproductive medicine we must think ‘genetics’: we must think of conditions that directly cause spermatogenic impairment/failure (AZFc microdeletions and Klinefelter syndrome), of conditions that indirectly cause infertility (cystic fibrosis mutations resulting in vasal agenesis), of conditions that will cause disease in the offspring (AZFc microdeletions and cystic fibrosis mutations), and of conditions that may cause disease in the offspring (pediatric cancers of various types that might have a genetic basis). We must stay aware of the expanding and explosive increase in genetic knowledge. We must not fall into the trap of just treating without regard to the education of our patients about their genetics, both for themselves and their children. Our goal should always be a happy, healthy family.
Declaration of Interest: The author reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
Abbreviations
| CFTR: | = | cystic fibrosis transmembrane conductance regulator |
| YCMD: | = | Y chromosomal microdeletion assay |
| CBAVD: | = | congenital bilateral absence of the vas deferens |
| NOA: | = | non-obstructive azoospermia |
| NAHR: | = | non-allelic homologous recombination |
| STSs: | = | sequence tagged sites |
| CF: | = | cystic fibrosis |
| TESE: | = | testis sperm extraction |
| ICSI: | = | intracytoplasmic sperm injection |
| PGD: | = | preimplantation genetic diagnosis. |
Related Research Data
References
- Bareil, C., Guittard, C., Altieri, J.P., Templin, C., Claustres, M. and des Georges, M. (2007) Comprehensive and rapid genotyping of mutations and haplotypes in congenital bilateral absence of the vas deferens and other cystic fibrosis transmembrane conductance regulator-related disorders. J Mol Diagn 9:582–588.
- Bojesen, A. and Gravholt, C.H. (2007) Klinefelter syndrome in clinical practice. Nat Clin Pract Urol 4:192–204.
- Brandell, R.A., Mielnik, A., Liotta, D., Ye, Z., Veeck, L.L., Palermo, G.D., (1998) AZFb deletions predict the absence of spermatozoa with testicular sperm extraction: preliminary report of a prognostic genetic test. Hum Reprod 13:2812–2815.
- Database, Cystic Fibrosis Mutation (CFM). http://www.genet.sickkids.on.ca/cftr.
- Fullerton, G., Hamilton, M. and Maheshwari, A. (2010) Should non-mosaic Klinefelter syndrome men be labelled as infertile in 2009? Hum Reprod 25:588–597.
- Hopps, C.V., Mielnik, A., Goldstein, M., Palermo, G.D., Rosenwaks, Z. and Schlegel, P.N. (2003) Detection of sperm in men with Y chromosome microdeletions of the AZFa, AZFb and AZFc regions. Hum Reprod 18:1660–1665.
- Kamp, C., Hirschmann, P., Voss, H., Huellen, K. and Vogt, P.H. (2000) Two long homologous retroviral sequence blocks in proximal Yq11 cause AZFa microdeletions as a result of intrachromosomal recombination events. Hum Mol Genet 9:2563–2572.
- Krausz, C. and Degl'Innocenti, S. (2006) Y chromosome and male infertility: update, 2006. Front Biosci 11:3049–3061.
- Krausz, C., Quintana-Murci, L. and McElreavey, K. (2000) Prognostic value of Y deletion analysis: what is the clinical prognostic value of Y chromosome microdeletion analysis? Hum Reprod 15:1431–1434.
- Kuroda-Kawaguchi, T., Skaletsky, H., Brown, L.G., Minx, P.J., Cordum, H.S., Waterston, R.H., (2001) The AZFc region of the Y chromosome features massive palindromes and uniform recurrent deletions in infertile men. Nat Genet 29:279–286.
- Lange, J., Skaletsky, H., van Daalen, S.K., Embry, S.L., Korver, C.M., Brown, L.G., (2009) Isodicentric Y chromosomes and sex disorders as byproducts of homologous recombination that maintains palindromes. Cell 138:855–869.
- Matzuk, M.M. and Lamb, D.J. (2008) The biology of infertility: research advances and clinical challenges. Nat Med 14:1197–1213.
- Nuti, F. and Krausz, C. (2008) Gene polymorphisms/mutations relevant to abnormal spermatogenesis. Reprod Biomed Online 16:504–513.
- Oates, R. and Lamb, D. (2009) Genetic aspects of infertility. In Infertility in the male eds. L. Lipshultz, , Cambridge University Press, New York, NY pp. 251–276.
- Oates, R.D. (2003) Clinical and diagnostic features of patients with suspected Klinefelter syndrome. J Androl 24:49–50.
- Oates, R.D. and Amos, J.A. (1994) The genetic basis of congenital bilateral absence of the vas deferens and cystic fibrosis. J Androl 15:1–8.
- Oates, R.D., Silber, S., Brown, L.G. and Page, D.C. (2002) Clinical characterization of 42 oligospermic or azoospermic men with microdeletion of the AZFc region of the Y chromosome, and of 18 children conceived via ICSI. Hum Reprod 17:2813–2824.
- Paduch, D.A., Fine, R.G., Bolyakov, A. and Kiper, J. (2008) New concepts in Klinefelter syndrome. Curr Opin Urol 18:621–627.
- Patrat, C., Bienvenu, T., Janny, L., Faure, A.K., Fauque, P., Aknin-Seifer, I., (2010) Clinical data and parenthood of 63 infertile and Y-microdeleted men. Fertil Steril 93:822–832.
- Ratbi, I., Legendre, M., Niel, F., Martin, J., Soufir, J. C., Izard, V., (2007) Detection of cystic fibrosis transmembrane conductance regulator (CFTR) gene rearrangements enriches the mutation spectrum in congenital bilateral absence of the vas deferens and impacts on genetic counselling. Hum Reprod 22:1285–1291.
- Reijo, R., Lee, T.Y., Salo, P., Alagappan, R., Brown, L. G., Rosenberg, M., (1995) Diverse spermatogenic defects in humans caused by Y chromosome deletions encompassing a novel RNA-binding protein gene. Nat Genet 10:383–393.
- Repping, S., Skaletsky, H., Lange, J., Silber, S., Van Der Veen, F., Oates, R.D., (2002) Recombination between palindromes P5 and P1 on the human Y chromosome causes massive deletions and spermatogenic failure. Am J Hum Genet 71:906–922.
- Samli, H., Samli, M.M., Yilmaz, E. and Imirzalioglu, N. (2006) Clinical, andrological and genetic characteristics of patients with congenital bilateral absence of vas deferens (CBAVD). Arch Androl 52:471–477.
- Schiff, J.D., Palermo, G.D., Veeck, L.L., Goldstein, M., Rosenwaks, Z. and Schlegel, P.N. (2005) Success of testicular sperm extraction [corrected] and intracytoplasmic sperm injection in men with Klinefelter syndrome. J Clin Endocrinol Metab 90:6263–6267.
- Sciurano, R.B., Luna Hisano, C.V., Rahn, M.I., Brugo Olmedo, S., Rey Valzacchi, G., Coco, R., (2009) Focal spermatogenesis originates in euploid germ cells in classical Klinefelter patients. Hum Reprod 24:2353–2360.
- Sigman, M., Lipshultz, L. and Howards, S. (2009) Office evaluation of the subfertile male. In Infertility in the male eds. L. Lipshultz, , Cambridge University Press, New York, NY pp. 153–176.
- Simoni, M., Bakker, E. and Krausz, C. (2004) EAA/EMQN best practice guidelines for molecular diagnosis of y-chromosomal microdeletions. State of the art 2004. Int J Androl 27:240–249.
- Simoni, M., Tuttelmann, F., Gromoll, J. and Nieschlag, E. (2008) Clinical consequences of microdeletions of the Y chromosome: the extended Munster experience. Reprod Biomed Online 16:289–303.
- Skaletsky, H., Kuroda-Kawaguchi, T., Minx, P.J., Cordum, H.S., Hillier, L., Brown, L. G., (2003) The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423:825–837.
- Staessen, C., Tournaye, H., Van Assche, E., Michiels, A., Van Landuyt, L., Devroey, P., (2003) PGD in 47,XXY Klinefelter's syndrome patients. Hum Reprod Update 9:319–330.
- Stouffs, K., Lissens, W., Tournaye, H., Van Steirteghem, A. and Liebaers, I. (2005) The choice and outcome of the fertility treatment of 38 couples in whom the male partner has a Yq microdeletion. Hum Reprod 20:1887–1896.
- Sun, C., Skaletsky, H., Rozen, S., Gromoll, J., Nieschlag, E., Oates, R., (2000) Deletion of azoospermia factor a (AZFa) region of human Y chromosome caused by recombination between HERV15 proviruses. Hum Mol Genet 9:2291–2296.
- Swerdlow, A.J., Schoemaker, M.J., Higgins, C.D., Wright, A.F. and Jacobs, P.A. (2005) Cancer incidence and mortality in men with Klinefelter syndrome: a cohort study. J Natl Cancer Inst 97:1204–1210.
- Turner, T. (2009) The epididymis and accessory sex organs. In Infertility in the male eds. L. Lipshultz, , Cambridge University Press, New York, NY pp. 90–103.
- Uzun, S., Gokce, S. and Wagner, K. (2005) Cystic fibrosis transmembrane conductance regulator gene mutations in infertile males with congenital bilateral absence of the vas deferens. Tohoku J Exp Med 207:279–285.
- Vogt, P.H. (2005) AZF deletions and Y chromosomal haplogroups: history and update based on sequence. Hum Reprod Update 11:319–336.
- Vorona, E., Zitzmann, M., Gromoll, J., Schuring, A.N. and Nieschlag, E. (2007) Clinical, endocrinological and epigenetic features of the 46, XX male syndrome compared to 47, XXY Klinefelter patients. J Clin Endocrinol Metab 92:3458–3465.
- Wang, T., Liu, J.H., Yang, J., Chen, J. and Ye, Z.Q. (2009) 46, XX male sex reversal syndrome: a case report and review of the genetic basis. Andrologia 41:59–62.
- Wilschanski, M., Dupuis, A., Ellis, L., Jarvi, K., Zielenski, J., Tullis, E., (2006) Mutations in the cystic fibrosis transmembrane regulator gene and in vivo transepithelial potentials. Am J Respir Crit Care Med 174:787–794.
- Yoshida, A., Miura, K., Nagao, K., Hara, H., Ishii, N. and Shirai, M. (1997) Sexual function and clinical features of patients with Klinefelter's syndrome with the chief complaint of male infertility. Int J Androl 20:80–85.