Abstract
The aim of this study was to investigate whether direct injection of nonviral DNA into the oviductal lumen and subsequent in vivo electroporation leads to in vivo gene transfer in mouse preimplantation embryos present within an oviduct, as an alternative to the pre-existing pronuclear microinjection-based transgenesis. With this technique, effects of expression of the gene of interest (GOI) on mouse preimplantation development can be monitored with relative ease. Superovulated 4-week-old B6C3F1 female mice (hybrids between C57BL/6N and C3H/HeN) were mated with adult B6C3F1 male mice. Two days later, females that had been identified as pregnant, based on the presence of copulation plugs, were injected with 1 µl of a solution containing an enhanced green fluorescent protein (EGFP) expression plasmid (0.5 µg) and 0.05% trypan blue. The entire oviduct was then electroporated using tweezer-type electrodes with 8 square-wave pulses of 50 V each with 50-ms duration. The next day, the 8-cell stage embryos were collected, and their number, morphology, and EGFP-derived fluorescence were recorded. Of the 12 oviducts (6 females used) examined, 3 contained fluorescent 8-cell stage embryos (33%, 19/58 tested), but the intensity of fluorescence varied among the embryos. In total, 10% (19/192 tested) of the embryos were fluorescent and the fluorescence was maintained in these embryos after 1 day of culture. However, the fluorescence disappeared in the late gestational stage fetuses, and the transgenes could not be detected. Our results indicate that it is possible to transfect in vivo preimplantation embryos, although the success rate appears to be relatively low and gene expression is transient. This technology may provide a new method for manipulating preimplantation embryos in vivo, by using, for example, Cre-mediated conditional DNA recombination.
Introduction
Methods used to transfer genes of interest (GOI) into cells/early embryos are powerful tools for studying the function of GOI and for the production of animal models of human diseases. Several methods have been proposed for performing gene transfer into preimplantation embryos, including pronuclear injection [Gordon et al. Citation1980; Palmiter et al. Citation1982; Gordon and Ruddle Citation1986], infection with retroviruses, lentiviruses, and adenoviruses [Jaenisch Citation1976; Stewart et al. Citation1987; Tsukui et al. Citation1995; Tsukui et al. Citation1996; Lois et al. Citation2002], in vitro electroporation [Grabarek et al. Citation2002], receptor-mediated transfection [Ivanova et al. Citation1999], and liposomal transfection [Carballada et al. Citation2000; Carballada et al. Citation2002]. All of these approaches involve early embryos isolated from the reproductive tracts of pregnant animals, with the in vitro transfected embryos being returned to the reproductive tract (oviduct or uterus) of pseudopregnant recipient females for further development. Direct gene delivery to preimplantation embryos in vivo would simplify the above ex vivo approach, because such an approach would not require the isolation of embryos, cultivation of transfected embryos, and their transfer to a recipient. This will in turn be helpful to reduce the number of animals used for experiments. However, to our knowledge, no attempt has been made to transfect preimplantation embryos in vivo, although successful gene transfer has been reported in post-implanted fetuses by direct gene delivery into the uterine horn of pregnant mice [Kimura et al. Citation2005; Koyama et al. Citation2006] or by indirect gene delivery to fetuses through intravenous administration of DNA [Tsukamoto et al. Citation1995; Kikuchi et al. Citation2002; O'Shea et al. 2006].
For effective gene transfer into preimplantation embryos in vivo, cleavage stage embryos would be the most appropriate target. Embryos at this stage are freely present in the oviductal lumen, into which we can easily introduce a transgene-containing solution, using a micropipette attached to a mouthpiece. Notably, in vivo electroporation, including gene delivery to eyes of newborn mice [Matsuda and Cepko Citation2004] and to fetal brain [Saito Citation2006], has been widely used and accepted by researchers as a powerful tool for efficient in vivo gene delivery [a review by Muramatsu et al. Citation1998]. Using this system, we previously found that direct injection of plasmid DNA [harboring an enhanced green fluorescent protein (EGFP) expression unit] into oviducts through the oviductal wall, and subsequent in vivo electroporation, resulted in transfection of the oviductal epithelium with an efficiency of approximately 40% [Sato Citation2005]. However, there were no fluorescent preimplantation embryos recovered from the DNA-injected females, and this technology was therefore termed ‘gene transfer to the oviductal epithelium’ (GTOVE) [Sato Citation2005]. Why were preimplantation embryos not successfully transfected using this method? In this case, gene transfer had been performed at Day 0.4 of pregnancy, when the vaginal plug first became apparent after mating. Notably, at this stage, fertilized single-cell eggs are still surrounded by a mass of cumulus cells, which could hamper efficient gene delivery. We therefore predicted that intraoviductal injection of DNA at Day 1.6 of pregnancy would result in successful transfection of 2-cell embryos, at which stage the cumulus cells are no longer present.
To test this hypothesis, we performed GTOVE, using plasmids coding for EGFP or β-galactosidase as reporter genes at Day 1.6 of pregnancy. In this study, we evaluated the feasibility, usefulness, and potential of this new approach of in vivo gene transfer into preimplantation embryos.
Results
Variation in EGFP fluorescence-intensity among GTOVE-treated oviducts
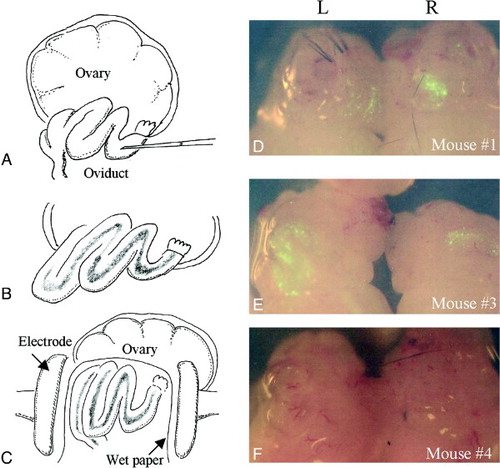
As we have previously shown, when the EGFP expression vector is introduced into oviducts by GTOVE (A–C), the oviductal epithelial lining and oviductal lumen are efficiently transfected [Sato Citation2005]. Six females (12 oviducts) were first subjected to GTOVE at Day 1.6 of pregnancy. The following day, the fluorescence in these cells was visible when the GTOVE-treated oviducts were dissected and observed under a fluorescence stereomicroscope (D–F). However, variations in the degree and intensity of fluorescence were observed among treated oviducts. For example, the oviduct classified as ++ (L in E) exhibited bright fluorescence spanning over 50% of the oviduct, while fluorescence in the oviduct classified as +/- (L in F) was very faint and was limited to a small area.
Figure 1. A–C) Schematic representation of the GTOVE procedure at Day 1.6 of pregnancy. One microliter of a solution, containing plasmid DNA and trypan blue dye, was injected into the oviductal lumen at the ampulla using a micropipette (A). After introduction, rapid flow of the injected material inside the oviducts was observed under a stereomicroscope (B). Immediately after injection, entire oviducts were covered with small pieces of wet paper, and were then gently grasped in tweezer-type electrodes (C), just before in vivo electroporation. D–F) Fluorescence in the oviducts 1 day after GTOVE. After the dissection of the ovary/oviduct/uterus from a GTOVE-treated female, fluorescence was inspected under a fluorescence stereomicroscope. In these panels, samples from mice #1, #3, and #4 (see Table 1) are shown. GTOVE: gene transfer to the oviductal epithelium and subsequent in vivo electroporation; L: left ovary/oviduct/uterus; R: right ovary/oviduct/uterus
Successful in vivo transfection of preimplantation embryos
After analyzing the fluorescence of oviducts dissected 1 day after surgery, embryos at the 8-cell stage were recovered by flushing the oviductal lumen. Fluorescence expression was then immediately monitored under an Olympus BX60 fluorescence microscope. Of 12 oviducts (6 females) examined, 3 (25%) possessed fluorescent embryos, although the levels of fluorescence were variable among the embryos (; A, B). For example, embryos marked as No. 1 to No. 3 exhibited relatively bright fluorescence, while the level of fluorescence in those marked No. 4 to No. 6 was weak or very faint (B). These differences in the intensity of EGFP expression suggest that variable amounts of the pCE-29 plasmids may have been taken up by the preimplantation embryos. In total, 10% (19/192 tested) of the embryos were fluorescent (). None of the control embryos, i.e., those recovered from females that had been injected with plasmid DNA, but had not been electroporated, exhibited fluorescence (; C, D). When the morphology of the embryos derived from GTOVE-treated mice was assessed, almost all (83%, 160/192) exhibited normal morphology (). For example, 85% (39/46) of the embryos obtained from the GTOVE-treated mouse No. 3 appeared normal, compared to control embryos (A vs. C in ).
Figure 2. Fluorescence in 8-cell embryos recovered from oviducts 1 day after GTOVE. A) Embryos recovered from both oviducts of GTOVE-treated mouse #3 were inspected for fluorescence under a fluorescence microscope. B) Magnified view of the box region shown in (A). Note the variation in fluorescence intensity among embryos. For example, embryos #1–3 exhibit brighter fluorescence than embryos #4–6. C) Embryos recovered from both oviducts of the control female mouse #7. D) Magnified view of the box region shown in (C). Note that no fluorescence is apparent in control embryos. GTOVE: gene transfer to the oviductal epithelium and subsequent in vivo electroporation; A–D) observed under light + UV illumination. Scale bars = 100 µm.
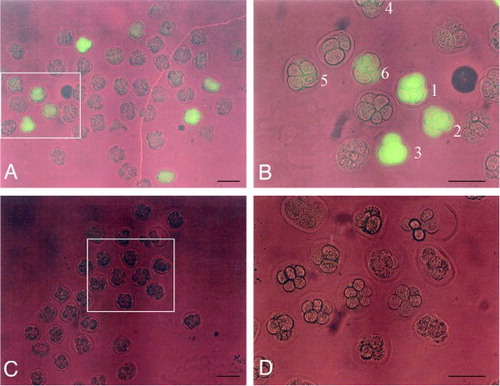
Table 1. Summary of the properties of females treated with intraoviductal gene transfer and their offspring (8-cell embryos).
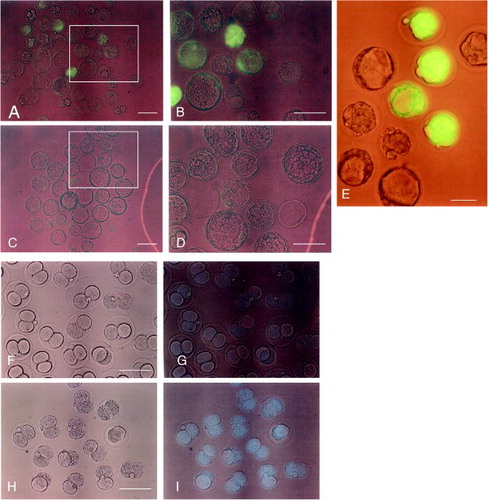
The fluorescent 8-cell stage embryos were cultured for an additional day to determine whether normal development could continue and if fluorescence could be maintained. As shown in A and B, the 8-cell stage embryos that were recovered from GTOVE-treated females successfully developed into early blastocysts. However, as expected, variation in fluorescence intensity was observed among these embryos. This was in contrast to blastocysts that were obtained from mating heterozygous MNCE-36 Tg mice and non-transgenic mice, which exhibited uniform and bright fluorescence (E). Control embryos also developed normally in vitro, but did not exhibit fluorescence (C, D).
Figure 3. Fluorescence at the blastocyst stage after 1 day culture of 8-cell embryos. A) Embryos derived from those shown in Figure 2. A, B. B) Magnified view of the box region shown in (A). C) Embryos derived from those shown in Figure 2. C, D. D) Magnified view of the box region shown in (C). E) Embryos derived from mating between a heterozygous MNCE-36 transgenic female and a non-transgenic male. Note the uniform, bright fluorescence among transgenic embryos. F–I) Two cell embryos recovered 6 h after instillation of Hoechst 33342 (H, I), or of water alone (F, G). F, H) observed under light; A–E, G, I) observed under light + UV illumination. Scale bars = 100 µm.
Confirmation that injected materials were uniformly distributed throughout the oviductal lumen
In this study, GTOVE was performed on 12 oviducts of 6 pregnant females, and preimplantation embryos were recovered from each female. However, only 3 oviducts delivered fluorescent embryos, while the remaining 9 did not (see ). This raises the possibility that the DNA solution introduced into the oviductal lumen may not have been uniformly distributed throughout each oviduct. To test this possibility, we injected Hoechst 33342, a fluorescent dye capable of staining the nuclei of cells, into the oviductal lumens of 2 pregnant normal females at Day 1.6 of pregnancy. After 6 h, a total of forty-six 2-cell embryos were collected, and the number of embryos that had been stained with the dye were determined. All of the recovered embryos exhibited Hoechst 33342-derived fluorescence (I), suggesting uniform distribution of the injected solution within each oviduct.
Confirmation that levels of fluorescence in GTOVE-treated embryos are correlated with levels of EGFP mRNA
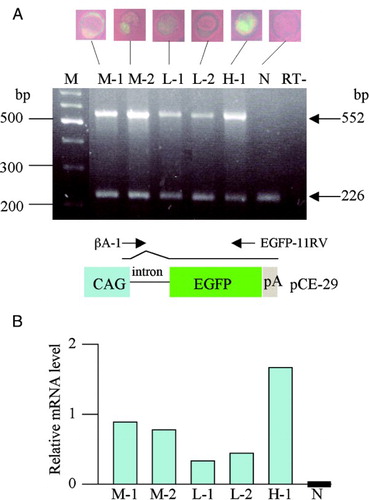
As mentioned above, we observed variations in the levels of EGFP-derived fluorescence among embryos recovered from GTOVE-treated females. To confirm that the levels of observed fluorescence are correlated with those of EGFP mRNA, semi-quantitative RT-PCR was performed using a single embryo. In this case, embryos exhibiting various levels of EGFP-derived fluorescence were selected and subjected to RT-PCR. The results are shown in A. As expected, the fluorescent embryos (including an embryo exhibiting relatively strong fluorescence, designated H-1), those exhibiting moderate fluorescence (designated M-1 to M-2), and those exhibiting faint fluorescence (designated L-1 to L-2) all had EGFP transcripts, but non-fluorescent control embryos (designated N) did not. Densitometric scanning of a band corresponding to EGFP mRNA also confirmed the above findings (B).
Figure 4. Semiquantitative RT-PCR analysis of EGFP and β-actin gene expression in GTOVE-derived blastocysts. A) A single blastocyst was subjected to lysis and subsequent RT-PCR for amplification of portions of EGFP (552 bp) and β-actin (226 bp) mRNAs, as described in Materials and Methods. M, 100-bp ladder markers; lane H-1, blastocyst strongly expressing EGFP; lanes M-1 to M-2, blastocysts moderately expressing EGFP; lane L-1 to L-2, blastocysts faintly expressing EGFP; lane N, non-transgenic blastocyst; lane RT-, distilled water (negative control). Below the RT-PCR data, a schematic representation of the structure of pCE-29 is shown. mRNA generated from the CAG promoter becomes mature after splicing at the 1st intron. The positions of βA-1 and EGFP-11RV primers are shown above the vector. CAG, cytomegalovirus enhancer and chicken β-actin promoter; EGFP, EGFP cDNA; pA, poly(A) site. B) Relative mRNA levels after normalization to levels of β-actin mRNA. Densitometric analysis of experiments was performed using Image J software and the data were shown as graphs. The EGFP mRNA band in the control (N) was set at ‘0’. GTOVE: gene transfer to the oviductal epithelium and subsequent in vivo electroporation
Inability to detect fluorescence or the presence of transgenes in late gestational fetuses
To monitor EGFP-derived fluorescence in post-GTOVE fetuses and to determine the presence of exogenous DNA, fetuses at Day 18.6 of pregnancy were dissected, inspected for fluorescence, and analyzed for the presence of pCE-29 plasmid by PCR-Southern analysis. Of 5 GTOVE-treated females, 4 were successfully pregnant and 60 fetuses were recovered. Unfortunately, none of the fetuses exhibited fluorescence, nor did they exhibit a positive PCR band corresponding to the transgenes (data not shown).
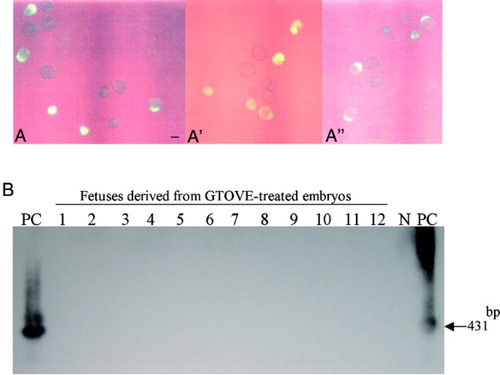
Since, as shown in , the transfection rate achieved by GTOVE was low, it was possible that no embryos had been transfected in these experiments. Therefore, we next set out to collect fluorescent embryos from GTOVE-treated females. Of 8 uteri (4 females used), 3 delivered blastocysts at Day 3.6 of pregnancy, 17 of which were fluorescent and 20 non-fluorescent (A, A', and A''). Fluorescent blastocysts were then transferred to the uteri of 3 pseudopregnant recipient ICR females for further development. When these females were inspected at Day 18.6 of pregnancy, 2 were pregnant, and subsequently 12 fetuses were recovered. As expected, all were negative for fluorescence (data not shown), and PCR-Southern analysis of their tail DNA failed to show the presence of transgenes in their genome (B).
Figure 5. A-A'') Blastocysts (Day 3.6 of pregnancy) recovered from 3 uteri of GTOVE-treated females. Of a total of 37 embryos recovered, 17 exhibited cytoplasmic EGFP-derived fluorescence. A, A', and A'' correspond to embryos isolated from each uterus. A-A'', observed under light + UV illumination. Scale bars = 100 µm. B) PCR-Southern analysis of tail DNA from fetuses developed from the blastocysts shown in A, A', and A''. No positive bands (indicated by the arrow at 431 bp) were detectable in these fetal samples (lanes 1 to 12). GTOVE: gene transfer to the oviductal epithelium and subsequent in vivo electroporation; PC: positive control (pCE-29); N: negative control (tail DNA from non-transgenic mouse)
Confirmation that preimplantation embryos can be successfully transfected in vivo
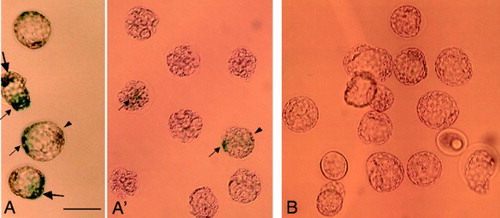
To confirm that another reporter gene can be successfully transfected into preimplantation embryos using GTOVE, we injected a lacZ expression plasmid pCAGGS-lacZ [Niwa et al. Citation1991] into the oviductal lumen of females at Day 1.6 of pregnancy. This plasmid expresses β-galactosidase upon transfection, and the transfected cells stain blue in the presence of an appropriate substrate, such as X-Gal [Sambrook et al. Citation1989]. After GTOVE, blastocysts at Day 3.6 of pregnancy were recovered from the uteri and cultured for 1 day prior to cytochemical staining for lacZ activity. Of a total of 8 oviducts (4 females used) injected, lacZ-expressing blastocysts were successfully recovered from 2 uteri (A and A'). Notably, almost all blastocysts exhibited a mosaic pattern of expression for lacZ activity; namely, some parts of an embryo were stained blue (arrows in A and A'), but other parts were not stained (arrowheads in A and A'). Control embryos that had been injected with plasmid DNA, but which had not been electroporated, were completely negative for lacZ activity (B).
Figure 6. LacZ activity in 1 day-cultured blastocysts, recovered from uteri 2 days after GTOVE. A, A') Embryos were cytochemically stained for lacZ activity, as described in Materials and Methods. Embryos in A and A' were isolated on different days. Note that almost all embryos exhibited mosaic staining (indicated by arrows). B) Control embryos isolated from mock-injected females. GTOVE: gene transfer to the oviductal epithelium and subsequent in vivo electroporation
Discussion
In our previous studies [Sato et al. 2005], 18 late morula/early blastocysts were recovered 3 days after GTOVE, but only 3 of these exhibited weak fluorescence in the blastocoel and 2 exhibited distinct fluorescence in the perivitelline space. However, EGFP fluorescence was completely lost after these EGFP-positive embryos were cultured for 1 day. On the basis of these findings, we concluded that plasmid DNA cannot be taken up by preimplantation embryos in vivo after GTOVE. However, as previously mentioned, this appears to be because GTOVE was performed at Day 0.4 of pregnancy, which corresponds to a stage during which fertilized single-cell eggs are still surrounded by cumulus cells, which may hamper the efficiency of gene delivery to the eggs. In the present study, we performed GTOVE at Day 1.6 of pregnancy, which corresponds to the 2-cell stage when cumulus cells have already been lost. Consequently, we confirmed the presence of fluorescent preimplantation embryos in oviducts with epithelia that exhibited bright fluorescence over a wide area.
It could be argued that fluorescent embryos recovered from GTOVE-treated oviducts may have been derived from a transgenic line that expresses EGFP constitutively. During the period that our GTOVE-related experiments were performed, we maintained EGFP-expressing transgenic lines, such as MNCE-36, in our laboratory. However, as shown in E, each transgenic preimplantation embryo derived from the MNCE-36 line exhibited uniform and bright fluorescence. This is in contrast to our current findings where fluorescence intensity varied among embryos, suggesting that the number of transgenes introduced into the oviductal lumen and taken up by each embryo is variable. Another possibility is that exogenous DNA is not easily taken up by zona pellucida (ZP)-intact embryos, because the ZP is thought to be a barrier that protects cells against invasion of foreign materials [Chen et al. 1989; Legge Citation1995]. However, successful introduction of plasmid DNA into ZP-intact preimplantation embryos has already been reported by others who used in vitro electroporation to enhance the uptake of exogenous DNA [Grabarek et al. Citation2002]. It would therefore be reasonable to hypothesize that GTOVE can confer gene delivery to preimplantation embryos in vivo, as well as to the oviductal epithelium.
In this study, of 12 DNA-injected oviducts, only 3 delivered fluorescent embryos (see ). Among embryos that exhibited successful transfection, variations in fluorescence intensity were observed (see ). This raised the possibility that injected DNA may not have been uniformly distributed within the oviducts. However, this possibility is not supported by experiments using Hoechst 33342 (see F–I). The most probable explanation for variation in gene transfer rate is that the strength of electric pulses provided by the pulse generator may vary among samples. Upon in vivo electroporation, oviducts must be held by tweezer-type electrodes. Since this is usually performed by hand, the distance between the sample and the electrodes may differ between experiments, which may in turn cause variation in the strength of electrical pulses.
Is there any indicator that enables transfection efficiency in GTOVE-mediated in vivo gene transfer to be predicted? We noted that there is a possible correlation between oviduct fluorescence and successful transfection of in vivo preimplantation embryos. As shown in , GTOVE-treated oviducts that express high or moderate fluorescence intensity contained fluorescent embryos, while those expressing weak, or no, fluorescence, did not.
The length of time that fluorescence can be maintained in GTOVE-treated embryos, and the degree of transgene integration into the mouse genome in the offspring of those embryos, may be of interest. We addressed these points using late-stage gestational fetuses at Day 18.6 of pregnancy. We obtained a total of 60 fetuses from 4 pregnant GTOVE-treated females. Unfortunately, they were all negative for both expression of fluorescence and the presence of transgenes. Thus, this present technique will be applicable to monitor effects of GOI on preimplantation development of mouse embryos within a limited period (probably 2-4 days after gene transfer). Furthermore, transfer of fluorescent blastocysts collected from the GTOVE-treated females to pseudopregnant recipient females yielded 12 fetuses at Day 18.6 of pregnancy, but all were negative for both expression of fluorescence and presence of the transgene (see ). This suggests that there was a failure of transgene integration into the host genome. This failure may be due, at least in part, to the use of circular plasmid DNA in GTOVE, since circular DNA has been shown to be less efficient with respect to gene integration than linear DNA [Chen et al. Citation2001; Chancham and Hughes Citation2001]. Alternatively, upon GTOVE, DNA might not have been uniformly incorporated into all blastomeres comprising 2-cell embryos. This idea is supported by the findings that when a lacZ expression vector was employed for GTOVE-mediated gene transfer, almost all of the resulting blastocysts exhibited a mosaic pattern of expression of lacZ upon cytochemical staining (see A, A'). This mosaic expression pattern was transient and was lost during embryonic development.
However, the transient nature of gene expression that occurs when using the in vivo gene delivery system described here can be useful when researchers want to perform Cre-mediated conditional DNA recombination [Lewandoski Citation2001] in vivo, to invert or delete DNA fragments in cis, or to switch DNA fragments in trans. For example, we generated a floxed transgenic mouse strain, known as CETZ-17 [Sato et al. Citation2000]. This strain carries a transgene comprising a CAG promoter, loxP-flanked EGFP cDNA, and the lacZ gene. The construct normally expresses EGFP-derived fluorescence, but not lacZ, systemically. When GTOVE is performed using a Cre expression vector in the CETZ-17 strain, the preimplantation embryos that are transiently transfected by the vector exhibit Cre-mediated excision of the floxed EGFP cDNA sequence in the integrated CETZ-17 transgene, and consequently expression of the lacZ gene commences.
This study has demonstrated that GTOVE confers in vivo gene transfer to preimplantation embryos, although the success rate is relatively low and gene expression is transient. Future studies will aim to improve transfection efficiency and to refine this novel technology so that it is suitable for the in vivo genetic manipulation of preimplantation embryos for transgenic studies.
Materials and Methods
Mice
Female B6C3F1 mice (hybrids between C57BL6/N and C3H/HeN; purchased from CLEA Japan Inc., Tokyo, Japan) aged 4 w were intraperitoneally (IP) injected with eCG (ASKA Pharmaceutical Co., Ltd., Tokyo, Japan) and hCG (ASKA Pharmaceutical Co., Ltd.) 48 h apart to induce superovulation. After hCG injection, each female was immediately mated with an adult B6C3F1 male (12 to 20 weeks old; CLEA Japan Inc.). Superovulated heterozygous MNCE-36 transgenic females [Sato et al. Citation2001], aged 4 w, were also mated with normal adult B6C3F1 males. This transgenic line was used as a control for EGFP expression, since the individuals expressed EGFP systemically under the control of the chicken β-actin promoter-based CAG [Niwa et al. Citation1991]. For all mated females, the day that the copulation plug was first observed in the morning, was designated D 0.4 of pregnancy. Pregnant females were subjected to surgery at D 1.6 of pregnancy, at which stage 2-cell embryos were present in the oviducts.
The mice were maintained on a 12-h light/12-h dark schedule (lights on from 0700 h to 1900 h) and were provided with food and water ad libitum. The experiments described were performed according to the Guide for the Care and Use of Laboratory Animals at Tokai University and were approved by the Animal Care Committee of Tokai University, School of Medicine. The experiments describing in vivo transfection of preimplantation mouse embryos by GTOVE are accompanied by surgery (exposure of ovary/oviducts) and operation/manipulation (intraoviductal injection of DNA and in vivo electroporation). All efforts were made to minimize the number of animals used and their suffering.
Plasmid DNA
The EGFP expression plasmid, pCE-29 ([Sato et al. Citation2002]; shown in the lower panel of A), was used in this study. The pCE-29 plasmid consists of a CAG promoter, introns of both the chicken β-actin and rabbit β-globin genes, EGFP cDNA (isolated from pEGFP-N1; Invitrogen Co., Carlsbad, CA, USA), and poly(A) sites of the SV40 virus. The lacZ expression plasmid, pCAGGS-lacZ [Niwa et al. Citation1991], was also used in this study. This plasmid consists of a CAG promoter, introns of both the chicken β-actin and rabbit β-globin genes, lacZ cDNA, and poly(A) sites of the rabbit β-globin gene. Circular plasmid DNA was dissolved in water to a concentration of 0.5 µg/μl. Just before GTOVE, trypan blue (Trypan Blue Stain 0.4%; Invitrogen Co.) was added to the DNA solution to a final concentration of 0.05% to enable visualization of the fate of the injected solution.
GTOVE
Intraoviductal injection of the DNA solution was performed as previously described [Sato Citation2005], and part of this procedure is schematically illustrated in A–C. Each pregnant female at E 1.6 was anesthetized with sodium pentobarbital. For injection of the DNA solution, 1 µl of solution, containing the pCE-29 plasmid and trypan blue, was slowly injected into the ampulla of an oviduct using a glass micropipette with a mouthpiece attached (A). After completing the injection procedure, the micropipette was rapidly removed. The fate of the trypan blue-containing solution was clearly visible under a stereomicroscope (B). Immediately after the DNA solution had been introduced, the oviductal regions were covered with a small piece of wet paper (Kimwipe; Jujo-Kimberly Co. Ltd., Tokyo, Japan), and were grasped in tweezer-type electrodes, with disc electrodes (diameter, 5 mm) at their tips (#CT-234; NEPA GENE Co. Ltd., Ichikawa, Chiba, Japan) (C). Eight square-wave pulses, with a pulse duration of 50 ms and an electric field intensity of 50 V, were administered from a square-wave pulse generator (T820; BTX Genetronics Inc., San Diego, CA, USA). The gaps between the electrodes used to hold the oviduct were approximately 2 mm. After gene transfer, the oviducts were returned to their original position to allow the preimplantation embryos to continue development. Females that were injected with the pCE-29 plasmid, but were not electroporated, were used as controls.
The following day or 2nd day, GTOVE-treated females were sacrificed for harvesting 8-cell, or blastocyst stage, embryos. In some cases, mice were maintained beyond the preimplantation stage, and were sacrificed at D 18.6 of pregnancy in order to check for fluorescence expression in the fetuses and for the presence of introduced transgenes in their genome. In some experiments, blastocysts recovered from GTOVE-treated females were inspected for fluorescence and immediately transferred to the uterine horn of pseudopregnant recipient ICR females (10 to 15 weeks old; CLEA Japan Inc.). The recipients were sacrificed at D 18.6 of pregnancy for assessment of transgene expression and integration.
Instillation of Hoechst 33342 into the oviductal lumen
Superovulated, normal B6C3F1 females (aged 4 w) that were proven to be pregnant were subjected to intraoviductal injection with 1 µl of water containing 1 mg/ml of Hoechst 33342 (Calbiochem Inc., San Diego, CA, USA) on D 1.6 of pregnancy. However, these mice were not electroporated in vivo. As controls, equivalent females were injected with Hoechst 33342-free water. To examine whether the preimplantation embryos were successfully stained with Hoechst 33342 in vivo, 2-cell stage embryos were collected 6 h after surgery and were immediately inspected for Hoechst 33342-derived fluorescence.
Collection and embryo culture
Eight-cell stage embryos were collected from oviducts at D 2.6 of pregnancy, 1 d after GTOVE had been carried out. The recovered embryos were inspected for expression of EGFP-derived green fluorescence, as described below, and their morphology was assessed. The embryos were then cultured with M16 medium [Whittingham Citation1971] in a Terasaki microtest plate (Nunc Inc., Naperville, IL, USA). Cultures were carried out for 1 d under paraffin oil and in an atmosphere of 5% CO2 at 37°C, until the early blastocyst stage.
Observation of EGFP fluorescence
Ovary-oviducts were excised from treated mice and were directly inspected for fluorescence under an Olympus SZX12 fluorescence stereomicroscope with DM505 filters (BP460-490 and BA510IF). The intensity of fluorescence was categorized as ++ (strong), + (moderate), +/- (faint), or – (no fluorescence). A rating of ++ indicates that more than 50% of an oviduct exhibited bright fluorescence (left (L) oviduct in E), while + indicates that 10%–40% of an oviduct exhibited bright fluorescence (L and right (R) oviducts in D), and +/- indicates that less than 10% of an oviduct exhibited fluorescence (L oviduct in F). A rating of – indicates that no fluorescence was observed (R oviduct in F).
EGFP-derived fluorescence in isolated preimplantation embryos was assessed using an Olympus BX60 fluorescence microscope with DM505 filters. Fluorescence ratings were determined by observing fluorescence intensity in 50% or more of an embryo. For example, ++ embryos exhibited strong fluorescence (as exemplified by the embryos marked as No. 1 to No. 3 in B); + embryos exhibited moderate fluorescence (as exemplified by the embryo marked as No. 6 in B); +/- embryos exhibited faint fluorescence (as exemplified by the embryos marked as No. 4 and No. 5 in B); – embryos exhibited no fluorescence. EGFP-derived fluorescence in isolated fetuses (D 18.6 of pregnancy) was assessed using the SZX12 fluorescence stereomicroscope. Hoechst 33342-derived fluorescence was also assessed using U-MWU filters (Olympus). Microphotographs were obtained using a digital camera (FUJIX HC-300/OL; Fuji Film, Tokyo, Japan) and were printed using a digital color printer (CP700DSA; Mitsubishi, Tokyo, Japan).
Cytochemical detection of lacZ activity
Blastocysts were first fixed in 4% paraformaldehyde in phosphate-buffered saline, without Ca2+ and Mg2+, pH 7.2 [PBS(-)] for 5 min at room temperature, and were then subjected to cytochemical staining for lacZ using the substrate, X-Gal, as described previously [Sato et al. Citation2000].
Detection of transgenes by polymerase chain reaction (PCR) and Southern blotting
Genomic DNA was isolated from the tail region of fetuses, as previously described [Blin and Stafford Citation1976], but with several modifications [Sato et al. Citation1994]. PCR amplification reactions were performed in a total volume of 10 µl reaction mixture, containing 10 mM Tris-HCl (pH 8.3); 50 mM KCl; 1.5 mM MgCl2; 0.25 mM each of dATP, dCTP, dGTP, and dTTP; 1 mM each of forward (5'-GCG ATG CCA CCT ACG GCA AGC-3') and reverse (5'-GAG CTG CAC GCT GCC GTC CTC-3') primers [Hyun et al. Citation2003], corresponding to the EGFP cDNA sequence (Accession No. U76561); 2 µl of genomic DNA; and 0.5 units of rTaq polymerase (#R001; TaKaRa Shuzo Co. Ltd., Tokyo, Japan). This primer set produces 431-bp fragments from the middle portion of the EGFP cDNA. PCR (40 cycles) was performed at 96°C for 10 s, 56°C for 1 min, and 72°C for 2 min. The resulting PCR products (5 µl) were separated on a 2% agarose gel and were stained with ethidium bromide (EtBr) for DNA visualization. In some cases, the gels were transferred to GeneScreenPlus nylon filters (#NEF976, PerkinElmer Life Sciences, Inc., Boston, MA, USA), and these filters were subjected to Southern hybridization, as described previously [Sato et al. Citation1994]. The probe used was a 32P-labeled 0.9-kb EGFP cDNA fragment, isolated from pEGFP-N1.
Isolation of total RNA and RT-PCR analysis
Total RNA was isolated from embryos, according to the method described by Klebe et al. [1996]. The first-strand cDNA was synthesized by RT (reverse transcription) using a Gene AmpR RNA PCR Kit (Applied Biosystems, Foster City, CA). The RT reaction mix included 6 µl of total RNA, 2 µl of 2 mM dNTPs, 1 µl of RNase inhibitor (1 U/μl), 1 µl of murine leukemia virus reverse transcriptase (2.5 U/μl), 4 µl of X5 RT-buffer, 1 µl of 2.5 nmol/mL oligo d(T)16 (Applied Biosystems), and 5 µl of RNase-free water in a total volume of 20 µl. The reaction conditions were as follows: 42°C for 60 min, 99°C for 5 min, and 4°C for 5 min. Finally, the RT mixture was stored at -20°C prior to use. For PCR analysis of the RT products, 5 µl of the RT mixture was mixed with 20 µl of a solution containing 2 µl of 2.5 mM MgC12, 2 µ l of 2 mM dNTPs, 2.5 µl of 10× PCR buffer, 0.2 µl of GeneAmpR AmpliTaq GoldR DNA Polymerase (5 U/μl; Applied Biosystems), 12.3 µl of water, and 1 µl of each primer (0.6 mM) in a total volume of 25 µl. The forward βA-1 primer (5'-TCT GAC TGA CCG CGT TAC TCC CAC A-3') and the reverse EGFP-11RV primer (5'-GTC CTC GAT GTT GTG GCG GAT-3'), corresponding to the region immediately downstream of the transcription initiation site of the chicken β-actin promoter in the CAG promoter (see pCE-29 construct in lower panel of A; Niwa et al. Citation1991), and the 3' end portion of EGFP cDNA, respectively, were used for detection of EGFP mRNA. This primer set yields a 552-base pair (bp) product. The forward mβA-3S primer (5'-GAG CTG CCT GAC GGC CAG GTC-3') and the reverse mβA-3RV primer (5'-CCT TCT GCA TCC TGT CAG CAA-3') correspond to the 3' region of endogenous mouse β-actin mRNA (Accession No. MN_007393.3). This primer set yields a 226-bp RT product. The PCR reaction conditions were as follows: 50 cycles of 95°C for 1 min, 56°C for 1 min, and 72°C for 1 min. The RT-PCR products were analyzed by electrophoresis on 2.0% agarose gels and photographed. A non-transgenic blastocyst was used as the negative control (N). Distilled water was also subjected to RT-PCR as negative control (RT-).
The intensity of the bands was quantitated by densitometry using Image J software (version 1.33U, a program inspired by NIH image; http://rsb.info.nih.gov/ij/index.html). Product intensities were then normalized by comparison to those of β-actin. Values from each embryo were expressed as graphs.
Declaration of interest: This study was supported by a grant from The Ministry of Education, Science, Sports and Culture, Japan. The authors have no conflicts of interest to report.
Author Contributions: Masahiro Sato was responsible for the conceptual design of this project, performed the cloning and a part of the GTOVE studies, and drafted the main part of the manuscript. Eri Akasaka and Makoto Saitoh performed molecular analyses of tail DNA and blastocyst samples. Masato Ohtsuka performed GTOVE, and Satoshi Watanabe helped to draft the manuscript.
Abbreviations
| E: | = | embryonic day |
| EGFP: | = | enhanced green fluorescent protein |
| GOI: | = | gene of interest |
| GTOVE: | = | gene transfer to the oviductal epithelium and subsequent in vivo electroporation |
| PCR: | = | polymerase chain reaction |
| Tg: | = | transgenic |
| ZP: | = | zona pellucida. |
References
- Blin, N. and Stafford, D.W. (1976) A general method for isolation of high molecular weight DNA from eukaryotes. Nucleic Acids Res 3:2303–2308.
- Carballada, R., Degefa, T. and Esponda, P. (2000) Transfection of mouse eggs and embryos using DNA combined to cationic liposomes. Mol Reprod Dev 56:360–365.
- Carballada, R., Relloso, M. and Esponda, P. (2002) Generation of transgenic mice by transfection of pronuclear embryos using lipid-DNA complexes. Zygote 10:209–216.
- Chancham, P. and Hughes, J.A.J. (2001) Relationship between plasmid DNA topological forms and in vitro transfection. Liposome Res 11:139–152.
- Chen, S.S. and Wrathall, A.E. (1989) The importance of the zona pellucida for disease control in livestock by embryo transfer. Br Vet J 145:129–140.
- Chen, Z.Y., Yant, S.R., He, C.Y., Meuse, L., Shen, S. and Kay, M.A. (2001) Linear DNAs concatemerize in vivo and result in sustained transgene expression in mouse liver. Mol Ther 3:403–410.
- Gordon, J.W., Scangos, G.A., Plotkin, D.J., Barbosa, J.A. and Ruddle, F.H. (1980) Genetic transformation of mouse embryos by microinjection of purified DNA. Proc Natl Acad Sci USA 77:7380–7384.
- Gordon, K. and Ruddle, F.H. (1986) Gene transfer into mouse embryos. Dev Biol 4:1–36.
- Grabarek, J.B.,Plusa, B., Glover, D.M. and Zernicka-Goetz, M. (2002) Efficient delivery of dsRNA into zona-enclosed mouse oocytes and preimplantation embryos by electroporation. Genesis 32:269–276.
- Hyun, S., Lee, G., Kim, D., Kim, H., Lee, S., Nam, D., (2003) Production of nuclear transfer-derived piglets using porcine fetal fibroblasts transfected with the enhanced green fluorescent protein. Biol Reprod 69:1060–1068.
- Ivanova, M.M., Rosenkranz, A.A., Smirnova, O.A., Nikitin, V.A., Sobolev, A.S., Landa, V., (1999) Receptor-mediated transport of foreign DNA into preimplantation mammalian embryos. Mol Reprod Dev 54:112–120.
- Jaenisch, R. (1976) Germ line integration and Mendelian transmission of the exogenous Moloney leukemia virus. Proc Natl Acad Sci USA 73:1260–1264.
- Kikuchi, N., Nakamura, S., Ohtsuka, M., Kimura, M. and Sato, M. (2002) Possible mechanism of gene transfer into early to mid-gestational mouse fetuses by tail vein injection. Gene Ther 9:1529–1541.
- Kimura, T., Nakamura, H., Koyama, S., Ogita, K., Tabata, C., Tsutsui, T., (2005) In vivo gene transfer into the mouse uterus: a powerful tool for investigating implantation physiology. J Reprod Immunol 67:13–20.
- Klebe, R.J., Grant, G.M., Grant, A.M., Garcia, M.A., Giambernardi, T.A. and Taylor, G.P. (1996) RT-PCR without RNA isolation. Biotechniques 21:1094–1100.
- Koyama, S., Kimura, T., Ogita, K., Nakamura, H., Tabata, C., Md Abu Hadi Noor Ali, K., (2006) Simple and highly efficient method for transient in vivo gene transfer to mid-late pregnant mouse uterus. J Reprod Immunol 70:59–69.
- Legge, M. (1995) Oocyte and zygote zona pellucida permeability to macromolecules. J Exp Zool 271:145–150.
- Lewandoski, M. (2001) Conditional control of gene expression in the mouse. Nat Rev Genet 2:743–755.
- Lois, C., Hong, E.J., Pease, S., Brown, E.J. and Baltimore, D. (2002) Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295:868–872.
- Niwa, H., Yamamura, K. and Miyazaki, J. (1991) Efficient selection for high-expression transformants with a novel eukaryotic vector. Gene 108:193–200.
- O'shea, K.S., De Boer, L.S., Slawny, N.A. and Gratsch, T.E. (2006) Transplacental RNAi: deciphering gene function in the postimplantation-staged embryo. J Biomed Biotechnol 2006:1–12.
- Matsuda, T. and Cepko, C.L. (2004) Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci USA 101:16–22.
- Muramatsu, T., Nakamura, A. and Park, H.M. (1998) In vivo electroporation: a powerful and convenient means of nonviral gene transfer to tissues of living animals (review). Int J Mol Med 1:55–62.
- Palmiter, R.D., Brinster, R.L., Hammer, R.E., Trumbauer, M.E., Rosenfeld, M.G., Birnberg, N.C. (1982) Dramatic growth of mice that develop from eggs microinjected with metallothionein-growth hormone fusion genes. Nature 300:611–615.
- Sambrook, J., Fritsche, E. and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Press, Cold Spring Harbor, New York, USA.
- Sato, M., Iwase, R., Kasai, K. and Tada, N. (1994) Direct injection of foreign DNA into mouse testis as a possible alternative of sperm-mediated gene transfer. Animal Biotech 5:19–31.
- Sato, M., Yasuoka, Y., Kodama, H., Watanabe, T., Miyazaki, J. and Kimura, M. (2000) New approach to cell lineage analysis in mammals using the Cre-loxP system. Mol Reprod Dev 56:34–44.
- Sato, M., Watanabe, T., Oshida, A., Nagashima, A., Miyazaki, J. and Kimura, M. (2001) Usefulness of double gene construct for rapid identification of transgenic mice exhibiting tissue-specific gene expression. Mol Reprod Dev 60:446–456.
- Sato, M., Ishikawa, A. and Kimura, M. (2002) Direct injection of foreign DNA into mouse testis as a possible in vivo gene transfer system via epididymal spermatozoa. Mol Reprod Dev 61: 49–56.
- Sato, M. (2005) Intraoviductal introduction of plasmid DNA and subsequent electroporation for efficient in vivo gene transfer to murine oviductal epithelium. Mol Reprod Dev 71:321–330.
- Saito, T. (2006) In vivo electroporation in the embryonic mouse central nervous system. Nat Prot 1:1552–1558.
- Stewart, C.L., Schuetze, S., Vanek, M. and Wagner, E.F. (1987) Expression of retroviral vectors in transgenic mice obtained by embryo infection. EMBO J 6:383–388.
- Tsukamoto, M., Ochiya, T., Yoshida, S., Sugimura, T. and Terada, M. (1995) Gene transfer and expression in progeny after intravenous DNA injection into pregnant mice. Nat Genet 9:243–248.
- Tsukui, T., Miyake, S., Azuma, S., Ichise, H., Saito, I. and Toyoda, Y. (1995) Gene transfer and expression in mouse preimplantation embryos by recombinant adenovirus vector. Mol Reprod Dev 42:291–297.
- Tsukui, T., Kanegae, Y., Saito, I. and Toyoda, Y. (1996) Transgenesis by adenovirus-mediated gene transfer into mouse zona-free eggs. Nat Biotechnol 14:982–985.
- Whittingham, D.G. (1971) Culture of mouse ova. J Reprod Fertil Suppl 14:7–21.