Abstract
The genetic diagnosis and screening of preimplantation embryos generated by assisted reproduction technology has been consolidated in the prenatal care framework. The rapid evolution of DNA technologies is tending to molecular approaches. Our intention is to present a detailed methodological view, showing different diagnostic strategies based on molecular techniques that are currently applied in preimplantation genetic diagnosis. The amount of DNA from one single, or a few cells, obtained by embryo biopsy is a limiting factor for the molecular analysis. In this sense, genetic laboratories have developed molecular protocols considering this restrictive condition. Nevertheless, the development of whole-genome amplification methods has allowed preimplantation genetic diagnosis for two or more indications simultaneously, like the selection of histocompatible embryos plus detection of monogenic diseases or aneuploidies. Moreover, molecular techniques have permitted preimplantation genetic screening to progress, by implementing microarray-based comparative genome hybridization. Finally, a future view of the embryo-genetics field based on molecular advances is proposed. The normalization, cost-effectiveness analysis, and new technological tools are the next topics for preimplantation genetic diagnosis and screening. Concomitantly, these additions to assisted reproduction technologies could have a positive effect on the schedules of preimplantation studies.
Introduction
Couples with a known genetic risk should start their reproductive project with accurate preconception genetic counselling. From the prevention point of view, all the information concerning different reproductive options should be exposed, ensuring that patients understand what is being explained. However, the rapid evolution of methodological approaches in genetic diagnosis makes this ideal situation difficult, particularly in the assisted reproductive field. In this context, the preimplantation genetic diagnosis (PGD) and preimplantation genetic screening (PGS) underwent a fast advance very recently. PGD was proposed as a very early form of genetic diagnosis in 1990 [Handyside et al. Citation1990] with the purpose of avoiding the transmission of genetic diseases to offspring. Nowadays, PGD represents a well-established alternative to prenatal diagnosis and termination of pregnancy, in couples at risk of transmitting disorders.
Since the first PGD application, it has been made available for a large number of rare genetic disorders [Preimplantation Genetic Diagnosis International Society Citation2008; Harper et al. Citation2012] and the number of cycles increases year by year (Harper et al. Citation2010b). The implementation of new technologies to PGD increases the diagnostic capacity and extends the application of PGD, not only for the most common monogenic diseases [Fiorentino et al. Citation2006], but also for screening aneuploidies on in vitro fertilization (IVF) patients (advanced maternal age or recurrent miscarriage factor) [Verlinsky et al. Citation1999; Munnè et al. 2002; Rubio et al. Citation2003]. Nevertheless, this last application has recently been subjected to a deep and interesting debate, reorienting the initial methodology [Harper et al. Citation2010a]. Also, this has permitted a new systematic analysis of the PGD indication [Harper and Harton Citation2010]. Moreover, PGD is also applied to more complex cases like single gene disorders combined with fluorescent in situ hybridization (FISH)-based aneuploidy studies [Verlinsky Citation2006] or human leukocyte antigen (HLA) matching [Verlinsky et al. Citation2001; Fiorentino et al. Citation2004; Fiorentino et al. Citation2005]. PGD has even been applied to HLA haplotyping combined with aneuploidy testing [Rechitsky Citation2006]. The combination of various PCR-based analyses on single cells, has permitted two indications simultaneously, for example testing two single gene disorders in the same couple [Altarescu et al. Citation2007; Alberola et al. Citation2009]. In addition, the recent application of microarray technologies in this field is quickly changing the diagnostic strategies. This new approach allows combining single gene PGD with aneuploidy screening of 23 autosomes and sexual chromosomes, simultaneously [Brezina et al. Citation2011]. Recently, there has been a clear tendency to perform molecular techniques. In this sense, aneuploidy screening and chromosomal rearrangement studies are quickly being replaced by molecular biology approaches based on microarray platforms (reviewed by [Harper and Harton Citation2010]), but also based on the analysis of short tandem repeat (STR) haplotypes [Fiorentino et al. Citation2010; Traversa et al. Citation2010].
In this fast-evolving scenario, a comprehensive review has been published recently [Harper and SenGupta 2012] revisiting the historical evolution of PGD/PGS, considering organization and methodological aspects. Our intention is to provide a detailed methodological view, explaining different diagnostic strategies based on molecular techniques that are currently applied in PGD/PGS from a genomic perspective.
Preconception Genetic Assessment
The referral of couples for PGD/PGS is not a spontaneous decision. Before PGD/PGS can be performed, there are some labor-intensive steps. In all cases, the accurate preconception genetic assessment is mandatory in order to clarify family history, compile clinical and genetic reports (if they exist), explain genetic disorders (if it applies), and evaluate the PGD/PGS request and IVF cycle in combination with biopsy procedures. The genetic risk, the success rates, the risk of misdiagnosis, and the importance of prenatal diagnosis in case of pregnancy must be specifically discussed. The complete PGD/PGS process must be explained to the patients and their queries must be answered. Finally, alternative reproductive options should be mentioned and a comprehensive informed consent form must be signed. Usually, these reproductive genetic studies are being offered to couples by physicians or reproductive medicine professionals from IVF clinics. In most cases, genetic studies are performed in collaboration with an external specialized genetics laboratory. The interaction of these two groups of experts is essential, not only for the coordination of PGD cycles but also for the exchange of knowledge in order to offer couples and families the best diagnostic options. The need for this interaction has been pointed out in various reports from the European Union (EU) and the rest of the world [Recommendations of the European Societies of Human Genetics and Human Reproduction and Embryology Citation2006; Soini et al. Citation2006; Harton et al. Citation2011a].
In Vitro Fertilization and Embryo Biopsy
A PGD/PGS working scheme includes, in all cases, performing an IVF cycle in order to generate embryos in the laboratory. Patients undergo a standard cycle of ovulation induction and each oocyte is microinjected with a single spermatozoon (intracytoplasmic sperm injection, ICSI). Mostly, the embryo biopsy is performed at day +3 of in vitro culture, when they are at the 6-8 cell stage, and one or two blastomeres are removed for study. Genetic testing of blastomeres allows extrapolating the embryo genetic status. Afterwards, euploid or free of the disease embryos are transferred to the maternal uterus. The timing for embryo biopsy is under a continuous discussion. Nowadays, in concordance with the application of microarray technology, other times for biopsy have been considered, like polar body [Geraedts et al. Citation2011] or trophectoderm analysis [Mamas et al. Citation2012].
Strategies in PGD for Single Gene Disorders
The essential advance in molecular PGD laboratories has been the incorporation of fluorescent polymerase chain reaction (f-PCR). Currently, the preferred technique is multiplex f-PCR, nested or heminested [Spits and Sermon Citation2009]. Before describing the different diagnostic approaches, it is important to state some particularities of the single cell PCR.
Single cell PCR
The most important limitation in molecular PGD is the amount of DNA in a single cell, of about 6 pg. Performing a nested or heminested PCR allows the reaction to be more specific. In this case, a first round PCR with outer primers followed by a second round PCR with fluorescently labelled inner primers allows good results.
There are problems inherent to single cell PCR, like contamination. The high number of PCR cycles performed in order to make the reaction more specific and the limited amount of DNA, make single cell PCR prone to contamination. This can be prevented by physically separating the pre-PCR, PCR, and post-PCR working areas, and having special conditions of sterility [Harton et al. Citation2011b]. Also, contamination can be from maternal or paternal origin. Removing the cumulus cells around the oocyte or performing ICSI, respectively, avoids this problem. Other sources of contamination can be from the PCR or biopsy technician, although it can be detected by making a blank for each blastomere and using multiplex PCR.
A third problem of single cell PCR is allele drop-out (ADO), the random non-amplification of one of the alleles in a heterozygous sample. It could be a cause of adverse misdiagnosis [Wilton et al. Citation2009] as ADO of the mutated allele could be interpreted as a normal embryo in autosomal dominant diseases. Also, in autosomal recessive diseases, ADO of the affected allele could alter the number of carrier embryos available for transfer, compromising the success of the PGD cycle. There are different hypotheses about the origin of ADO. Nevertheless, there is not a consensus about its origin, but the limiting number of DNA copies (two) available in the blastomere appears to be the most accepted cause. Concerning the ways of reducing ADO, increasing the denaturing temperature and choosing alkaline lysis or proteinase K-SDS method can help [Piyamongkol et al. Citation2003; Thornhill et al. Citation2005].
Preclinical informativity studies and validation
A particularity of PGD for single gene disorders is the need of an informativity study before starting the IVF cycle for each couple [Alberola et al. Citation2009, Alberola et al. Citation2011; Bautista-Llácer et al. Citation2010; Vendrell et al. Citation2011]. Preclinical studies are performed on single cells (lymphocytes, fibroblasts, or buccal mucosa, depending on the laboratory) from both members of the couple and relatives. The purpose of the informativity study is different depending on the type of diagnostic approach (direct or indirect PGD, see below). Specifically, the informativity test is intended to confirm the disease-causing mutation/s and to find the informative polymorphic STR markers linked to the mutation/s or genic regions implicated. STR markers are sequences of two, three, four, or even five nucleotides repeated in tandem, typically found in intron regions throughout the genome. The number of repeats can vary between individuals. These STR markers would support the diagnosis in case of direct diagnosis PGD. Besides, they are used to establish haplotypes in case of indirect analysis. Moreover, PCR amplification efficiency and ADO rates on single cells are evaluated as the same optimized conditions are expected on blastomeres [Thornhill et al. Citation2005; Harton et al. Citation2011b].
These preclinical tests allow establishing the inheritance of these disorders in each couple and give robustness to the diagnostic procedure. However, there are couples applying for PGD with no family history, with a de novo disease-causing mutation. In these particular cases, informativity work-ups based on sperm or oocytes analysis have recently been assessed [Rechitsky et al. Citation2011].
Indirect PGD
The indirect approach is based on haplotype studies [Renwick Citation2006; Renwick et al. Citation2010]. Basically, a set of STR markers linked to the disease-causing gene are studied in the family. STR markers should be, ideally, physically close (less than 1 Mb) to the gene of interest. Also, they must be at both sides of the gene and be fully informative in order to be able to detect eventual recombination. The objective is to identify ‘high risk’ and ‘low risk’ haplotypes in the family, with the purpose of inferring the embryo genetic status ().
Figure 1. Indirect preimplantation genetic diagnosis (PGD) for spinal muscular atrophy. A) Location and distances to the SMN1 gene of the short tandem repeat microsatellite markers used. B) Family tree showing the relatives involved in the informativity study and PGD cycle. Both members of the couple are carriers of a deletion involving exons 7 and 8 in the SMN1 gene. The dotted line represents the paternal disease-bearing haplotype. The thick black line illustrates the maternal disease-bearing haplotype. C) Electropherograms of the PGD for spinal muscular atrophy. Examples for two of the embryos analyzed. The numbers represent fragment sizes in base pairs for every allele. Numbers in bold stand for the alleles associated to the maternal mutation; numbers in bold and italics correspond to the alleles associated to the paternal mutation. Cen: centromere; tel: telomere; Mb: megabases; ADO: allele drop-out.
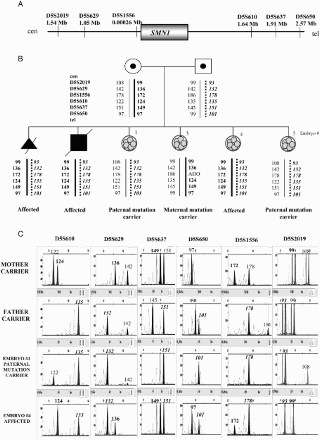
This haplotyping strategy allows one to carry out PGD when the disease-causing gene is identified but not the mutation; when, due to the nature of the mutation, direct genotyping cannot be performed (expansions, duplications, and deletions bigger than 500 base pairs, GC enriched regions) or even when there exist homologous pseudogenes. Furthermore, in dynamic mutations due to triplet expansions, the general strategy consists of amplifying the region of expansion [Sermon et al. Citation2001]. Sometimes (e.g., myotonic dystrophy) expanded alleles cannot be detected by standard PCR due to the size of the mutation and only the healthy allele amplifies. In these cases, the informativity study prior to PGD is essential in order to determine the high and low-risk haplotypes.
One interesting advantage of this methodology is the possibility of standardization for a particular single gene disorder. A few standardized applications have been proposed [Gigarel et al. Citation2008, Fassihi et al. Citation2010]. This wide-scope approach is very useful in cases where DNA from the parents and at least one affected member, or sufficient unaffected members of a family are available in order to identify low and high-risk haplotypes.
Indirect PGD is limited in particular cases. Occasionally, there are couples/families with a non-informative STR marker panel. Moreover, there are genetic regions where it is impossible to find enough STR markers flanking both sides of the gene of interest. This is an indispensable requirement in indirect studies in order to detect recombination events and avoid adverse misdiagnosis. Also, families where the affected members are dead, presenting more than one disease simultaneously, or de novo mutations, require a specific direct approach. In cases of extremely rare genetic disorders with a wide genetic heterogeneity, the setting-up and validation of a generic strategy is a laborious task for the very few cases expected. In our opinion, a case by case strategy is the gold standard option for couples at risk of extremely rare disorders [Vendrell et al. Citation2011].
Direct PGD
The direct approach allows interrogating the mutation(s) involved in the disease directly on the blastomere. Most of the genetic disorders diagnosed by PGD involve a heterogeneous spectrum of mutations. This has influenced the progress of direct strategies on single cell to fluorescent-based, computer-assisted, and standardized PCR protocols. In this sense, it is possible to establish a general strategy depending on the mutation type: small deletions, duplications, expansions, or insertions (below 500 base pairs) or point mutations. Informative STR markers flanking the region involving the mutation, located at both sides of the gene are included in all cases. This confers robustness, as it supports the direct genotyping and allows detecting ADO, contamination, and eventual recombination.
PGD for small expansions, deletions, duplications, or insertions is performed by fragment analysis (), generally by means of fluorescent multiplex PCR [Thornhill et al. Citation2005]. In the case of point mutations, minisequencing has permitted the analysis of specific mutation/s without sequencing the entire PCR product [Fiorentino et al. Citation2003]. The region involving the mutation is amplified by PCR and the PCR product is analyzed by minisequencing. Only one nucleotide is elongated, as the minisequencing primer is designed to terminate one nucleotide before the mutation. Therefore, only one dideoxynucleotide of the four intervening in the reaction is incorporated. Each dideoxynucleotide is fluorescently labelled with different fluorochromes emitting in different length waves detected by capillary electrophoresis ().
Figure 2. Direct preimplantation genetic diagnosis (PGD) for Huntington disease. A) Position and distances to the HTT gene of the short tandem repeat microsatellite markers involved in the informativity study and PGD cycle. B) Genealogic tree of the family taking part in the study and PGD results. The arrow designates the consultant affected patient. The thick black line illustrates the expansion-bearing haplotype. C) Electropherograms of the PGD for Huntington disease. Some examples of the embryos analyzed are displayed. The numbers represent fragment sizes (in base pairs) for every allele. Numbers corresponding to the disease-bearing allele are in bold. Cen: centromere; tel: telomere; Mb: megabases; ADO: allele drop-out.
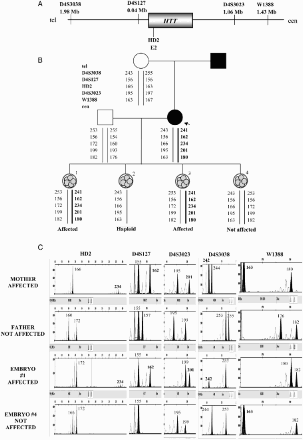
Figure 3. Direct PGD for cystic fibrosis. A) Location and distances to the CFTR gene of the short tandem repeat microsatellite markers used in the preclinical study and preimplantation genetic diagnosis (PGD) cycle. B) Pedigree of the family involved in the study and PGD results. The mother carries the mutation c.2988 + 1G > A in intron 16 of the CFTR gene. The father carries the mutation c.489 + 1G > T in intron 4 of the CFTR gene. The affected daughter carries both mutations in compound heterozygosis. These two positions were minisequenced in the embryos. The dotted line represents the paternal disease-bearing haplotype. The thick black line represents the maternal disease-bearing haplotype. C) Electropherograms of the PGD for cystic fibrosis. Some examples are shown for embryos 1 and 4. The numbers represent fragment sizes for every allele in base pairs. Numbers in bold represent the alleles associated to the maternal mutation; numbers in bold and italics correspond for the alleles associated to the paternal mutation. Cen: centromere; tel: telomere; Mb: megabases; G:guanine; A: adenine; T: timine.
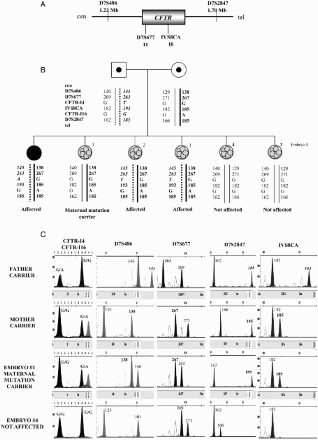
There are some other techniques to study point mutations in PGD, like digestion with restriction endonucleases, amplification refractory mutation system (ARMS) or double-ARMS, and real-time PCR. Restriction endonucleases allow distinguishing between two alleles, normal and mutated, when the mutation is inside the restriction site [Goossens et al. Citation2000]. The ARMS technique uses one or two highly specific primers to detect the mutation of interest by PCR; if there is amplification, the mutation is present [Moutou et al. Citation2007]. However, these two techniques are being replaced by minisequencing, as they need very strict controls to be able to distinguish between ADO and non-amplification. Finally, real-time PCR strategy combined with STR-based haplotype analysis has been proposed [Traeger-Synodinos Citation2006]. Briefly, this kind of PCR uses fluorescent probes or dyes and the accumulation of amplicons produced throughout the progress of PCR reaction is monitored.
Direct approaches in PGD present a series of advantages: i) it allows PGD in couples with de novo mutations or in non-informative couples where standardized methodology is not possible, ii) it also allows the combination of different techniques in recessive diseases and in combined indications (i.e., single gene disorders plus aneuploidy screening by microarray technology), and iii) it decreases the possibility of adverse misdiagnosis in cases where the disease-bearing haplotypes are wrongly assigned [Altarescu et al. Citation2008] or where a few STR markers are available, or when they are poorly distributed along the region of study [Wilton et al. Citation2009]. Finally, the application of different direct methodologies allows increasing the number of diseases for which PGD is possible. Nevertheless, direct analysis presents an important challenge, which is to know the mutation responsible for a single gene disorder. This is complicated in disorders of very low prevalence or in polygenic diseases.
Whole Genome Amplification
The PCR-based methods presented have many advantages, but there are still some limitations. These methods cannot meet the needs of whole genome research. It is well-known that the limiting factor of any technique applied to PGD is the amount of DNA available. One approach that increases the quantity of DNA from one single cell is whole genome amplification (WGA) technologies [Coskun and Alsmadi Citation2007]. Basically, it is based on the general amplification of the whole genomic sequence, yielding microgram quantities of DNA while respecting the original sequence. There are PCR and non-PCR methods of WGA, thoroughly reviewed by Zheng and coworkers [2011]. WGA allows combining different indications in the same PGD cycle, like HLA haplotyping or microarray technology like comparative genomic hybridization (CGH) plus detection of a single gene disorder [Handyside et al. Citation2004; Brezina 2011].
HLA-Compatible Embryos
The PGD strategy used by couples with a son requiring hematopoietic transplantation from a HLA compatible donor is particularly remarkable. Moreover, if the disorder is genetic and parents are carriers of the mutation-causing disease, PGD is more complex. In these cases, PGD for the monogenic disorder plus HLA haplotyping is performed, in order to select healthy and compatible embryos with the previous affected sibling [Fiorentino et al. Citation2004; Fiorentino et al. Citation2005; Rechitsky Citation2006; Van de Velde et al. Citation2009). An informativity study is required in order to identify the informative STR markers. Eight to 13 informative STRs are selected (according to different groups), distributed along the HLA region, spanning 40 Mb on chromosome 6p. Multiple displacement amplification (MDA) or multiplex PCR (depending on the group) follows, and the work-up is validated on single cells isolated from the couple, together with mutation detection of the disease plus STRs supporting the diagnosis or haplotypes ().
Figure 4. Combined preimplantation genetic diagnosis (PGD) for Fanconi anemia combined with human leukocyte antigen (HLA) matching. A) Location of the short tandem repeat microsatellite marker D16S520 with respect to the FANCA gene on chromosome 16. B) Genealogic tree of the family involved in the preclinical study and PGD results. The arrow indicates the consultant patient. Haplotypes for the HLA region on chromosome 6 plus Fanconi anemia mutations positions on chromosome 16 were established. The discontinuous line and the fine line represent the HLA haplotypes from the mother and the father, respectively, that embryos should inherit in order to be HLA compatible with the affected son. The thick black line symbolizes the disease-bearing haplotype for the paternal mutation (c.3788_3790delTCT) and the dotted line denotes the disease-bearing haplotype for the maternal mutation (c.4130C > G). Mutation c.3788_3790delTCT was determined by fragment analysis and position c.4130 of the FANCA gene was minisequenced. The genetic status for every embryo obtained is differentiated between HLA compatibility and Fanconi anemia diagnosis. Cen: centromere; tel: telomere; Mb: megabases; C: cytosine; G:guanine.
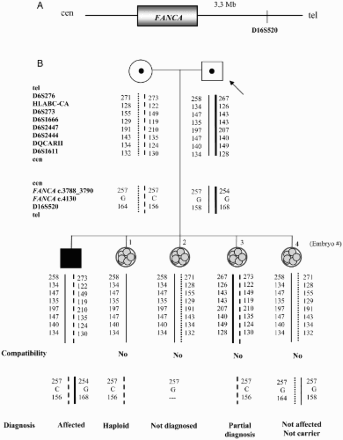
Molecular Strategies for Chromosomal Studies
Nowadays, molecular biology approaches are systematically replacing FISH techniques for some PGD/PGS indications. There has been a great debate about the usefulness of PGS, where up to 12 chromosomes are tested. This debate is still open. However, the main conclusion is that FISH-based PGS does not increase the pregnancy rate for advanced maternal age, as there might be other chromosomes involved causing aneuploidy that are not checked [Harper et al. Citation2010a]. CGH has been proposed as an alternative to FISH, where all chromosomes are analyzed by using metaphase chromosomes as the template [Wilton Citation2005; Obradors et al. Citation2008]. However, it is time-consuming, labor intensive, and requires the appropriate skills and expertise in order to interpret the results. Recently, two types of microarray-based techniques are being clinically used in PGS and PGD for chromosomal rearrangements: i) array-CGH (aCGH) [Gutiérrez-Mateo et al. 2011] and ii) single-nucleotide polymorphism arrays (aSNP) [Johnson et al. Citation2010; Treff et al. Citation2010].
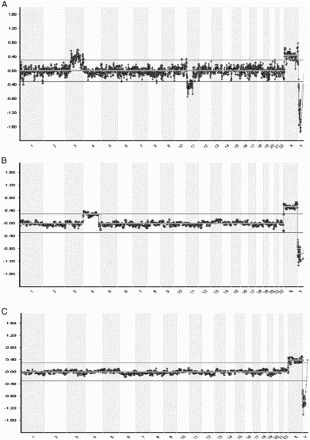
In aCGH, whole DNA pre-amplified from single blastomere (or trophectodermic cells) and normal reference samples are labelled using different fluorophores (red and green) and competitively hybridized to bacterial artificial chromosomes (BAC) clones printed on a glass slide. Each BAC clone corresponds to a specific segment of a chromosome. The ratio of the fluorescence intensity is calculated in order to find copy number variations. Computer software analyzes the ratio of red/green fluorescence on each clone calculating loss or gain of the particular region as the log2 ratio. Technical improvements have allowed reducing the time, and a diagnosis is given in approximately 24 hours. The software shows the information corresponding to the signal of the sample across the whole chromosome (). This application yields a wide-scope information at chromosome level, detecting numerical and structural chromosome abnormalities [Fiorentino et al. Citation2011], even small losses in chromosome arms. The limitation of aCGH is that polyploidy, haploidy, and balanced structural rearrangements cannot be detected. Simultaneously, a PCR-based strategy has been developed to detect reciprocal and Robertsonian translocation carriers in PGD [Fiorentino et al. Citation2010; Traversa et al. Citation2010]. Nevertheless, normal, balanced, and unbalanced translocations carrier embryos can be distinguished with a novel molecular approach [Shamash et al. Citation2011] by using preimplantation genetic haplotyping [Renwick et al. Citation2010].
Figure 5. Example of blastomere array- comparative genomic hybridization charts. Chromosomal position is represented on the X-axis and the Log2 ratio Ch1/Ch2 (the ratio of red/green fluorescence intensity on each clone) on the Y-axis. A) Female blastomere with gain of chromosome 3(p14.2→qter) and loss of chromosome 11(q12.3→qter). The father carries a balanced translocation with chromosome formula: 46,XY;t(3;11)(p21;q13). B) Array profile showing a female cell with gain of chromosome 4. C) Normal female profile.
aSNP platforms let us obtain information concerning how much of each chromosome was inherited. SNPs are changes in only one nucleotide in the DNA sequence within the population. These changes permit one to differentiate one chromosome from another in any person. In this sense, aSNP have been introduced for aneuploidy detection in the context of PGS, giving extra information in relation to parent-of-origin and, in some cases, it offers information related to uniparental disomy. However, the major limitation of this approach is that data analysis is labour-intensive and requires optimized protocols. In this sense, a very interesting debate comparing BACs versus SNPs-based arrays has been discussed recently [Bisignano et al. Citation2011; Handyside Citation2011]. Unfortunately, publications of clinical applications in the field of PGS are still scarce.
What's Next?
The diagnostic strategies in PGS/PGD have experienced a rapid evolution. Nowadays, the different strategies represent a consolidated approach for single (or a few) cell genetic analysis. As it has occurred in other genetic applications, three basic points should be considered in the near future: i) standardization of the analytical methods, ii) analysis of cost-effectiveness, and iii) application of emerging technological advances. Related to standardization, the application of international quality standards to PGD laboratories, as the ISO 9001:2008 certification [Vendrell et al. Citation2009] and the ISO 15189 [Harper et al. Citation2010c] or equivalent accreditation, are extremely important in an under-regulated diagnostic approach like PGD/PGS. In this sense, organization is essential. Aspects as licensing, professional qualification of personnel, and inclusion/exclusion criteria for patient referrals or genetic conditions that can be diagnosed with PGD have recently been thoroughly revised [Harton et al. Citation2011a]. From the regulatory point of view, every country regulates these practices in a more or less specific way and in some cases, there are no explicit laws. In addition, major differences have generated a significant flow of patients between different countries. In this scenario, it is very complex to determine what actions must be taken to ensure quality standards in PGD/PGS. Various scientific societies [The Practice Committee of American Society for Reproductive Medicine Citation2004; The Preimplantation Genetic Diagnosis International Society (PGDIS) Citation2004; 2008; Thornhill et al. Citation2005; Harton et al. Citation2011a; Harton et al. Citation2011b; Harton et al. Citation2011c; Harton et al. Citation2011d] have proposed different guidelines in order to standardize and achieve the highest levels of optimization, reproducibility, and control of risks in genetic diagnosis on single cells.
Accreditation by national bodies is the formal recognition of technical competence and the compliance of the international quality standards. Concretely, the norm ISO 15189 treats two main aspects concerning medical laboratories: i) management and ii) technical requirements. The global process is externally audited by recognized experts, considering pre-analytical, analytical, and post-analytical phases, in the context of a quality management system and combined with the participation in inter-laboratory ring tests. The aim is to implement and analyze the ‘quality indicators’, systematically monitored and documented. The final objective is to continue improving.
The cost-effectiveness of these analytical processes is not irrelevant. PGD/PGS is a part of a complex and costly process, with highly trained professionals (clinicians, embryologists, and geneticists experts), and with a high technological component. Globally, there are several strategies related to funding the IVF-PGD/PGS treatment: national public programs, private clinics, private/public copayment, and insurance coverage. Different countries maintain medical, ethical, and legal discussions concerning IVF-PGD/PGS treatments. However, there are few studies about the economic impact of PGD applications on the healthcare systems. Tur-Kaspa and co-workers [2010] presented a very interesting study in this sense, taking cystic fibrosis as an example. They conclude that “a national IVF-PGD program is a highly cost effective novel strategy of modern preventive medicine.” In agreement with the authors, IVF-PGD could play a significant role reducing incidence of inherited disorders, and perhaps, it should be a principal issue in the debate concerning early diagnosis and preventive medicine.
Regarding emerging techniques, the next step presents new challenges. Working with single cells is difficult due to the limiting quantity of DNA. WGA technologies have permitted applying haplotyping to aSNP for PGD [Handyside et al. Citation2010; Handyside Citation2011]. This new ‘karyomap’ identifies the parental origin of each chromosome and permits analysis of recombination patterns. Besides, this technique offers the possibility of detecting numerical and structural chromosomal imbalances. This new SNP-based karyomapping combined with WGA in PGD is at the initial stage and forthcoming validation in single cell is necessary in order to be established as a new tool in diagnosis.
Combining indications as single-gene disorders detection plus aneuploidy screening has been suggested. Recently, this new PGD indication has been proposed by trophectoderm biopsy [Brezina et al. Citation2011]. This approach introduces a double novelty: the timing of biopsy (on day +5) and the combined genetic study. In this direction, our group presented, very recently (in the past meeting of The Spanish Association for the Study of Reproductive Biology, ASEBIR), the use of WGA methods including specific primers to amplify the genetic regions of interest, permitting the simultaneous application of aCGH and PCR-based technologies in one single blastomere.
Furthermore, the application of genomic strategies based on high throughput sequencing platforms is a very promising approach [Mardis Citation2011]. Nowadays, Next Generation Sequencing (NGS) is revolutionizing the classical concept of genetic diagnosis, offering a ‘genomic view’ in the comprehension of gene expression profiles, DNA methylation changes, and identification of non-coding RNA expression profiles. From the ‘embryo-genomics’ point of view, the whole-transcriptome analysis on single mouse blastomere [Tang et al. Citation2009], and the comprehension of methylome (methylation patterns) based on high throughput sequencing from preimplantation embryos [Ma et al. Citation2012] represent the first step in this new genomic era.
Final Comments
Historically, PGD/PGS have been closely linked to the development of assisted reproduction technologies. In fact, the first publications appeared in the context of IVF units. Currently, most IVF clinics offer this highly specialized genetic study in association with a team of geneticists. This successful union will continue in order to achieve new milestones. In fact, from the assisted reproduction point of view, the emergence of new methods of cryopreservation (vitrification), new embryo culture conditions, different timing of biopsy (polar body or trophectoderm), and embryo transfer strategies, could change the classical schedules of PGD/PGS. In conclusion, rapid advances in genetics and genomics are changing genetic testing and screening in preconception, prenatal, and newborn care. It is fundamentally important that assisted reproduction clinicians and experts who are recommending such studies have a deep knowledge of the current sensitivity and specificity of tests, thus they could perform a correct referral for complex results and preconception genetic assessment.
Author Contributions: The authors contributed equally in designing and performing the experiments, analysis of data and writing of the manuscript.
Declaration of interest: The authors declare the absence of any financial or commercial interest in reference to the submitted material. The authors would not economically benefit from publication of the manuscript. All data are presented accomplishing a balanced, independent, objective, and scientifically rigorous manner. Any other commercial interest related with the activity of our Company is absolutely out of our intention.Notice of CorrectionThe version of this article published online ahead of print on 3 AUG 2012 contained a number of minor typographical errors. These have been corrected for this version
Abbreviations
| PGD: | = | preimplantation genetic diagnosis |
| PGS: | = | preimplantation genetic screening |
| IVF: | = | in vitro fertilization |
| FISH: | = | fluorescent in situ hybridization |
| HLA: | = | human leukocyte antigen |
| STR: | = | short tandem repeat |
| ICSI: | = | intracytoplasmic sperm injection |
| ADO: | = | allele drop-out; f-PCR: fluorescent polymerase chain reaction |
| ARMS: | = | amplification refractory mutation system |
| WGA: | = | whole genome amplification |
| CGH: | = | comparative genomic hybridization |
| aSNP: | = | single-nucleotide polymorphism arrays |
| BAC: | = | bacterial artificial chromosomes. |
Related Research Data
References
- Alberola, T.M., Bautista-Llácer, R., Fernández, E., Vendrell, X. and Pérez-Alonso, M. (2009) Preimplantation genetic diagnosis of P450 oxidoreductase deficiency and Huntington Disease using three different molecular approaches simultaneously. J Assist Reprod Genet 26:263–271.
- Alberola, T.M., Bautista-Llácer, R., Vendrell, X., García-Mengual, E., Pardo, M., Vila, M., (2011) Case report: birth of healthy twins after preimplantation genetic diagnosis of propionic acidemia. J Assist Reprod Genet 28:211–216.
- Altarescu, G., Brooks, B., Margalioth, E., Eldar Geva, T., Levy-Lahad, E. and Renbaum, P. (2007) Simultaneous preimplantation genetic diagnosis for Tay-Sachs and Gaucher disease. Reprod Biomed Online 15:83–88.
- Altarescu, G., Eldar Geva, T., Brooks, B., Margalioth, E., Levy-Lahad, E. and Renbaum, P. (2008) PGD on a recombinant allele: crossover between the TSC2 gene and ‘linked’ markers impairs accurate diagnosis. Prenat Diagn 28:929–933.
- Bautista-Llácer, R., Alberola, T.M., Vendrell, X., Fernández, E. and Pérez-Alonso, M. (2010) Case report: first successful application of preimplantation genetic diagnosis for hereditary angiooedema. Reprod Biomed Online 21:658–662.
- Bisignano, A., Wells, D., Harton, G. and Munné, S. (2011) PGD and aneuploidy screening for 24 chromosomes: advantages and disadvantages of competing platforms. Reprod Biomed Online 23:677–685.
- Brezina, P.R., Benner, A., Rechitsky, S., Kuliev, A., Pomerantseva, E., Pauling, D., (2011) Single-gene testing combined with single nucleotide polymorphism microarray preimplantation genetic diagnosis for aneuploidy: a novel approach in optimizing pregnancy outcome. Fertil Steril 95:1786.e5–8.
- Coskun, S. and Alsmadi, O. (2007) Whole genome amplification from a single cell: a new era for preimplantation genetic diagnosis. Prenat Diagn 27:297–302.
- Fassihi, H., Liu, L., Renwick, P.J. and McGrath, J.A. (2010) Development and successful clinical application of preimplantation genetic haplotyping for Herlitz junctional epidermolysis bullosa. Br J Dermatol 162:1330–1336.
- Fiorentino, F., Magli, M., Podini, D., Ferraretti, A.P., Nuccitelli, A., Vitale, N., (2003) The minisequencing method: an alternative strategy for preimplantation genetic diagnosis of single gene disorders. Mol Hum Reprod 9:399–410.
- Fiorentino, F., Biricik, A., Karadayi, H., Berkil, H., Karlikaya, G., Sertyel, S., (2004) Development and clinical application of a strategy for preimplantation genetic diagnosis of single gene disorders combined with HLA matching. Mol Hum Reprod 10:445–460.
- Fiorentino, F., Kahraman, S., Karadayi, H., Biricik, A., Sertyel, S., Karlikaya, G., (2005) Short tandem repeats haplotyping of the HLA region in preimplatation HLA matching. Eur J Hum Genet 13:953–958.
- Fiorentino, F., Biricik1, A., Nuccitelli, A., De Palma, R., Kahraman, S., Iacobelli, M., (2006) Strategies and clinical outcome of 250 cycles of Preimplantation Genetic Diagnosis for single gene disorders. Hum Reprod 21:670–684.
- Fiorentino, F., Kokkali, G., Biricik, A., Stavrou, D., Ismailoglu, B., De Palma, R., (2010) Polymerase chain reaction-based detection of chromosomal imbalances on embryos: the evolution of preimplantation genetic diagnosis for chromosomal translocations. Fertil Steril 94:2001–2011.
- Fiorentino, F., Spizzichino, L., Bono, S., Biricik, A., Kokkali, G., Rienzi, L., (2011) PGD for reciprocal and Robertsonian translocations using array comparative genomic hybridization. Hum Reprod 26:1925–1935.
- Geraedts, J., Montag, M., Magli, M.C., Repping, S., Handyside, A., Staessen, C., (2011) Polar body array CGH for prediction of the status of the corresponding oocyte. Part I: clinical results. Hum Reprod 26:3173–3180.
- Gigarel, N., Frydman, N., Burlet, P., Kerbrat, V., Tachdjian, G., Fanchin, R., (2008) Preimplantation genetic diagnosis for autosomal recessive polycystic kidney disease. Reprod Biomed Online 16:152–158.
- Goossens, V., Sermon, K., Lissens, W., Vandervorst, M., Vanderfaeillie, A., De Rijcke, M., (2000) Clinical application of preimplantation genetic diagnosis for cystic fibrosis. Prenat Diagn 20:571–581.
- Gutiérrez-Mateo, C., Colls, P., Sánchez-García, J., Escudero, T., Prates, R., Ketterson, K., (2011) Validation of microarray comparative genomic hybridization for comprehensive chromosome analysis of embryos. Fertil Steril 95:953–958.
- Handyside, A.H., Kontogianni, E.H., Hardy, K. and Winston RM. (1990) Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature 344:768–770.
- Handyside, A.H., Robinson, M.D., Simpson, R.J., Omar, M.B., Shaw, M.A., Grudzinskas, J.G., (2004) Isothermal whole genome amplification from single and small number cells: a new era for preimplantation genetic diagnosis of inherited disease. Mol Hum Reprod 10:767–772.
- Handyside, A.H, Harton, G.L, Mariani, B., Thornhill1, A.R, Affara, N., Shaw, M., (2010) Karyomapping: a universal method for genome wide analysis of genetic disease based on mapping crossovers between parental haplotypes. J Med Genet 47:651–658.
- Handyside, A.H. (2011) PGD and aneuploidy screening for 24 chromosomes by genome-wide SNP analysis: seeing the wood and the trees. Reprod Biomed Online 23:686–691.
- Harper, J.C., Coonen, E., De Rycke, M., Fiorentino, F., Geraedts, J., Goossens, V., (2010a) What next for preimplantation genetic screening (PGS)? A position statement from the ESHRE PGD Consortium steering committee. Hum Reprod 25:821–823.
- Harper, J.C., Coonen, E., De Rycke, M., Harton, G., Moutou, C., Pehlivan, T., (2010b) ESHRE PGD Consortium data collection X: cycles from January to December 2007 with pregnancy follow-up to October 2008. Hum Reprod 25:2685–2707.
- Harper, J.C., Sengupta, S., Vesela, K., Thornhill, A., Dequeker, E., Coonen, E., (2010c) Accreditation of the PGD laboratory. Hum Reprod 25:1051–1065.
- Harper, J.C. and Harton, G. (2010) The use of arrays in preimplantation genetic diagnosis and screening. Fertil Steril 94:1173–1177.
- Harper, J.C. and Sengupta, S.B. (2012) Preimplantation genetic diagnosis: State of the ART 2011. Hum Genet 131:175–186.
- Harper, J.C., Wilton, L., Traeger-Synodinos, J., Goossens, V., Moutou, C., SenGupta, S.B., (2012) The ESHRE PGD Consortium: 10 years of data collection. Hum Reprod Update Feb 16 [Epub ahead of print].
- Harton, G., Braude, P., Lashwood, A., Schmutzler, A., Traeger-Synodinos, J., Wilton, L., (2011a). ESHRE PGD consortium best practice guidelines for organization of a PGD center for PGD/preimplantation gentic screening. Hum Reprod 26(1):14–24.
- Harton, G.L., De Rycke, M., Fiorentino, F., Moutou, C., SenGupta, S., Traeger-Synodinos, J., (2011b) ESHRE PGD consortium best practice guidelines for amplification-based PGD. Hum Reprod 26:33–40.
- Harton, G.L., Magli, M.C., Lundin, K., Montag, M., Lemmen, J. and Harper J.C (2011c) ESHRE PGD Consortium/Embryology Special Interest Group—best practice guidelines for polar body and embryo biopsy for preimplantation genetic diagnosis/screening (PGD/PGS). Hum Reprod 26 (1):41–46.
- Harton, G.L., Harper, J.C., Coonen, E., Pehlivan, T., Vesela, K. and Wilton, L. (2011d) ESHRE PGD consortium best practice guidelines for fluorescence in situ hybridization-based PGD. Hum Reprod 26(1):25–32.
- Johnson, D.S., Gemelos, G., Baner, J., Ryan, A., Cinnioglu, C., Banjevic, M., (2010) Preclinical validation of a microarray method for full molecular karyotyping of blastomeres in a 24-h protocol. Hum Reprod 25:1066–1075.
- Ma, J.Y., Liang, X.W., Schatten, H. and Sun Q. (2012) Active DNA demethylation in mammalian preimplantation embryos: new insights and new perspectives. Mol Hum Reprod Mar 23 [Epub ahead of print].
- Mamas, T., Gordon, A., Brown, A., Harper, J. and Sengupta S. (2012) Detection of aneuploidy by array comparative genomic hybridization using cell lines to mimic a mosaic trophectoderm biopsy. Fertil Steril Jan 24 [Epub ahead of print].
- Mardis, E.R. (2011) A decade's perspective on DNA sequencing technology. Nature 470:198–203.
- Moutou, C., Gardes, N., Nicod, J.C. and Viville, S. (2007) Strategies and outcomes of PGD of familial adenomatous polyposis. Mol Hum Reprod 13:95–101.
- Munnè, S., Cohen, J. and Sable, D. (2002) Preimplantation genetic diagnosis for advanced maternal age and other indications. Fertil Steril 78:234–236.
- Obradors, A., Fernández, E., Oliver-Bonet, M., Rius, M., de la Fuente, A., Wells, D., (2008) Birth of a healthy boy after a double factor PGD in a couple carrying a genetic disease and at risk for aneuploidy: case report. Hum Reprod 23:1949–1956.
- Piyamongkol, W., Bermudez, M., Harper, J. and Wells, D. (2003) Detailed investigation of factors influencing amplification efficiency and allele drop-out in single cell PCR: implications for preimplantation genetic diagnosis. Mol Hum Reprod 9:411–420.
- The Practice Committee of American Society for Reproductive Medicine (2004) Preimplantation Genetic Diagnosis. Fertil Steril 82:S120–122.
- The Preimplantation Genetic Diagnosis International Society (PGDIS) (2004) Guidelines for good practice in PGD. Reprod Biomed Online 9:430–434.
- Preimplantation Genetic Diagnosis International Society (2008) Guidelines for good practice in PGD: programme requirements and laboratory quality assurance. Reprod Biomed Online 16:134–147.
- Rechitsky, S. (2006) Preimplantation HLA typing with aneuploidy testing. Reprod Biomed Online 12:89–100.
- Rechitsky, S., Pomerantseva, E., Pakhalchuk, T., Pauling, D., Verlinsky, O. and Kuliev, A. (2011) First systematic experience of preimplantation genetic diagnosis for de-novo mutations. Reprod Biomed Online 22:350–361.
- Recommendations of the European Societies of Human Genetics and Human Reproduction and Embryology (2006) The need for interaction between assisted reproduction technology and genetics. Eur J Hum Genet 14:509–511.
- Renwick, P. (2006) Proof of principle and first cases using preimplantation genetic haplotyping - a paradigm shift for embryo diagnosis. Reprod Biomed Online 13:110–119.
- Renwick, P., Trussler, J., Lashwood, A., Braude, P. and Ogilvie C.M. (2010) Preimplantation genetic haplotyping: 127 diagnostic cycles demonstrating a robust, efficient alternative to direct mutation testing on single cells. Reprod Biomed Online 20:470–476.
- Rubio, C., Simon, C., Vidal, F., Rodrigo, L., Pehlivan, T., Remohí, J., (2003) Chromosomal abnormalities and embryo development in recurrent miscarriage couples. Hum Reprod 18:182–188.
- Sermon, K., Seneca, S., de Rycke, M., Goossens, V., Van de Velde, H., De Vos, A., (2001) PGD in the lab for triplet repeat diseases - myotonic dystrophy, Huntington's disease and Fragile-X syndrome. Mol Cell Endocrinol 183:S77–85.
- Shamash, J., Rienstein, S., Wolf-Reznik, H., Pras, E., Dekel, M., Litmanovitch, T., (2011) Preimplantation genetic haplotyping a new application for diagnosis of translocation carrier's embryos- preliminary observations of two robertsonian translocation carrier families. J Assist Reprod Genet 28:77–83.
- Soini, S., Ibarreta, D., Anastasiadou, V., Aymé, S., Braga, S., Cornel, M., (2006). The interface between assisted reproductive technologies and genetics: technical, social, ethical and legal issues Eur J Hum Genet 14:588–645.
- Spits, C. and Sermon, K. (2009) PGD for monogenic disorders: aspects of molecular biology. Prenatal Diagn 29:50–56.
- Tang, F., Barbacioru, C., Wang, Y., Nordman, E., Lee, C., Xu, N., (2009) mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 6:377–382.
- Thornhill, A.R., deDie-Smulders, C.E., Geraedts, J.P. and ESHRE PGD Consortium (2005) Best practice guidelines for clinical preimplantation genetic diagnosis (PGD) and preimplantation genetic screening (PGS). Hum Reprod 20:35–48.
- Traeger-Synodinos, J. (2006) Real-time PCR for prenatal and preimplantation genetic diagnosis of monogenic diseases. Mol Aspects Med 27:176–191.
- Traversa, M.V., Carey, L. and Leigh D. (2010) A molecular strategy for routine preimplantation genetic diagnosis in both reciprocal and Robertsonian translocation carriers. Mol Hum Reprod 16:329–337.
- Treff, N.R., Su, J., Tao, X., Levy, B. and Scott, R.T. (2010) Accurate single cell 24 chromosome aneuploidy screening using whole genome amplification and single nucleotide polymorphism microarrays. Fertil Steril 94:2017–2021.
- Tur-Kaspa, I., Aljadeff, G., Rechitsky, S., Grotjan, H.E. and Verlinsky, Y. (2010) PGD for all cystic fibrosis carrier couples: novel strategy for preventive medicine and cost analysis. Reprod Biomed Online 21:186–195.
- Van de Velde, H., De Rycke, M., De Man, C., De Hauwere K., Fiorentino F., Kahraman, S., (2009) The experience of two European preimplantation genetic diagnosis centres on human leukocyte antigen typing. Hum Reprod 1:1–9.
- Vendrell, X., Carrero, R., Alberola, T., Bautista-Llácer, R., García-Mengual, E., Claramunt, R., (2009) Quality management system in PGD/PGS: now is the time. J Assist Reprod Genet 26:197–204.
- Vendrell, X., Bautista-Llácer, R., Alberola, T.M., García-Mengual, E., Pardo, M., Urries, A., (2011) Pregnancy after PGD for recessive dystrophic epidermolysis bullosa inversa: genetics and preimplantation genetics J Assist Reprod Genet 28:825–832.
- Verlinsky, Y., Cieslak, J., Ivakhnenko, V., Evsikov, S., Wolf, G., White, M., (1999) Prevention of age-related aneuploidies by polar body testing of oocytes. J Assist Reprod Genet 16:65–69.
- Verlinsky, Y., Rechitsky, S., Schoolcraft, W., Strom, C. and Kuliev, A. (2001) Preimplantation diagnosis for Fanconi anemia combined with HLA matching. J Am Med Assoc 285:3130–3133.
- Verlinsky, Y. (2006) Preimplantation genetic diagnosis for Pelizaeus-Merzbacher disease with testing for age-related aneuploidies. Reprod Biomed Online 12:83–88.
- Wilton, L. (2005) Preimplantation genetic diagnosis and chromosome analysis of blastomeres using comparative genomic hybridization. Hum Reprod Update 11:33–41.
- Wilton, L., Thornhill, A., Traeger-Synodinos, J., Sermon, K.D. and Harper, J.C. (2009) The causes of misdiagnosis and adverse outcomes in PGD. Hum Reprod 24:1221–1228.
- Zheng, Y.M., Wang, N., Li, L. and Jin, F. (2011) Whole genome amplification in preimplantation genetic diagnosis. J Zhejiang Univ Sci B 12:1–11.