
Abstract
Patients with Turner syndrome are generally characterized by having short stature with no secondary sexual characteristics. Some abnormalities, such as webbed neck, renal malformations (>50%) and cardiac defects (10%) are less common. The intelligence of these patients is considered normal. Non-mosaic monosomy X is observed in approximately 45% of postnatal patients with Turner syndrome and the rest of the patients have structural abnormalities or mosaicism involving 46,X,i(Xq), 45,X/46,XX, 45,X and other variants. The phenotype of 45,X/46,X,+mar individuals varies by the genetic continent and degree of the mosaicism. The gene content of the marker chromosome is the most important when correlating the phenotype with the genotype. Here we present an 11-year-old female who was referred for evaluation of her short stature and learning disabilities. Conventional cytogenetic investigation showed a mosaic 45,X/46,X,+mar karyotype. Fluorescence in situ hybridization showed that the marker chromosome originated from the X chromosome within the androgen receptor (AR) and X-inactive specific transcript (XIST) genes. Therefore, it is possible that aberrant activation of the marker chromosome, compromising the AR and XIST genes, may modify the Turner syndrome phenotype.
Introduction
Turner syndrome is characterized by developmental problems of the secondary sexual characteristics [Gardner et al. Citation2011], gonodal dysgenesis [Alvarez-Nava et al. Citation2003], unusual facial features, webbed neck, low posterior hairline, broad chest with widely spaced nipples, as well as renal and cardiovascular abnormalities [Batch Citation2002]. Short stature is the most common feature and is present in at least 95% of individuals with Turner syndrome [Batch Citation2002; Gardner et al. Citation2011]. This characteristic depends on the deletion of the short stature Homeobox (SHOX gene) on chromosome X [Hutson et al. Citation2014]. The height of adult females with Turner syndrome who have not received any growth-promoting therapy is reduced by approximately 20 cm or 3.0 SD below the mean [Saenger et al. Citation2001].
Approximately 45% of postnatal patients with Turner syndrome are characterized with apparent non-mosaic monosomy X and the rest have structural abnormalities or mosaicism [Jhang et al. Citation2014]. The phenotypic variability in cases with X derived marker chromosomes generally depends on the size of the marker chromosome and the presence of a functioning X-inactive specific transcript (XIST). Turner syndrome mosaics are subcategorized according to whether the second cell line contains the whole or part of the X chromosome [Mazzaschi et al. Citation2014]. Generally, the deleted part of the X chromosome determines the phenotype. For example, terminal X chromosome deletions are often associated with functional ovarian phenotypes [Gardner et al. Citation2011] and proximal Xq (such as Xq13) deletions often result in absent breast development, primary amenorrhea, and gonadal failure. In fact, the Xq26-q28 region has been associated with premature ovarian failure 1 (POF1) while Xq13.3-q21.1 with POF2, as both of these regions were proposed to compromise ovarian determinant genes [Zhong and Leyman Citation2012]. Thus, in Turner syndrome the variants X,abn(X) cause a wide phenotypic range as shown in [Gardner et al. Citation2011].
Figure 1. Schematic diagram of the X chromosome showing ovarian function as a function of non-mosaic terminal deletions. There are a large number of phenotypic conditions that have been associated with the deleted/duplicated/inactivate or mutated regions on the X chromosome. The terminal X chromosome deletions are often associated with functional ovarian phenotypes and proximal Xq (such as Xq13) deletions often result in absent breast development, primary amenorrhea, and gonadal failure. Therefore, the non-classical form of Turner syndrome, which is called variant Turner syndrome, shows a highly variable phenotype due to the genotypic background of the patients. (a): the region of the marker chromosome [Simpson and Rajkovic Citation1999]. The schematic demonstration of the marker chromosome gene continent, which contained the Xq11.2q13.2 region with the androgen receptor and the X-inactive specific transcript genes and with a deletion of the region distal to Xq13.2.
![Figure 1. Schematic diagram of the X chromosome showing ovarian function as a function of non-mosaic terminal deletions. There are a large number of phenotypic conditions that have been associated with the deleted/duplicated/inactivate or mutated regions on the X chromosome. The terminal X chromosome deletions are often associated with functional ovarian phenotypes and proximal Xq (such as Xq13) deletions often result in absent breast development, primary amenorrhea, and gonadal failure. Therefore, the non-classical form of Turner syndrome, which is called variant Turner syndrome, shows a highly variable phenotype due to the genotypic background of the patients. (a): the region of the marker chromosome [Simpson and Rajkovic Citation1999]. The schematic demonstration of the marker chromosome gene continent, which contained the Xq11.2q13.2 region with the androgen receptor and the X-inactive specific transcript genes and with a deletion of the region distal to Xq13.2.](/cms/asset/6606d820-ebcb-4815-9fe6-5dd277024976/iaan_a_1109007_f0001_c.jpg)
Here, we report on a patient who had a severe phenotype with mild mental retardation, learning disabilities, and several dysmorphic features such as short stature, hypertelorism, anteverted nares, downslanting eyes, with abnormal hormonal levels of follicle stimulating hormone (FSH), luteinizing hormone (LH), and unconjugated estriol (UE). Cytogenetic analysis showed that the patient had a marker chromosome with 45,X/46,X,+mar karyotype. Although X chromosome activation is yet to be confirmed, the marker chromosome compromised the XIST region, and the phenotypic severity of the patient supports this hypothesis.
Case Presentation
The proband was an 11-year-old girl, born in a twin pregnancy to non-consanguineous parents. The pregnancy and delivery were uncomplicated and her twin brother was normal. The birth weight was 900 gm and she exhibited severe signs of being small for her gestational age.
The patient was referred for genetic counseling at the age of 11 due to her short stature (128 cm) and poor school performance. Her FSH level was 106.1 uU/mL, LH level was 21.38 uU/mL, UE level was <5.00 and the glucose-6-phosphate dehydrogenase (G6PD) level was 0,4u/g Hb. Her weight was 35.5 kg at the time of referral. She also exhibited dysmorphic features, such as a simian crease on both hands and clinodactly on the right hand. Furthermore, short and hypoplastic nails, low posterior hairline, downslanting eyes, hypertelorism, anteverted nares, a short-bridged nose, and long philtrum were observed. Early psychomotor development was reported as normal. Although overall learning disabilities and mild mental retardation were observed, further analysis for mental retardation was not performed due to the refusal of the patient. Abdominal ultrasound was performed and ovaries were not observed. Following a convulsion attack, she was treated with Depakine and Ritalin (methylphenidate HCl).
Results
GTL banded chromosome analysis on peripheral lymphocytes revealed that the patient was 95% mosaic (45,X [95]/46,X,+mar[5] (). Subsequently, multicolor FISH (M-FISH) was carried out in order to identify the marker chromosome found in a fraction of the metaphases. This revealed that the marker chromosome was derived from the X chromosome (). In order to characterize the marker chromosome, the presence of three genes was analyzed using locus specific FISH with three LSI probes representing different parts of the X chromosome. The probes targeted the androgen receptor (AR) gene, the SHOX region, and the XIST gene. These investigations revealed that the marker chromosome was derived from Xq since it comprised DXZ1, the AR region (Xq11.2- Xq12), and the XIST region (Xq13.2) (). The data thus suggests a mosaic marker chromosome was comprised of an X centromere and pericentromeric euchromatin from the long arm of the X chromosome, including the androgen receptor and XIST region. Karyotype analysis was performed independently two times.
Figure 2. G-band chromosomal analysis of peripheral blood lymphocytes revealed 45,X/46,X,+mar and 45,X. (A) G-band chromosomal analysis of peripheral blood lymphocytes revealed 45,X/46,X,+mar. A GTL banded karyotype of the patient showing an 45,X cell line. All autosomes appear normal in their banding patterns. (B) G-band chromosomal analysis of peripheral blood lymphocytes revealed 45,X. A GTL banded karyotype of the patient showing an 46,X,+marker. All autosomes appear normal in their banding patterns.
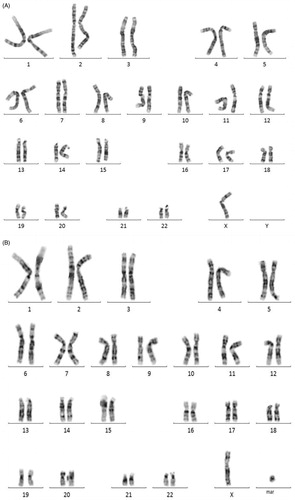
Figure 3. Multicolor FISH (M-FISH) analysis showing a marker chromosome is derived from the chromosome X. M-FISH preparation of a karyotype showing the normal X-chromosome and the marker chromosome which was derived from the X chromosome.
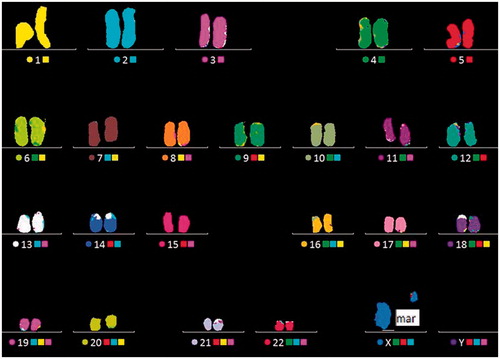
Figure 4. Fluorescence in situ hybridization (FISH) image of metaphase spread using a short stature homeobox (SHOX), androgen receptor (AR), and X-inactive specific transcript (XIST) probes. (A) FISH image of metaphase spread using a SHOX probe. Dual color FISH with the SHOX gene specific probes showing the normal X chromosome and the marker chromosome. The X chromosome centromere probe (Spectrum Aqua; sp aqua; DXZ1) on an inverted grey scale DAPI-stained metaphase spread shows hybridization to both the X chromosome and the marker chromosome. SHOX region (Spectrum Red; sp red) probe on an inverted grey scale DAPI-stained metaphase spread shows hybridization to the X chromosome and deleted in the marker chromosome. (B) FISH image of metaphase spread using an AR probe. Locus specific FISH with the AR gene specific probes showing the normal X chromosome and the marker chromosome. The AR probe (sp red) on an inverted grey scale DAPI-stained metaphase spread shows hybridization to both the X chromosome and the marker chromosome. (C) FISH image of metaphase spread using an XIST probe. Locus-specific FISH with the XIST gene specific probes showing the normal X chromosome and the marker chromosome. The XIST probe (sp red) on an inverted grey scale DAPI-stained metaphase spread shows hybridization to both the X chromosome and the marker chromosome.

Discussion
We report on a patient with the karyotype 45,X/46,X,+mar, who exhibited mild mental retardation and dysmorphic features. The phenotype of patients with Turner syndrome varies substantially depending on the level of the mosaicism and the genetic content of the marker chromosome. The marker chromosome detected for the patient reported presented chromosomes comprising the Xq11.2q13.2 region with the AR and the XIST genes and with a deletion of the region distal to Xq13.2. The deleted regions contain a number of genes, which are associated with various physical features of Turner syndrome and with ovarian dysfunction [Wolff et al. Citation2010].
Both the clinical features and the severity of Turner syndrome patients can vary depending on the deleted region of the X chromosome [Gardner et al. Citation2011] or the presence of a marker or r(X) chromosome [Dennis et al. Citation1993; Gardner et al. Citation2011]. The additional small marker or r(X) chromosome with the 45,X karyotype may cause mental retardation (MR) and/or developmental delay (DD) with the full Turner syndrome phenotype (). Dennis and colleagues [Citation1993] reported MR/DD in addition to the physical findings of typical Turner syndrome in 26 of 41 individuals with a small (5 size of a G group chromosome) marker or r(X) chromosomes. Other findings described in these 26 cases included significant growth retardation (22 cases) and unusual facial features (13 cases). In three cases with ring (X) chromosomes, a number of additional features were found such as relative hyperteloroid eye problems [Dennis et al. Citation1993], hypothyroidism and glucose intolerance [Batch Citation2002; Sybert and McCauley Citation2004], heart abnormalities [Cohen et al. Citation1967], seizures [Callen et al. Citation1991], hypotonia [Buhler Citation2011], simian creases [Buhler Citation1980], and facial hemangiomata [Bishop et al. Citation1966].
Table 1. Clinical findings for small marker or ring X chromosomes.
Cole and colleagues [Citation1994] reported four cases with mosaicism levels varying from 70% to 16% and karyotypes of 45,X/46,X,+r. They had minor anomalies and/or short stature. Unusual facial appearance, MR/DD, and syndactylies were observed. They noted that among their patients, mental retardation was noted most frequently in the cases having at least 50% mosaicism for the marker [Cole et al. Citation1994]. Similar features were found in our patient, namely a Turner syndrome picture with abnormal facial appearance, MR, short stature, and DD. However, in our patient the mosaicism level for the marker chromosome was only 5%. Thus, our patient exhibited a more severe phenotype in spite of the fact that the mosaicism level was lower than in the cases described by Cole and colleagues. This, however, may be due to different gene content of the marker chromosome. It may be of relevance here that the marker chromosome in our patient contained both the AR and the XIST genes.
The AR gene is an important gene in male reproductive development and the function of testosterone also acts as the estradiol precursor as in females. Thus, AR is important for both male and female reproductive development. The expression of AR is present at most stages of follicular development and the AR protein has been localized to the theca cells of preantral follicles and the granulos cells of antral follicles [Walters et al. Citation2008]. Previous animal studies have shown that AR exon 1 deleted females with loss of AR protein exhibited increased atresia, which is positively correlated with an early decline in follicle number leading to accelerated ovarian failure [Shiina et al. Citation2006]. Furthermore, it is known that a partial deletion of the long arm X chromosome, with the critical region Xq13-q26, causes significant ovarian insufficiency and gonodal dysgenesis [Moka et al. Citation2013]. This concurs with the observations of the patient presented here with the absence of ovaries detected by the USG examination.
Cole et al. [Citation1994] and Liehr et al. [Citation2007] reported MR for these Turner syndrome variant patients. Badeschi and colleagues [Citation2008] described a male with severe MR with duplication on the X chromosome Xq12 to Xq12.1, which contains the OPHN1 gene. In the same way as in the literature, we detected the partial Xq11.2- Xq13.2 duplication in our patient with mild MR.
The severe phenotype observed for the patient reported here can be explained by the marker chromosome escaping the inactivation of functional disomy of the X chromosome. When one X chromosome has an imbalance that does not involve an autosome, the X chromosome inactivation centre (XIC) on the abnormal X chromosome is activated. This activation leads to nonrandom skewing of X chromosome inactivation, with the XIST transcript inactivating the abnormal chromosome. Consequently, the phenotype of this group of patients is generally a phenotype variant of mild Turner syndrome [Gardner et al. Citation2011]. Distinctively from the literature, our patient shows MR/DD and at the same time, the marker chromosome also contains the XIST region. Similar to our case, Migeon and colleagues [Citation2000] reported two Turner syndrome variants with MR, who had intact XIST regions on their marker chromosomes. It can thus be hypothesized that the mosaic marker results in a gain of X chromosome material (from Xq11.11q13.2) against a Turner syndrome genotype background.
In light of the previous literature, we conclude that our index case with 45,X/46,X,+mar, with AR and XIST genes selectively give the phenotype due to functional disomy for these genes carried on the marker chromosomes. A marker chromosome containing euchromatic sequences along with mild MR and severe dismorphic features with abnormal hormonal levels were observed. The mild MR with Turner syndrome stigmata also makes this variant Turner syndrome patient unique.
Methods
Metaphase chromosomes were prepared from PHA stimulated cultured lymphocytes and analyzed with standard GTL, C-banding, and NOR banding. Chromosome analysis of the patient was carried out by applying GTL banding at a 600-band level according to ISCN [2009] (100 metaphases).
FISH was performed to characterize the marker chromosome. The chromosome slides were prepared as described elsewhere [Jalal et al. Citation2003]. M-FISH (VYSIS, Inc, Downers Grove, IL, USA) was performed according to the manufacturer’s instructions (24XCyte Human Multicolor FISH Probe Kit, MetaSystems).
A standard protocol as directed in the probe kit (VYSIS, Inc) was followed for locus specific FISH. The slides with metaphase chromosomes were prepared following the standard cytogenetic protocol and were dehydrated in ethanol series (70%, 80%, and 100%) for 1 min each at room temperature and then air-dried. The FISH for locus specific probes (XIST, SHOX, and AR) (VYSIS, Inc) was performed according to the manufacturer’s protocol (VYSIS, Inc). The co-denaturation of metaphase chromosomes and the probes were performed using Hybrite at 73°C for 3 min and hybridization was carried out for 24 h at 37°C. The post-hybridization washes were performed according to the manufacturer’s protocol (VYSIS, Inc). The slides were counterstained with 4',6-diamidino-2-phenylindole (DAPI) (10μl/slide), covered with coverslip, and stored in the dark prior to the analysis. The metaphases were analyzed using a Zeiss Axioplan fluorescence microscope (Zeiss, Jena, Germany) with MetaSystems (Isis) software (Altlussheim, German). Ethical approval was granted and informed written consent was obtained from both the patient and the family.
| Abbreviations | ||
| AR | = | androgen receptor |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| DD | = | developmental delay |
| FISH | = | fluorescence in situ hybridization |
| FSH | = | follicle stimulating hormone |
| G6PD | = | glucose-6-phosphate dehydrogenase |
| LH | = | luteinizing hormone |
| M-FISH | = | multicolor FISH |
| MR | = | mental retardation |
| NP | = | nonyl henoxypolyethoxylethanol |
| PHA | = | phytohemagglutinin |
| UE | = | unconjugated estriol |
| SHOX gene | = | short stature homeobox |
| SSC | = | saline-sodium citrate buffer |
| XIST | = | X-inactive specific transcript |
Declaration of interest
All authors certify that they have no affiliations with or involvement in any organization or entity with any financial or non-financial interest in the subject matter or materials discussed in this manuscript.
Author contribution
Performed the study, undertook data collection, and took primary responsibility for data analyses and write-up: NS, RK. Contributed with data generation and assisted in data coding: RK, NO. Designed and supervised the study: NS, RK. G-Banded Metaphase check in: AC, NO. Evaluated for endocrinological aspects: RB. All authors contributed to the writing and preparation of this manuscript.
References
- Abdelmoula, N.B., Amouri, A., Portnoi, M.F., Saad, A., Boudawara, T., Mhiri, M.N., et al. (2004) Cytogenetics and fluorescence in situ hybridization assessment of sex-chromosome mosaicism in Klinefelter's syndrome. Ann Genet 47(2):163–175
- Alvarez-Nava, F., Soto, M., Sanchez, M.A., Fernandez, E. and Lanes R. (2003) Molecular Analysis in Turner Syndrome. J Pediatr 142(3):336–340
- Bandeschi, M.F., Novelli, A., Bernardini, L., Parazzini, C., Bianchi, V., Torres, B., et al. (2008) Association of syndromic mental retardation with an Xq12q13.1 duplication encompassing the oligophrenin 1 gene. Am J Med Genet A 146A(13):1718–1724
- Batch, J. (2002) Turner syndrome in childhood and adolescence. Best Practice & Research Clinical Endocrinology and Metabolism 16(3):465–482
- Bishop, A.M., Blank, C.E., Simpson, K. and Dewhurst, C.J. (1966) An XO-X ring X chromosome mosaicism in an individual with normal secondary sexual development. J Med Genet 3(2):129–133
- Bühler, E.M. (1980) A synopsis of the Human Y chromosome. Hum Genet 55(2):145–175
- Bühler, E., Bachmann, C., Goyert, H., Heinzel-Gutenbrunner, M., Kamp-Becker, I.J. (2011) Differential diagnosis of autism spectrum disorder and attention deficit hyperactivity disorder by means of inhibitory control and ‘theory of mind.’. Hum Genet 55(2):145–175
- Callen, D.F., Eyre, H.J., Baker, E., Ringenbergs, M., Freemantle, C.J. and Haan, E.A. (1991) Chromosomal origin of small ring marker chromosomes in man: Characterization by molecular genetics. Am J Hum Genet 48(4):769–782
- Cantú, E.S., Jacobs, D.F. and Pai, G.S. (1995) An atypical Turner syndrome patient with ring X chromosome mosaicism. Ann Clin Lab Sci 25(1):60–65
- Chomczyk, I., Panasiuk, B., Wojda, A., Hubert, E. and Midro, A.T. (1999) Turner syndrome in a girl with marker chromosome in karyotype. Ginekol Pol 70(5):348–353
- Cohen, M.M., Sandberg, A.A., Takagi, N. and MacGillivray, M.H. (1967) Autoradiographic investigations of centric fragments and rings in patients with stigmata of gonadal dysgenesis. Cytogenetics 6(3):254–267
- Cole, H., Huang, B., Salbert, B.A., Brown, J., Howard-Peebles, P.N., Black, S.H., et al. (1994) Mental retardation and Ullrich- Turner syndrome in cases with 45,X/46,X,+mar:Additional support for the loss of the X inactivation center hypothesis. Am J Med Genet 52(2):136–146
- Collins, A.L., Cockwell, A.E., Jacobs, P.A. and Dennis N.R. (1994) A comparison of the clinical and cytogenetic finding in nine patients with a rind (X) cell line and 16 45,X patients. J Med Genet 31(7):528–533
- Crolla, J.A. and Llerena J.C. Jr (1988) A mosaic 45,X/46,X,r(?) karyotype investigated with X and Y centromere-specific probes using a non-autoradiographic in situ hybridization technique. Hum Genet 81(1):81–84
- Dennis, N.R., Collins, A.L., Crolla, J.A., Cockwell, A.E., Fisher, A.M. and Jacobs P.A. (1993) Three patients with ring (X) chromosomes and a severe phenotype. J Med Genet 30(6):482–486
- Gardner, R.J., Sutherland, G.R. and Shaffer L.G. (2011) Chromosome abnormalities and Genetic counseling, Fourth Edition, Oxford University Press, pp. 479–480
- González-del-Angel, A., Blanco, B., del Castillo, V. and Carnevale, A. (1995) Identification of sex chromosome markers using fluorescence in situ hybridization (FISH). Rev Invest Clin 47(2):117–1125
- Hutson, J.M., Grover, S.R., O’Connell, M. and Pennell, S.D. (2014) Malformation Syndromes associated with disorders of sex development. Nat Rev Endocrinol 10(8):476–487
- Jalal, S.M., Totowa, N.J. and Law, M.E. (2003) Multicolor FISH In Methods in Molecular Biology, Vol. 204: Molecular Cytogenetics: Protocols and Applications. ed. Fan, Y.S. © Humana Press Inc., pp.105–120
- Jhang, K.M., Chang, T.M., Chen, M. and Liu, C.S. (2014) Generalized epilepsy in a patient with mosaic Turner Syndrome: A case report. J Med Case Rep 8:109
- Liehr, T., Mrasek, K., Hinreiner Reich, D., Ewers, E., Bartels, I., Seidel J., et al. (2007) Small Supernumary Marker Chromosomes (sSCM) in patients with a 45,X/46,X,+mar Karyotype-17 new cases and a review of the literature. Sex Dev 1(6):353–362
- Lindgren, V., Chen, C.P., Bryke, C.R., Lichter, P., Page, D.C. and Yang- Feng, L.T. (1992) Cytogenetic and molecular characterization of marker chromosomes in patients with mosaic 45,X karyotypes. Hum Genet 88(4):393–398
- Mazzaschi, P.L.P., Taylor, J., Robertson, S.P., Love, D.R. and George A.M. (2014) A Turner Syndrome Patient Carrying a mosaic Distal X chromosome Marker. Case Rep Genet 2014:597314
- Migeon, B.R., Ausems, M., Giltay, J., Hasley-Royster, C., Kazi, E., Lydon, T.J., et al. (2000) Severe phenotypes associated with inactive ring X chromosomes. Am J Med Genet 93(1):52–57
- Moka, R., Sewwlakshmi, K., Gopinath, P.M. and Satyamoorthy, K. (2013) Cytogenetic evaluation of patients with clinical spectrum of Turner syndrome. J Hum Reprod Sci 6(2):129–132
- Saenger, P., Wikland, K.A., Conway, G.S., Davenport, M., Gravholt, C.H., Hintz, R., et al. (2001) Fifth International Symposium on Turner Syndrome Recommendations for the diagnosis and management of Turner syndrome. J Clin Endocrinol Metab 86(7):3061–3069
- Schwartz, S., Depinet, T.W., Leana-Cox, J., Isada, N.B., Karson,E.M., Park, V.M., et al. (1997) Sex chromosome markers: Characterization using fluorescence in situ hybridization and review of the literature. Am J Med Genet 71(1):1–7
- Shaffer, L.G., Slovack, M.L., Campbell, L.J. (eds). (2009) An International System for Human Cytogenetic Nomenclature: Recommendations of the International Standing Committee on Human Cytogenetic Nomenclature. Cytogenetic and Genome Research
- Shiina, H., Matsumuto, T., Sato, T., Igarashi, K., Miyamoto, J., Takemasa, S., et al. (2006) Premature ovarian failure in androgen receptor deficient mice. Proc Natl Acad Sci USA 103(1):224–229
- Simpson, J. (2011) Ovarian dysgenesis and premature ovarian failure caused by X chromosomal abnormalities. Glob libr women's med ISSN: 1756–2228. DOI 10.3843/GLOWM.10354
- Simpson, J.L. and Rajkovic A. (1999) Ovarian differentiation and gonadal failure. Am J Med Genet 89(4):186–200
- Sybert,V.P. and McCauley, E. (2004) Turner's syndrome. N Engl J Med 351(12):1227–1238
- Thong, M.K., Manonmani, V. and Norlasiah, I.S. (1996) Supernumerary chromosomes in mosaic Turner syndrome. Med J Malaysia 51(4):487–490
- Van Dyke, D.L., Wiktor, A., Palmer, E.G., Miller, D.A., Witt, M., Babu, V.R., et al. (1992) Ulrich syndrome with a small ring X chromosome and presence of mental retardation. Am J Med Genet 43:996–1005
- Walters, K.A., Allan, C.M. and Handelsman D.J. (2008) Androgen Actions and the Ovary. Biol Reprod 78(3):380–389
- Wolff, J.D., Dyke, D.L., Powell, C.M. and Working Group of the ACMG Laboratory Quality Assurance Committee (2010) Laboratory guideline for Turner Syndrome. Genet Med 12(1):52–55
- Wolff, D.J., Miller, A.P.,Van dyke, D.L., Schwartz, S. and Willard, H.F. (1996) Molecular definition of break points with human Xq isochromosmes: Implications for mechanisms of formation. Am J Hum Genet 58(1):154–160
- Ye, Z.C., Cai, J.G., Zhu, X.Y., Zhao, R., He, X.Y., Zhong, Y., et al. (2009) Analysis of the small supernumerary marker chromosome in Turner syndrome with 45, X/46, X, + mar karyotype. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 26(4):461–464
- Zhong, Q. and Layman, L.C. (2012) Genetic Considerations in the Patient with Turner Syndrome—45,X with or without Mosaicism. Fertil Steril 98(4):775–779