
Abstract
We reconstructed the complete mitochondrial genomes of six higher termite species from metagenomic datasets of their isolated hindgut compartments. The sequencing reads were retrieved and assembled with the mitochondrial-baiting and iterative-mapping algorithm (MITObim), which yielded closed mitogenomes without additional finishing efforts (average coverage ranging from 2300- to 17,000-fold). The genomes ranged from 16.1 to 17.6 kbp in size and had G+C contents between 32 and 35 mol%; each contained the same 37 genes present also in the mitochondria of other termite species. Our study substantially increases the number of termite mitogenomes available for phylogenetic studies and offers a facile strategy for identifying host species in metagenomic studies of their associated microbiota.
Keywords:
Termites are a diverse and ecologically important group of insects that originated from an ancestral cockroach (Inward et al., Citation2007). The detailed analysis of the evolutionary relationships among termites requires multi-gene phylogenies, which can be obtained from entire mitochondrial genomes (Cameron et al., Citation2012). However, despite the scientific interest in termites and their role in lignocellulose digestion (Brune, Citation2014), only few mitochondrial genomes have been sequenced. While most previous studies used primer walking as the sequencing strategy (Cameron et al., Citation2012; Cameron & Whiting, Citation2007; Tokuda et al., Citation2012), Qian (Citation2014) assembled the mitogenome of Zootermopsis nevadensis from the data of the recently published first termite genome (Terrapon et al., Citation2014). Here, we show that this strategy can be employed to reconstruct complete mitochondrial genomes from the host information contained in metagenomic datasets of their associated microbiota.
Using the quality-trimmed sequencing reads obtained from the metagenomes of six higher termite species, we assembled the mitogenomes using the mitochondrial baiting and iterative mapping algorithm (MITObim; Hahn et al., Citation2013) with default parameters and the mitogenome of Nasutitermes triodiae (accession number JX144940) as initial template. The assemblies yielded high-quality mitogenomes with coverage ranging from 2300- to 17,000-fold (). The genome length ranged from 16.0 to 17.6 kbp, and the G + C contents varied between 32 and 35 mol%. The nucleotide compositions of all mitogenomes were asymmetric (A: 41.4–43.6; C: 19.8–22.4; G: 11.6–12.9; T: 23.5–26.1); the values were in the same range as that of the four other mitogenomes of higher termites sequenced to date. The sequences were analyzed using the Improved de novo Metazoan Mitochondrial Genome Annotation (MITOS) webserver (Bernt et al. Citation2013; http://mitos.bioinf.uni-leipzig.de/), which provides a convenient and accurate means of gene prediction and annotation of mitochondrial genomes, including the necessary quality control.
Figure 1. Characteristics of the mitogenomes of six higher termite species. (a) Maximum likelihood tree based on the amino acid sequences of all protein-coding genes (PCG) of the mitogenomes (•, > 90% bootstrap support) and a brief description of their associated metadata. Source, dataset from the gut compartment with the highest proportion of host sequences; IMG ID, taxon object identifier of the raw read data in the Integrated Microbial Genomes database (330000xxxx; http://img.jgi.doe.gov/). (b) Linearized Schematic gene map (identical for all species); trn genes encoding tRNAs are abbreviated.
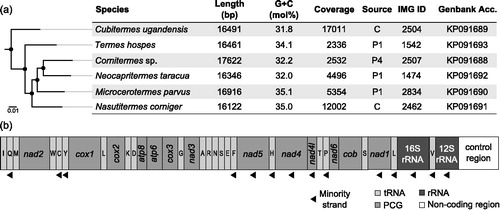
All mitogenomes contained the same 37 genes that are present in all termite mitogenomes sequenced to date; the genes varied slightly in length: 13 protein-coding genes (159–1729), a 12S rRNA (805–817 bp) gene, a 16S rRNA (1357–1388 bp) gene, and 22 tRNA genes (62–75 bp). Gene order and directionality were identical in all genomes; as in other termites, the rRNAs and several subunits of respiratory complex I were encoded on the minority strand (). Also the intergenic regions (1–94 bp), intergenic overlapping regions (1–41 bp), and A + T-rich control region contributed to the length heterogeneity of the genomes.
In addition to illustrating that mitochondrial genomes can be reconstructed from metagenomic datasets, our study substantially increases the number of available mitogenomes of higher termites, which will be important for future phylogenetic studies of this important insect group. Furthermore, the study contributes novel sequences to the sets of molecular markers, such as cytochrome oxidase and 16S rRNA genes that are frequently used for identifying host species via DNA barcoding methods in large-scale surveys of microbial symbionts (Dietrich et al., Citation2014).
Declaration of interest
This study was financially supported by the Max Planck Society and a grant of the Deutsche Forschungsgemeinschaft (SFB 987). The metagenomic datasets were obtained in the context of a collaborative project on termite gut microbiota between A. Brune and the Joint Genome Institute, Walnut Creek, California, in the Community Sequencing Program 2013 financed by the US Department of Energy.
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, et al. (2013). MITOS: Improved de novo Metazoan Mitochondrial Genome Annotation. Mol Phylogenet Evol 69:313–19
- Brune A. (2014). Symbiotic digestion of lignocellulose in termite guts. Nat Rev Microbiol 12:168–80
- Cameron SL, Lo N, Bourguignon T, Svenson GJ, Evans TA. (2012). A mitochondrial genome phylogeny of termites (Blattodea: Termitoidae): Robust support for interfamilial relationships and molecular synapomorphies define major clades. Mol Phylogenet Evol 65:163–73
- Cameron SL, Whiting MF. (2007). Mitochondrial genomic comparisons of the subterranean termites from the Genus Reticulitermes (Insecta: Isoptera: Rhinotermitidae). Genome 50:188–202
- Dietrich C, Köhler T, Brune A. (2014). The cockroach origin of the termite gut microbiota: Patterns in bacterial community structure reflect major evolutionary events. Appl Environ Microbiol 80:2261–9
- Hahn C, Bachmann L, Chevreux B. (2013). Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—A baiting and iterative mapping approach. Nucl Acids Res 41:e129
- Inward D, Beccaloni G, Eggleton P. (2007). Death of an order: A comprehensive molecular phylogenetic study confirms that termites are eusocial cockroaches. Biol Lett 3:331–5
- Qian ZQ. (2014). The complete mitogenome of the dampwood termite Zootermopsis nevadensis (Insecta: Isoptera: Termopsidae). Mitochondrial DNA 10:1–2. doi:10.3109/19401736.2014.936419
- Terrapon N, Li C, Robertson HM, Ji L, Meng X, Booth W, Chen Z, et al. (2014). Molecular traces of alternative social organization in a termite genome. Nat Commun 5:3636
- Tokuda G, Isagawa H, Sugio K. (2012). The complete mitogenome of the Formosan termite, Coptotermes formosanus Shiraki. Insect Soc 59:17–24