
Abstract
Objective:
To evaluate the bioequivalence of five 0.1 mg dutasteride capsules to one 0.5 mg dutasteride capsule in healthy adult male subjects under fasting conditions.
Methods:
This was a single-center, open-label, randomized, single dose, two-way cross-over study (ClinicalTrials.gov identifier NCT01929330). Thirty-six healthy male subjects aged 18–65 years received 5 × 0.1 mg dutasteride softgel capsules and 1 × 0.5 mg dutasteride softgel capsule in a randomized order, with a minimum washout of 28 days between each drug administration. Serial blood samples were collected for the measurement of serum dutasteride concentrations by a validated HPLC-MS/MS method. Dutasteride pharmacokinetic parameters were calculated using non-compartmental analysis. Maximum concentration (Cmax) and area under the concentration-time curve to the last quantifiable concentration (AUC[0–t]) were compared between treatments. Safety and tolerability were monitored throughout the study.
Results:
Five 0.1 mg dutasteride capsules were demonstrated to be bioequivalent to 1 × 0.5 mg dutasteride capsule, as the 90% confidence intervals for Cmax and AUC were within the accepted bioequivalence range of 0.80–1.25. The geometric least squares means ratios and associated 90% confidence intervals for 5 × 0.1 mg capsules vs 1 × 0.5 mg capsule were 1.01 (0.97–1.05) for Cmax and 0.91 (0.84–1.00) for AUC(0–t). Adverse events (AEs) were reported for 42% (15/36) and 36% (12/33) of subjects in the 5 × 0.1 mg and 1 × 0.5 mg dosing sessions, respectively. The most frequent AE for both treatments was headache. No subject had a serious AE.
Conclusions:
Five 0.1 mg dutasteride capsules were shown to be bioequivalent to one 0.5 mg dutasteride capsule in healthy adult male subjects under fasted conditions, suggesting that the two dose strengths can be interchanged. Both treatments were generally well tolerated in healthy male subjects.
Introduction
Androgenetic alopecia (AGA) is a common, androgen-induced, pattern of progressive loss of scalp hair with an onset at any age after puberty in genetically pre-disposed individualsCitation1. The influence of androgens on scalp hair growth is mediated by local and systemic conversion of testosterone to dihydrotestosterone (DHT) by the enzyme 5alpha-reductase (5AR)Citation1. DHT binds to the same androgen receptors as the parent compound, but up to 5-times more avidly, and is thought to influence hair growth via the activation of follicular androgen receptors in genetically susceptible individualsCitation2. 5AR has been shown to exist as two isoenzyme formsCitation2,Citation3. The presence of both isoenzymes in the hair follicles suggests that both are likely to be important in the pathogenesis and treatment of AGACitation4–7.
Dutasteride, a Δ′-4-azasteroid, is a competitive and specific inhibitor of both isoenzymes of 5ARCitation8. It is indicated at a dose of 0.5 mg daily to treat and prevent progression of benign prostatic hyperplasia (BPH) as monotherapy and in combination with tamsulosinCitation9. Additionally, dutasteride has been shown to increase hair growth and restoration in men with AGACitation10–12. Approval of dutasteride 0.5 mg daily for the treatment of male AGA was granted in Korea in July 2009 and approvals are now being sought in other countries.
The objective of the current study was to determine the bioequivalence of five 0.1 mg soft gelatin capsules of dutasteride compared to one 0.5 mg soft gelatin capsule of dutasteride in healthy male subjects. Secondary objectives were to characterize the pharmacokinetics (PK) of dutasteride following administration of the two formulations and to assess safety and tolerability.
Participants and methods
Participants
Male subjects aged between 18–65 years, weighing at least 50 kg, with a body mass index between 19–32 kg/m2, and in good general health as determined by medical history, clinical examination, 12-lead electrocardiogram (ECG), and clinical laboratory tests, were eligible for the study. Screening was conducted within 30 days prior to the first dosing.
Study design and ethics
This was an open-label, randomized, single dose, two-way cross-over study enrolling 36 subjects under fasting conditions (ClinicalTrials.gov identifier NCT01929330). It was undertaken at a single center, GlaxoSmithKline Medicines Research Unit, Prince of Wales Hospital, Randwick, New South Wales, Australia, between September 2013 and January 2014. The study was conducted in compliance with Good Clinical Practice guidelines after the protocol was approved by the relevant independent ethics committee. Written informed consent was obtained from each subject in accordance with the Declaration of Helsinki.
Each subject participated in two treatment periods. Eligible subjects were allocated to one of two sequences in accordance with the randomization schedule, which was generated by GlaxoSmithKline using validated internal software. In the first treatment period, each subject received either 1 × 0.5 mg dutasteride softgel capsule or 5 × 0.1 mg dutasteride softgel capsules orally under fasting conditions and received the alternate treatment in the second treatment period after a washout of 28 days. Participants remained in the clinical unit until completion of all assessments at 24 h post-dose in each treatment period and returned to the unit for the collection of remaining PK samples at 36, 48, and 72 h. Upon completion of the second treatment period, or early withdrawal, subjects returned to the clinical unit within 10–14 days for a follow-up visit and were also contacted via telephone for a second follow-up visit within 50–54 days.
Dutasteride soft gelatin capsules at strengths of 0.5 mg and 0.1 mg were manufactured by Catalent Pharma Solutions, Beinheim, France for GlaxoSmithKline.
Pharmacokinetic assessment
Blood samples for pharmacokinetic analysis were collected up to 72 h post-dose in each treatment period. Blood samples were collected into a 2 mL clot activator tube and left at room temperature for at least 20 min, but no more than 40 min to permit clot formation. Samples were centrifuged at 2500–3000 rpm (∼650–1450 × g) for 10–15 min to achieve a clear serum layer. Serum was transferred to a secondary, labeled container and stored at −20 °C until analysis.
Concentrations of dutasteride in serum were measured by high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) under the direction of Bioanalytical Science and Toxicokinetics, GlaxoSmithKline. Briefly, the analytical system consisted of a Leap Technologies autosampler (Carrboro, NC), a Thermo Scientific, BETASIL Silica-100 analytical column (Waltham, MA), and an API-4000 mass spectrometer (Applied Biosystems/MDS Sciex, Canada). Dutasteride was extracted from serum after the addition of an isotopically labeled internal standard ([Citation13C6]-dutasteride). Extracts were analyzed by HPLC-MS/MS using a TurboIonSpray interface and multiple reaction monitoring. Computer systems used to acquire and quantify data included Analyst software (version 1.4; Applied Biosystems/MDS Sciex, Canada) and Study Management System 2000 (version 2.3; GlaxoSmithKline). Dutasteride was extracted from serum samples using a validated liquid–liquid extraction using hexane/methyl tert-butyl ether/methylene chloride. Using a 100 µl aliquot, the lower and upper limits of quantification for dutasteride in serum were 25.0 pg/mL and 25 000 pg/mL, respectively. The standard curves for dutasteride were linear over their respective concentration ranges.
In the validation, the inter-assay accuracy (percent bias) ranged from −5.2% to 1.2% for dutasteride. The intra-assay precision and accuracy values for dutasteride were 6.7% and 8.9%, respectively. The quality control (QC) samples were prepared at four different analyte concentrations and analyzed with each batch of sample against separately prepared calibration standards. For the analysis to be acceptable, no more than one-third of the total QC results were to deviate from the nominal concentration by more than 15%, and at least 50% of the results from each QC concentration should be within 15% of nominal.
Along with all the required validation experiments as outlined in regulatory guidelines, validation experiments also tested the assay selectivity in the presence of caffeine, acetaminophen, acetylsalicylic acid, chlorpheniramine maleate, ibuprofen, naproxen, pseudoephedrine, salicylic acid, ethinyl estradiol, levonorgenstrel, and norethindrone. None of these possible co-administered drugs had any effect on the quantification of dutasteride in serum. The applicable analytical runs met all pre-defined run acceptance criteria.
Dutasteride serum concentration-time data were analyzed by non-compartmental methods using WinNonlin 6.3 (Pharsight Corporation, Mountain View, CA). All calculations were based on actual sampling times. The primary dutasteride PK parameters were maximum concentration (Cmax) and area under the serum concentration-time curve to the last quantifiable concentration [AUC(0–t)]. Time to maximum concentration (tmax) was a secondary parameter.
Safety assessments
Adverse event (AE) information volunteered by the subject, obtained from investigator questioning, or detected by other means was collected from the start of study treatment until the last follow-up contact. Vital signs and 12-lead ECG were recorded at screening, pre-dose, 3 h and 24 h post-dose in each treatment period, and at follow-up. Clinical laboratory tests were done at screening and on Day −1 of each treatment period.
Statistical methods
The study was designed to test the bioequivalence of 5 × 0.1 mg dutasteride capsules (Test) relative to 1 × 0.5 mg dutasteride capsule (Reference). The null hypothesis was that the true ratio of the geometric mean of the Test to the geometric mean of the Reference, µ(Test)/µ(Reference), for each primary PK end-point, was either less than or equal to 0.80 or greater than or equal to 1.25. The alternate hypothesis was that the true ratio of the Test geometric mean to the Reference geometric mean was greater than 0.80 and less than 1.25. For each primary PK parameter, a two one-sided t-test procedure with α = 0.05 for each one-sided test was used to test this set of hypotheses. This was equivalent to requiring that the 90% confidence interval for the true ratio of Test to Reference geometric means fell entirely within the range of 0.80–1.25.
The sample size was estimated on the basis of the largest within-subject coefficient of variance (CVw) for AUC(0–t) and Cmax observed in previous studies (24.8%), which translates to a standard deviation of 0.2442 on the natural log scale. On this basis, assuming that the actual ratio of geometric means would not differ from unity, a sample size of 28 statistically evaluable subjects would ensure 90% power to demonstrate bioequivalence. This calculation assumed a true ratio of 1.00 and that the CVw from the current study was not larger than that used in the sample size calculation. To ensure that 28 subjects completed dosing and critical assessments for both treatment periods, ∼36 subjects were to be enrolled.
Following loge-transformation, AUC(0–t) and Cmax of dutasteride were analysed separately using a mixed effects model with fixed effect terms for sequence, period, and treatment. Subject within sequence was treated as a random effect in the model. Point estimates and their associated 90% confidence intervals were constructed for the differences, Test–Reference. The point estimates and their associated 90% confidence intervals were then back-transformed to provide point estimates and 90% confidence intervals for the ratios, Test:Reference. Tmax of dutasteride was analyzed separately using a non-parametric procedure to compute point estimates and associated 90% confidence intervals for the median differences, Test–Reference.
Results
Study population
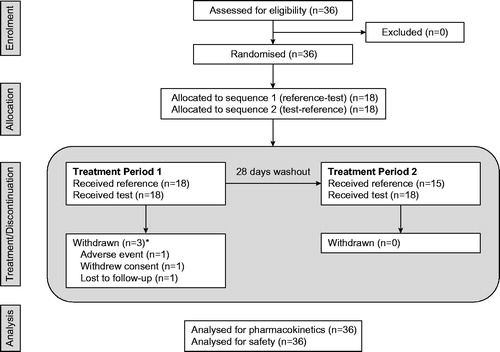
A total of 36 subjects were enrolled in the study (). Three subjects were withdrawn during treatment period 1 (after receiving 5 × 0.1 mg dutasteride capsules). The remaining 33 subjects proceeded to treatment period 2. Overall, 36 subjects received 5 × 0.1 mg dutasteride capsules and 33 subjects received 1 × 0.5 mg dutasteride capsule. All 36 subjects were healthy adult males. Mean (SD) age was 29.5 (8.47) years; mean (SD) BMI was 24.4 (2.33) kg/m2; 24 (67%) and 11 (31%) subjects were of White and Asian racial origin, respectively, and one (3%) subject was of mixed race.
Figure 1. Flow of participants through the study. Reference = 1 × 0.5 mg dutasteride capsule. Test = 5 × 0.1 mg dutasteride capsules. *The three withdrawn subjects each received the test formulation in Treatment period 1.
Pharmacokinetics
Peak serum and elimination phase concentrations of dutasteride were similar following single dose administration of each formulation (). Geometric least squares mean values for 5 × 0.1 mg and 1 × 0.5 mg formulations, respectively, were 2804 pg/mL and 3067 pg/mL for Cmax and 48507 pg.h/mL and 48048 pg.h/mL for AUC(0–t) (). The median time to peak concentration was 1.5 h for each formulation (range = 0.75–6 h).
Figure 2. Mean serum dutasteride concentration by planned relative time. (a) Reference = 1 × 0.5 mg dutasteride capsule. (b) Test = 5 × 0.1 mg capsules. The circles represent the mean plasma concentration at each time point. The grey shaded area represents ± 1 SD around the mean. The lower limit of quantification was 25.0 pg/mL.
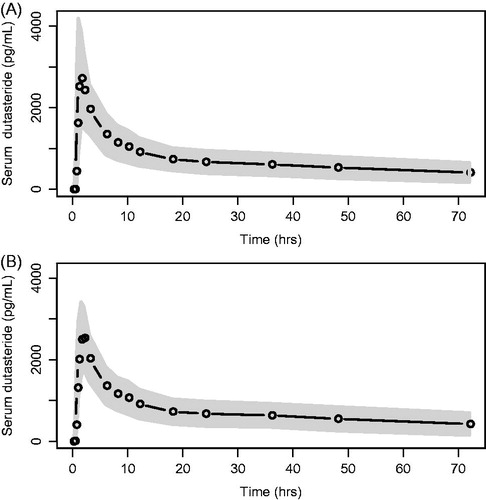
Table 1. Statistical analysis of loge transformed primary serum pharmacokinetic parameters.
The 90% confidence intervals for the ratio (test:reference) of geometric means for AUC(0–t) (ratio = 1.01; 90% CI = 0.97–1.05) and Cmax (ratio = 0.91; 90% CI = 0.84–1.00) were within the limits of 0.80–1.25 (), indicating that 5 × 0.1 mg dutasteride capsules are bioequivalent to 1 × 0.5 mg dutasteride capsule. Similarly, the median tmax values for the two treatments were not significantly different (median difference = 0.25 h; 90% CI = 0.00–0.50).
Within-subject variability was ∼22% for Cmax and 9% for AUC, which was lower than that used in sample size calculations.
Safety and tolerability
A total of 39 AEs were reported by 21 (58%) subjects over the course of the study. All AEs were of mild or moderate intensity; no severe AEs were reported. AEs were reported for a similar proportion of subjects after single dose administration of 5 × 0.1 mg dutasteride capsules and 1 × 0.5 mg dutasteride capsule (42% and 36%, respectively) ().
Table 2. Summary of adverse events.
The proportion of subjects who reported headache was higher after the administration of 1 × 0.5 mg dutasteride capsule (24%) compared to the administration of 5 × 0.1 mg dutasteride capsules (14%) (). All episodes of headache were of mild intensity. There was no pattern to the temporal occurrence of headache, with onset ranging from 4 h to 25 days after dosing. Two subjects reported nausea after administration of 5 × 0.1 mg dutasteride capsules, whereas there were no reports following dosing with 1 × 0.5 mg dutasteride capsule (). Onset of nausea was from 10–20 min after dosing; all episodes were mild in intensity and resolved within 20 min, with no further action required.
No subject had an SAE. One subject was withdrawn from the study due to an AE of upper-airway cough syndrome after dosing with 5 × 0.1 mg capsules in treatment period 1; this AE was not considered by the investigator to be related to study drug.
No clinically significant changes from baseline in blood pressure or heart rate were observed after single administration of either formulation. No clinically significant ECG abnormalities were recorded during the study. No clinical laboratory result was reported as an AE.
Discussion
The objective of this study was to examine the relative bioavailability and bioequivalence of the two strengths of dutasteride to be used for the treatment of androgenetic alopecia. The results show that five 0.1 mg dutasteride capsules are bioequivalent to one 0.5 mg dutasteride capsule, suggesting that the two dose strengths can be interchanged. The mean concentration vs time profiles for each treatment were nearly identical. Both peak and total exposures were very similar, as evidenced by the geometric mean ratios being close to unity. The 90% confidence intervals for both peak and total exposure were well within the 0.80–1.25 regulatory threshold for bioequivalence. These results were expected, as both formulations are soft gelatin capsules containing dutasteride dissolved in a mixture of mono-di-glycerides of caprylic/capric acidCitation13, and are very similar except for the size and strength of the capsules. Both capsule strengths have similar in vitro dissolution profiles (data on file at GlaxoSmithKline).
Consistent with previously reported dataCitation14, single dose administration of dutasteride was relatively well tolerated in the healthy male subjects participating in this study. A similar tolerability profile was observed for both formulations. Any observed differences in frequencies of individual AEs following administration of each formulation are likely chance findings due to the relatively small number of subjects participating in the study, rather than actual differences in the tolerability of the two formulations.
The small sample size and single dose design of this study prevent definitive conclusions being made regarding the pharmacokinetics and safety of dutasuteride capsules in the AGA patient population. However, dutasteride at a dose of 0.5 mg daily has been approved for more than 10 years for the treatment of BPH and considerable clinical trials and post-marketing experience is availableCitation9,Citation14. In large, well-controlled clinical trials in patients with BPH, dutasteride had a tolerability profile comparable with that of placebo, with the exception of a small increase in the incidence of impotence and decreased libido during the first year of therapyCitation14. Dutasteride was also well tolerated in placebo-controlled clinical trials in men with AGA, and no new safety concerns were raisedCitation10–12.
Conclusion
Bioequivalence was demonstrated for one 0.5 mg dutasteride capsule and five 0.1 mg dutasteride capsules following single dose administration to healthy male subjects. Both treatments were generally well tolerated in healthy male subjects.
Transparency
Declaration of funding
GlaxoSmithKline sponsored the study and provided funding for the study and for the development and publishing of the present manuscript.
Declaration of financial/other relationships
All authors are employees of GlaxoSmithKline. MJF, DAC, NS, CLB, and OB have stock/stock options in the company.
Acknowledgments
The authors thank study volunteers for their participation and the staff at GlaxoSmithKline Medicines Research Unit, Australia for support in conducting the study. Writing assistance was provided by Julie Taylor of Peak Biomedical Ltd, paid for and on behalf of GlaxoSmithKline.
References
- Kaufman KD. Androgen metabolism as it affects hair growth in androgenetic alopecia. Dermatol Clin 1996;14:697–711
- Sinclair R. Male pattern androgenetic alopecia. BMJ 1998;317:865–9
- Olsen EA. Androgenetic alopecia. In: Olsen EA, ed. Disorders of hair growth: diagnosis and treatment. New York, NY: McGraw-Hill, 1994. pp 257–83
- Jenkins EP, Andersson S, Imperato-McGinley J, et al. Genetic and pharmacological evidence for more than one human steroid 5 alpha-reductase. J Clin Invest 1992;89:293–300
- Russell DW, Wilson JD. Steroid 5 alpha-reductase: two genes/two enzymes. Annu Rev Biochem 1994;63:25–61
- Sawaya ME, Price VH. Different levels of 5alpha-reductase type I and II, aromatase, and androgen receptor in hair follicles of women and men with androgenetic alopecia. J Invest Dermatol 1997;109:296--300
- Thigpen AE, Silver RI, Guileyardo JM, et al. Tissue distribution and ontogeny of steroid 5 alphareductase isozyme expression. J Clin Invest 1993;92:903–10
- Frye SV. Discovery and clinical development of dutasteride, a potent dual 5alpha-reductase inhibitor. Curr Top Med Chem 2006;6:405–21
- Wu C, Kapoor A. Dutasteride for the treatment of benign prostatic hyperplasia. Expert Opin Pharmacother 2013;14:1399–408
- Olsen EA, Hordinsky M, Whiting D, et al. The importance of dual 5α-reductase inhibition in the treatment of male pattern hair loss: results of a randomized placebo-controlled study of dutasteride versus finasteride. J Am Acad Dermatol 2006;55:1014–23
- Eun HC, Kwon OS, Yeon JH, et al. Efficacy, safety, and tolerability of dutasteride 0.5 mg once daily in male patients with male pattern hair loss: a randomized, double-blind, placebo-controlled, phase III study. J Am Acad Dermatol 2010;63:252–8
- Gubelin Harcha W, Barboza Martínez J, Tsai TF, et al. A randomized, active- and placebo-controlled study of the efficacy and safety of different doses of dutasteride versus placebo and finasteride in the treatment of male subjects with androgenetic alopecia. J Am Acad Dermatol 2014;70:489–98
- AVODART® (dutasteride) Soft Gelatin Capsules. Prescribing Information. GlaxoSmithKline, The GSK group of companies, 2014. https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Avodart/pdf/AVODART-PI-PIL.PDF. Accessed July 13, 2015
- Andriole GL, Kirby R. Safety and tolerability of the dual 5alpha-reductase inhibitor dutasteride in the treatment of benign prostatic hyperplasia. Eur Urol 2003;44:82–8