
Abstract
A new series of tetrazole-, oxadiazole- and cyanosubstituted 1,4-dihydropyrimidinone compounds were synthesized, and their inhibitory effects on the activity of purified human carbonic anhydrase (hCA) I were evaluated. 4-Cyanophenyl-1,4-dihydropyrimidinone compounds were prepared with 1,3-diketone, cyanobenzaldehyde and urea. The compounds were reacted with sodium azide and then with anhydride to get the final products. The results showed that all the synthesized compounds inhibited the CA isoenzyme activity. The compound 4-(1,7,7-trimethyl-2,5-dioxo-1,2,3,4,5,6,7,8-octahydroquinazoline-4-yl)benzonitrile 6c (IC50 = 0.0547 mM) has the most inhibitory effect.
Introduction
Carbonic anhydrases (CAs, EC 4.2.1.1) are widespread zinc metalloenzymes that catalyse the reversible hydration of carbon dioxide (CO2) to bicarbonate (HCO3−) and a proton (H+) with water (Gilmour and Perrry 2009). CAs are ubiquitous enzymes present in prokaryotes and eukaryotes, which are encoded by four evolutionarily unrelated gen families (α-, β-, γ- and ξ-CAs) (Hen et al. 2011). Up to now, 16 human CA (hCA) isoforms have been identified exhibiting significant differences in catalytic activity, subcelluler localization and tissues expression. They play important roles in many of the physiological processes such as several cell proliferation, intra and extracellular pH homeostasis and differentiation, modulation of neuronal transmission and biochemical pathways (Gitto et al. 2012, Supuran 2011). In human, CAs are found in a variety of tissues such as lungs, skins, eyes, kidneys, the nerves systems and the gastrointestinal tract (Supuran 2011). Biological activities of this metalloenzyme family have several medicinal applications such as treatment for glaucoma, diuretics, management of several neurological disorders, whereas several agents are in clinical evaluations as antiobesity or antidrug (Ekinci et al. 2012).
Nowadays, 1,4-dihydropyrimidinone (DHPM) compounds have much attention due to their significant biological activities. The compounds have various therapeutic and pharmacological properties such as calcium channel modulators, antihypertensive agents, α1a-adrenergic receptor antagonists (Kappe 2000), antiviral, antitumour, antibacterial and anti-inflammatory activities (Kappe 2000). The dihydropyrimidinone cores are also found in many natural products and marine alkaloids, and have been found to be potent HIV gp-120CD4 inhibitors (Snider et al. 1996).
The simple and direct method for the synthesis of dihydropyrimidinones (DHPMs) first reported by Biginelli (1893) in 1893 was synthesized using an aldehyde, a β-ketoester and urea (or thiourea) under strongly acidic conditions, but the reaction suffered from backs such as long reaction time and low yields. For this transformation, several methods were improved such as using zirconium hydrogen phosphate (Besoluk et al. 2010), alumina sulphuric acid (Besoluk et al. 2008) and heteropoly acids (Rafiee and Jafari 2006).
Heterocyclic compounds, containing several nitrogen atoms, are scaffolds that are frequently considered when designing bioactive compounds (Sabbah et al. 2012). Tetrazoles have a wide range of applications in medicinal chemistry especially in drug in isosteric replacement of carboxylic acid moiety (Patil et al. 2012, Herr 2002). The tetrazole ring is found in well-known medicines such as Diovan, Aprovel, Cozaar and Benicar used for the treatment of cardiovascular diseases and hypertension (Katritzky et al. 2010). The substituted derivatives of the compounds have also been used in a wide range of applications in material sciences (Singh et al. 2006), antibiotics, tuberculostatics, analgesics and fungicides (Ichikawa et al. 2001, Rajasekaran and Thampi 2004, Waisser et al. 2004).
In this study, a new series of tetrazole-, oxadiazole- and cyanosubstituted 1,4-dihydropyrimidinone compounds were synthesized, and their inhibitory effects on the activity of purified human carbonic anhydrase (hCA) I were evaluated.
Materials and methods
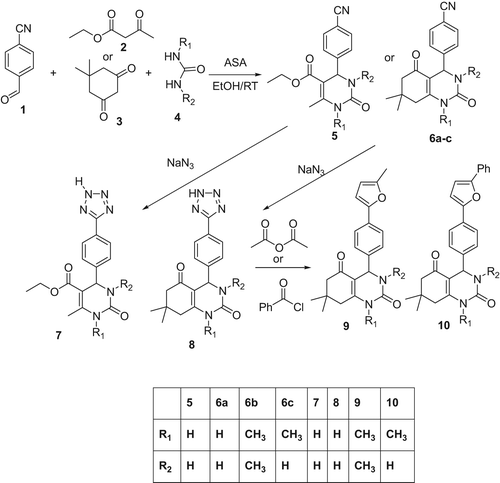
Tetrazole-, oxadiazole- and cyanosubstituted 1,4-dihydropyrimidinone compounds shown in were synthesized and examined the effects on carbonic anhydrase I. 4-Cyanophenyl-1,4-dihydropyrimidinone was prepared with 1,3-diketone, cyanobenzaldehyde and urea. The compound was reacted with sodium azide and then with anhydride to get the products (5–10) at high yields.
Scheme 1. Synthesis of tetrazole- and oxadiazole-substituted 1,4-dihydropyrimidinone derivatives.
General
All starting materials and reagents were purchased from commercial suppliers. Reactions were monitored by TLC and TLC plates visualized with short-wave UV fluorescence (k = 254 nm). Melting points were taken on a Yanagimoto micro-melting point apparatus and were uncorrected. IR spectra were measured on a SHIMADZU Prestige-21 (200 VCE) spectrometer and 1H and 13C NMR spectra on a spectrometer at VARIAN Infinity Plus 300 and at 75 Hz, respectively. 1H and 13C chemical shifts were referenced to the internal deuterated solvent. The elemental analysis was carried out with a Leco CHNS-932 instrument. Flash column chromatography was performed using Merck silica gel 60 (230–400 mesh ASTM).
Synthesis of 1,4-dihydropyrimidinone (5 or 6)
A mixture of 4-cyanobenzaldehyde (3 mmol), dimedone or ethylasetoasetate (3 mmol), urea (4.5 mmol) and alumina sulphuric acid (ASA) catalyst (7% mmol) in ethanol were finely mixed together in a flask at room temperature for two hours. After cooling at room temperature, the reaction mixture was poured onto crushed ice (50 g) and stirred for 10 min. The precipitate was filtered under suction and washed with cold water (20 ml) to remove excess urea. Then, the solid was dissolved in ethanol, filtered to remove the catalyst and purified further by recrystallization (hot ethanol).
Synthesis of tetrazole-substituted 1,4-dihydropyrimidinone (7 or 8)
The compounds (5 or 6) (5 mmol), sodium azide (20 mmol) and ammonium chloride (20 mmol) in 5 ml DMF, were finely mixed together in a flask at 150°C for 20 h. After cooling, the reaction mixture was poured into iced cold water (200 ml) and stirred. The pH was adjusted to 1.0 with HCl. Then, the solid was filtered and dried.
Synthesis of 1,3,4-okzadiazole-substituted 1, 4-dihydropyrimidinone (9 or 10)
The tetrazole derivative (1 mmol) in 2 ml of acetic anhydride was heated at 150°C for 20 h. After cooling, the reaction mixture was poured into iced cold water (100 ml) and extracted with dichloromethane. Then, the solvent was evaporated and adduct was crystallized from the mixture of dichloromethane-hexane (1:1 ratio).
Ethyl 4-(4-cyanophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate(5): yield: 88%; m.p.: 168–69°C; 1 H NMR (DMSO-d6)(300 mHz): 9.35(H,s), 7.80(2H,d), 7.40(2H,d), 5.25(H,s), 3.95(2H,q), 2.25(3H,s), and 1.10(3H,t); 13CNMR (DMSO-d6)(75mHz): 164.8, 151.7, 149.9, 149.1, 132.8, 127.4, 188.6, 109.8, 98.9, 59.5, 53.3, 17.4, and 13.4; and IR (υ, cm− 1): 3358, 3226, 3103, 2976, 2229. Anal. Calcd. For C15H15N3O3: C, 63; H, 5.30; and N, 14.73. Found: C, 63.54; H, 5.72; and N, 14,21.
4-(7,7-dimethyl-2,5-dioxo-1,2,3,4,5,6,7,8-octahydroquinazoline-4-yl)benzonitrile (6a): yield: 88%; m.p.: 258–60°C; 1H NMR (DMSO-d6)(300mHz): 9.61(H,s), 7.91(H,s), 7.82(2H,d), 7.40(2H,d), 5.21(H,s), 2.40(2H,d,d), 2.20(2H,d,d), 0.95(3H,s), and 1.01(3H,s); 13CNMR(DMSO-d6)(75mHz): 193.6, 185.7, 153.7, 152.3, 133.1, 127.9, 118.6, 110.6, 110.0, 103.4, 52.6, 32.9, 32.6, 29.3, and 27.5; and IR (υ, cm− 1): 3334, 3207, 3089, 2962, 2227, 1683, and 1614. Anal. Calcd. For C17H19 N3O2: C, 68.67; H, 6.44; and N, 14.13. Found: C, 68.14; H, 5.78; and N, 14,45.
4-(1,3,7,7-tetramethyl-2,5-dioxo-1,2,3,4,5,6,7,8-octahydroquinazoline-4-yl)benzonitrile (6b): yield: 88%; m.p.: 169–70°C; 1H NMR (DMSO-d6)(300mHz): 7.62(2H,d), 7.42(2H,d), 5.44(H,s), 3.25(3H,s), 2.97(3H,s), 2.40(2H,d,d), 2.20(2H,d,d), and 1.10(3H,s), 1.00(3H,s); 13CNMR(DMSO-d6)(75mHz):194.4, 153.3, 153.5, 146.8, 132.4, 127.5, 123.0, 118.7, 111.8, 110.1, 58.4, 49.9, 40.3, 35.4, 33.3, 31.0, and 28.9; and IR (υ, cm− 1): 2956, 2229, 1674, and 1604. Anal. Calcd. For C19H23N3O2: C, 70.13; H, 7.12; and N, 12.91. Found: C, 70.77; H, 7.58; and N, 13.51.
4-(1,7,7-trimethyl-2,5-dioxo-1,2,3,4,5,6,7, 8-octahydroquinazoline-4-yl)benzonitrile (6c): yield: 80%; m.p.: 207–09°C; 1HNMR(DMSO-d6)(300mHz): 8,18(H,s), 7.80(2H,d), 7.40(2H,d), 5.22(H,s), 3.17(3H,s), 2.60(2H,d,d), 2.25(2H,d,d), 1.10(3H,s), and 0.98(3H,s); 13CNMR(DMSO-d6)(75mHz):194.0, 155.3, 153.0, 150.0, 133.1, 127.7, 119.5, 110.6, 109.8, 60.4, 49.5, 32.8, 30.1, 29.7, and 28.4; and IR (υ, cm− 1): 3244, 3132, 3061, 2225, 1693, and 1600. Anal. Calcd. For C18H22N3O2: C, 69.21; H, 7.10; and N, 13.45. Found: C, 70.37; H, 7.48; and N, 13.21.
Ethyl 4-(4-(1H-tetrazole-5-yl)phenyl)-6-methyl-2-oxo-1, 2,3,4-tetrahydropyrimidine-5-carboxylate (7): yield: 90%; m.p.: 251–52°C; 1H NMR (DMSO-d6)(300mHz): 9.35(H,s), 8.01(2H,d), 7.83(H,s), 7.51(H,d), 5.20(H,s), 4.01(2H,q), 2.25(3H,S), and 1.10(3H,t); 13C NMR (DMSO-d6)(300mHz): 165.1, 151.8, 148.8, 147.8, 127.2, 127.3, 98.5, 53.7, 17.7, and 13.9; and IR (υ, cm− 1): 3217, 3088, 2914, 1697, and 1643. Anal. Calcd. For C15H16N6O3: C, 54.87; H, 4.91; and N, 25.60. Found: C, 55.33; H, 5.25; and N, 26.10.
4-(4-(1H-tetrazole-5-yl)phenyl)-7,7-dimethyl-3,4,7, 8-tetrahydroquinazoline-2,5(1H,6H)-dione (8): yield: 88%; m.p.: 243–45°C;1HNMR(DMSO-d6)(300mHz):9.62(H,s), 8.00(2H,d), 7.85(H,s), 7.45(2H,d), 5.20(H,s), 2.80(2H,d,d), 2.42(2H,d,d), 1.10(3H,s), and 0.90(3H,s); 13CNMR (DMSO-d6)(75mHz):193.7, 153.4, 152.5, 148.3, 127.9, 127.8, 107.5, 92.6, 92.6, 52.6, 50.4, 33.2, 29.3, 28.3, and 27.4; and IR (υ, cm− 1): 3406, 3244, 2954, 1678, 1647, and 1620. Anal. Calcd. For C17H20N6O2: C, 59.99; H, 5.92; and N, 24.69. Found: C, 60.33; H, 6.26; and N, 25.10.
1,3,7,7-tetramethyl-4-(4-(5-methyl-1,3,4-oxadiazole- 2-yl)phenyl)-3,4,7,8-tetrahydroquin azoline-2,5(1H,6H)-dione (9): yield: 68%; m.p.: 178–79°C; 1HNMR(DMSO-d6)(300mHz): 7.85(2H,d), 7.40(2H,d), 5.20(H,s), 3.30(3H,s), 2.85(3H,s), 2.40(2H,d,d), 2.20(2H,d,d), 1.10(3H,S), and 0.98(3H,s); 13CNMR(DMSO-d6)(75mHz): 198,9, 164.8, 165.2, 157.9, 150.2, 138.4, 128.6, 127.6, 105.6, 66.7, 50.8, 39.3, 34.1, 33.9, 31.5, 27.6, and 14.3; and IR (υ, cm− 1): 3334, 2978, 2229, 1697, and 1651. Anal. Calcd. For C21H24N4O3: C, 66.30; H, 6.36; and N, 14.73. Found: C, 66.88; H, 6.56; and N, 15.10.
1,7,7-trimethyl-4-(4-(5-phenyl-1,3,4-oxadiazole-2-yl phenyl)-3,4,7,8-tetrahydroquinazoli ne-2,5- (1H,6H)- dione (10): yield: 67%; m.p.: 276–77°C; 1H NMR (CDCl3 + DMSO-d6)(300mHz): 8.10(2H,d), 8.00(2H,d), 7.90(H,s), 7.68 (2H,t), 7.60(H,t), 7.45(2H,d), 5.41(H,s), 3.20(3H,s), 2.60(2H,d,d), 2.40(2H,d,d), 1.10(3H,s), and 0.98(3H,s); 13C NMR(DMSO-d6)(75mHz):199.2, 167.3, 156.8, 148.3, 142.9, 129.2, 128.6, 127.3, 127.9, 127.6, 126.7, 124.8, 105.9, 50.3, 48.6, 39.8, 33.5, 31.6, and 27.5; IR (υ, cm− 1): 3319, 3062, 2956, 1685, and 1622. Anal. Calcd. For C25H24N4O3: C, 70.08; H, 5.65; and N, 13.08. Found: C, 70.38; H, 6.06; and N, 13.48.
Preparation of haemolysate and purification from blood red cells
Blood samples (25 mL) were taken from healthy human volunteers. They were anticoagulated with acid–citrate–dextrose, centrifuged at 2000 g for 20 min at 4°C, and the supernatant was removed. The packed erythrocytes were washed three times with 0.9% NaCl and then haemolysed in cold water. The ghosts and any intact cells were removed using centrifugation at 2000 g for 25 min at 4°C, and the pH of the haemolysate was adjusted to 8.5 with solid Tris base. The 25-mL haemolysate was applied to an affinity column containing L-tyrosine-sulphonamide-Sepharose-4B (Arslan et al. 1996) equilibrated with 25 mM Tris–HCl/0.1 M Na2SO4 (pH, 8.5). The affinity gel was washed with 50 mL of 25 mM Tris–HCl/22 mM Na2SO4 (pH, 8.5). The hCA I isozyme was then eluted with 0.1 M NaCl/25 mM Na2HPO4 (pH 6.3) and recovered hCA I. Fractions of 3 mL were collected and their absorbance measured at 280 nm.
CA enzyme assay
CA activity was measured using the Maren method based on the determination of the time required for the pH to decrease from 10.0 to 7.4 due to CO2 hydration (Maren 1960). The assay solution was 0.5 M Na2CO3/0.1 M NaHCO3 (pH, 10.0), and phenol red was added as the pH indicator. CO2–hydratase activity (enzyme units (EU)) was calculated using the equation t0-tc/tc where t0 and tc are the times for pH change of the nonenzymatic and the enzymatic reactions, respectively.
In vitro inhibition studies
For the inhibition studies of sulphonamide, different concentrations of these compounds were added to the enzyme. Activity percentage values of CA for different concentrations of each sulphonamide were determined by regression analysis using Microsoft Office 2000 Excel. CA enzyme activity without a synthesized compounds solution was accepted as to be 100%.
Results and discussion
For evaluating the physiologically relevant human CA isozymes hCA I activity, several new tetrazole-, oxadiazole- and cyanosubstituted 1,4-dihydropyrimidinone compounds were subjected to CA inhibition assay with CO2 as a substrate.
The prepared compounds were characterized by 1H NMR, 13C NMR, IR and elemental analysis. The amide hydrogen resonances between 8.00 and 9.70 ppm and was indicated from the 1H NMR spectra. The signals for aromatic hydrogen are between 7.40 and 8.50 ppm. The hydrogen next to the phenyl ring was observed at around 5.20 ppm. From the 13C NMR spectra, carbonyl carbons are seen between 200 and 150 ppm. In the infrared spectra of compounds, it was possible to observe the absorptions between 3250 and 3450 cm− 1 relating to NH stretching and absorptions in 1650–1750 cm− 1 from carbonyl moiety stretching. CN stretching was observed around 2220 cm− 1.
In this study, we have examined the effects of the compounds (5–10) on hCA I. The results showed that all the synthesized compounds inhibited the hCA I activity. The IC50 values of (5–10) analogues against hCA I are summarized in . It is determined that the inhibition values are in between 0.0547 and 0.1473 mM for hCA I. Among the compounds, 6c and 8 were found to be the most active for CAs with the values of 0.0547 mM and 0.062 mM, respectively.
Table I. IC50 values of the synthesized compounds.
CA inhibitors lower intraocular pressure by reducing bicarbonate formation in the ciliary process, thus lowering Na+ transport and flow of aqueous humour. Unfortunately, systemic therapy with parenteral sulphonamides and their derivatives leads to significant side effects, many of them being probably due to the inhibition of CA isoforms in other tissues. Acetazolamide is the most widely used inhibitor and has advantages over the others because it is 20 times less active against hCA I than against hCA II in erythrocytes. But the inhibition of various CA isoforms that are present in tissues other than eye leads to an entire range of side effects, the most prominent being numbness and tingling of extremities, metallic taste, depression, fatigue, malaise, weight loss, decreased libido, gastrointestinal irritation, metabolic acidosis, renal calculi and transient myopia (Maren 1960, Arslan et al. 1997, Supuran and Scozzafava 2000). For similar reasons, designing of new drugs is essential for clinical application.
Sulphonamides and phenols represent classes of effective CAIs, with the sulphonamides and their bioisoesterase (sulphamates and sulphamides) having clinical applications. Sulphonamide compounds are coordinated to the zinc (II) ion within the hCAs active site, whereas its organic scaffold fills the entire enzyme cavity, making an extensive series of van der Waals and polar interactions with amino acid residues at the bottom, in the middle and at the entrance of the active site cavity (Maresca et al. 2010). The other classes of CAIs are the coumarins, and their inhibition mechanisms are different from those of the other CAIs due to their binding at the entrance of the enzyme active site. Coumarins have bulky pendant group and cannot bind enzyme effectively in the restricted space near Zn2+ ion. The compounds exhibit unusual binding mode not interacting with the metal ion of the enzyme (Maresca et al. 2009, Maresca and Supuran 2010).
The slow cytosolic isoform hCA I was weakly inhibited by the synthesized compounds (5–10). This is an extremely desirable feature because hCA I is not a drug target, but an off-target, being a widely expressed isoform in many tissues and cell types and possessing house-keeping physiological functions (Supuran 2008). We assume that the synthesized compounds have similar interactions with enzyme as the coumarins. They are big compounds to interfere with the binding to the enzyme active site (zinc ion).
Enzyme activity studies are important issues for drug design and biochemical applications (Aydemir and Kavrayan 2009, Arslan et al. 2012, Gencer et al. 2012, Cicek et al. 2012, Demir et al. 2012, Demirel and Tarhan 2004, Sayin et al. 2012, Gökce et al. 2012, Bytyqi-Damoni et al. 2012, Ozensoy et al. 2008, Supuran and Scozzafawa 2007). The results showed that the synthesized compounds inhibited the hCA I enzyme activity. The compounds have weak inhibitory effects, and they may be taken for further evaluation in vivo studies.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
This work was supported by Research Fund of the Sakarya University. Project Number: 2010-02-04–013.
References
- Arslan M, Gencer N, Arslan O, Guler OO. 2012. In vitro efficacy of some cattle drugs on bovine serum paraoxonase 1 (PON1) activity. J Enzyme Inhib Med Chem. 27:722–729.
- Arslan O, Nalbantoglu B, Demir N, Ozdemir H, Küfrevioglu OI. 1996. A new method for the purification of carbonic anhydrase isozymes by affinity chromatography. Turk J Med Sci. 26:163–166.
- Arslan O, Kufrevioglu OI, Nalbantoglu B. 1997. Synthesis and investigation of inhibition effects of new carbonic anhydrase inhibitors. Bioorg Med Chem. 3:515–518.
- Aydemir T, Kavrayan D. 2009. Purification and characterization of glutathione-S-transferase from chicken erythrocyte. Artif Cells Blood Substit Immobil Biotechnol. 37:92–100.
- Besoluk S, Kucukislamoglu M, Zengin M, Arslan M, Nebioglu M. 2010. An efficient one-pot synthesis of dihydropyrimidinones catalyzed by zirconium hydrogen phosphate under solvent-free conditions. Turk J Chem 34:411–416.
- Besoluk S, Kucukislamoglu M, Nebioglu M, Zengin M, Arslan M. 2008. Solvent-free synthesis of dihydropyrimidinones catalyzed by alumina sulfuric acid at room temperature. J Iran Chem Soc. 5:62–66.
- Biginelli P. 1893. Ital synthesis of 3,4-dihydropyrimidin-2(1H)-ones. Gazz Chem. 23:360–362.
- Bytyqi-Damoni A, Genç H, Zengin M, Beyaztas S, Gençer N, Arslan O. 2012. In vitro effect of novel β-lactam compounds on xanthine oxidase enzyme activity. Artif Cells Blood Substit Immobil Biotechnol. 40:369–377.
- Cicek B, Ergun A, Gencer N. 2012. Synthesis and evaluation in vitro effects of some macro cyclic thiocrown ethers on erythrocyte carbonic anhydrase I and II. Asian J Chem. 24:3729–3731.
- Demir D, Gencer N, Er A. 2012. Purification and characterization of prophenoloxidase from Galleria mellonella L. Artif Cells Blood Substit Biotechnol. 40:391–395.
- Demirel LAB, Tarhan L. 2004. Dismutation properties of purified and GDA modified CuZnSOD from chicken heart. Artif Cells Blood Substit Immobil Biotechnol. 32:609–624.
- Ekinci D, Çavdar H, Durdagi S, Talaz O, Sentürk M, Supuran CT. 2012. Structure-activity relationships for the interaction of 5, 10-dihydroindeno[1,2-b] indole derivatives with human and bovine carbonic anhydrase isoforms I, II, III, IV and VI. Europ J Med Chem. 49:68–73.
- Gencer N, Ergun A, Demir D. 2012. In vitro Effects of Some Anabolic Compounds on Erythrocyte Carbonic Anhydrase I and II. J Enzyme Inhib Med Chem. 27:208–210.
- Gocke B, Gencer N, Arslan O, Turkoglu SA, Alper M, Kockar F. 2012. Evaluation of in vitro effects of some analgesic drugs on erythrocyte and recombinant carbonic anhydrase I and II. J Enzyme Inhib Med Chem. 27:37–42.
- Gilmour KM, Perrry SF. 2009. Carbonic anhydrase and acid-base regulation in fish. J Exp Biol. 212:1647–1661.
- Gitto R, Damiano FM, Mader P, De Luca L, Ferro S, Supuran CT, et al. 2012. Synthesis, structure-activity relationship studies, and X-ray crystallographic analysis of arylsulfonamides as potent carbonic anhydrase inhibitors J Med Chem. 55:3891–3899.
- Hen N, Bialer M, Yagen B, Maresca A, Aggarwal M, Robbins AH, et al. 2011. Anticonvulsant 4-aminobenzenesulfonamide derivatives with branched-alkylamide moieties: X-ray crystallography and inhibition studies of human carbonic anhydrase isoforms I, II, VII, and XIV. J Med Chem. 54:3977–3981.
- Herr RJ. 2002. 5-Substituted-1H-tetrazoles as carboxylic acid isosteres: medicinal chemistry of synthetic methods. Bioorg Med Chem. 10:3379–3391.
- Ichikawa T, Yamada M, Yamaguchi M, Kitazaki T, Matsushita Y, Higashikawa K, Itoh K. 2001. Optically active antifungal azoles. XIII. Synthesis of stereoisomers and metabolites of 1-[(1R,2R)-2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3(1H-1,2,4-triazol-1-yl)propyl]-3-[4-(1H–1-tetrazolyl)phenyl]-2-imidazolidinone (TAK-456)Chem Pharm Bull. 49:1110.
- Kappe CO. 2000. Biologically active dihydropyrimidones of the Biginelli-type - a literature survey. Eur J Med Chem. 35:1043–1052.
- Kappe CO. 2000. Recent advances in the biginelli dihydropyrimidine synthesis. New tricks from an old dog. Accounts Chem Res. 33: 879–888.
- Katritzky AR, El-Gendy BDEM, Draghici B, Hall CD, Steel PJ. 2010. NMR Study of the Tautomeric Behavior of N-(α-Aminoalkyl)tetrazoles. J Org Chem. 75:6468–6476.
- Maren TH. 1960. A simplified micromethod for the determination of carbonic anhydrase and its inhibitors. J Pharm Exp Ther. 130: 2629–34.
- Maresca A, Temperini C, Pochet L, Masereel B, Scozzafava A, Supuran CT. 2010. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem. 53:335–344.
- Maresca A, Temperini C, Vu H, Pham NB, Poulsen SA, Scozzafava A, et al. 2009. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc. 131:3057–3062.
- Maresca A, Supuran CT. 2010. Coumarins incorporating hydroxy- and chloro-moieties selectively inhibit the transmembrane, tumor- associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II. Bioorg Med Chem Lett. 20:4511–4514.
- Ozensoy O, Arslan O, Kockar F. 2008. Differential in vitro inhibition effects of some antibiotics on tumor associated carbonic anhydrase isozymes of hCA-IX and hCA-XII. J Enzyme Inh Med Chem. 23: 579–585.
- Patil UB, Kumthekar KR, Nagarkar JM. 2012. A novel for synthesis of 5-substituted 1H-tetrazole from oxime and sodium azide. Tetrahedron Lett. 53:3706–3709.
- Rafiee E, Jafari H. 2006. A practical and green approach towards synthesis of dihydropyrimidinones: using heteropoly acids as efficient catalysts. Bioorg Med Chem Lett. 16:2463–2466.
- Rajasekaran A, Thampi PP. 2004. Synthesis and analgesic evaluation of some 5-[beta-(10-phenothiazinyl)ethyl]-1-(acyl)-1,2,3,4-tetrazoles. Eur J Med Chem. 39:273–279.
- Sabbah M, Fontaine F, Grand L, Boukraa M, Efrit ML, Doutheau A, et al. 2012. Synthesis and biological evaluation of new N-acyl-homoserine-lactone analogues, based on triazole and tetrazole scaffolds, acting as LuxR-dependent quorum sensing modulators. Bioorg Med Chem. 20:4727–4736.
- Sayin D, Cakir DT, Gencer N, Arslan O. 2012. Effects of some metals on Paraoxonase activity from shark Scyliorhinus canicula. J Enzyme Inhib Med Chem. 27:595–598.
- Singh RP, Verma RD, Meshri DT, Shreeve JM. 2006. Energetic nitrogen-rich salts and ionic liquids Angew Chem Int Ed Engl. 45:3584–3601.
- Snider BB, Chen J, Patil AD, Freyer AJ. 1996. Synthesis of the tricyclic portions of batzelladines A, B and D. Revision of the stereochemistry of batzelladines A and D Tetrahedron Lett. 37: 6977–6980.
- Supuran CT, Scozzafawa A. 2007. Carbonic anhydrases as targets for medicinal chemistry Bioorg Med Chem. 15:4336–4350.
- Supuran CT. 2011. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem. 3:1165–1180.
- Supuran CT, Scozzafava A. 2000. Carbonic anhydrase inhibitors and their therapeutic potential. Exp Opta Ther Patents. 10:575–579.
- Supuran CT. 2008. CA inhibition mechanism by sulfonamides (a) and anions (b). Nature Rev Drug Discov. 7:168–181.
- Waisser K, Adamec J, Kunes J. Kaustova J. 2004. Antimicrobacterial 1-aryl-5-benzylsulfanyltetrazoles. Chem Pap. 58:214–219.