
Abstract
In this study, in vitro and in vivo effects of some commonly used fungicides, antibiotics, and various chemicals on isolated and purified catalase from Phanerochaete chrysosporium were investigated. The catalase was purified 129.10-fold by using 60% ammonium sulfate and 60% ethanol precipitations, DEAE-cellulose anion exchange and Sephacryl-S-200 gel filtration chromatographies from P. chrysosporium growth in carbon- and nitrogen-limited medium for 12 days. The molecular weight of native purified catalase from P. chrysosporium was found to be 290 ± 10 kDa, and sodium dodecyl sulfate (SDS)–PAGE results indicated that enzyme consisted of four apparently identical subunits, with a molecular weight of 72.5 ± 2.5 kDa. Kinetic characterization studies showed that optimum pH and temperature, Km and Vmax values of the purified catalase which were stable in basic region and at comparatively high temperatures were 7.5, 30°C, 289.86 mM, and 250,000 U/mg, respectively. The activity of purified catalase from P. chrysosporium was significantly inhibited by dithiothreitol (DTT), 2-mercaptoethanol, iodoacetamide, EDTA, and sodium dodecyl sulfate (SDS). It was found that while antibiotics had no inhibitory effects, 45 ppm benomyl, 144 ppm captan, and 47.5 ppm chlorothalonil caused 14.52, 10.82, and 38.86% inhibition of purified catalase, respectively. The inhibition types of these three fungicides were found to be non-competitive inhibition with the Ki values of 1.158, 0.638, and 0.145 mM and IC50 values of 0.573, 0.158, 0.010 mM, respectively. The results of in vivo experiments also showed that benomyl, captan and chlorothalonil caused 15.25, 1.96, and 36.70% activity decreases after 24-h treatments compared to that of the control.
Introduction
Fungicides can be identified as chemical compounds or biological organisms used extensively in agriculture to increase yield and quality of the crops by killing or inhibiting fungi and fungal spores (Haverkate et al. Citation1969). When considered from this point of view, it can be said that fungicides are required in agricultural production, but at the same time the widespread use of them generates a series of environmental problems. In addition, fungicides constitute potential risks to humans exposing to them directly or indirectly through diet (Muri et al. Citation2009). In vivo and in vitro techniques may help to understand metabolic effects of fungicides as well as their kinetic properties. This is especially important in terms of evaluating the alteration effects of fungicides on activities of diverse enzymes. For instance, it was found that deltamethrin and cypermethrin had non-competitive and propoxur had uncompetitive inhibitory effects on glucose-6-phosphate dehydrogenase activity purified from rainbow trout blood with the Ki values of 1.84, 2.63, and 16.55, respectively (Şentürk et al. Citation2009). Besides, in vivo studies of deltamethrin by the same research group showed that carbonic anhydrase activity was significantly inhibited at 24 and 48 h (Ceyhun et al. Citation2010).
Catalase (EC 1.11.1.6) is widely distributed in nature, and it is one of the antioxidant enzymes that protects cells from the toxic effects of hydrogen peroxide by catalyzing its decomposition to water and dioxygen (Aydemir and Kuru Citation2003). Numerous studies have focused on catalase levels during various diseases because of its therapeutic usability (Al-Abrash et al. Citation2000). On the other hand, catalase has many industrial applications instead of its medicinal importance (Kirk et al. Citation2002). Catalase is commonly used with the purpose of the elimination of H2O2 after sterilization process of milk (Akertek and Tarhan Citation1995; Tarhan Citation1995) and textile bleaching (Costa et al. Citation2002), production of gluconic acid and phenylpyruvic acid, and synthesis of dihydroxyacetone and various biosensor applications (Horst et al. Citation2006). Production and purification of catalase from easily obtainable and producible sources with economic techniques are of great importance because of the above-mentioned uses of the enzyme. Until recently, different research groups have purified catalase from a wide variety of sources such as photosynthetic bacterium Rhodospirillum rubrum S1 (Kang et al. Citation2006), dog erythrocytes (Nakamura et al. Citation1998), castor bean (Ota et al. Citation1992), thermophilic bacterium Metallosphaera hakonensis (Ebara and Shigemori Citation2008), soil bacterium Comamonas terrigena N3H (Zámocký et al. Citation2004), pichia pastoris (Shi et al. Citation2007), Thermoascus aurantiacus (Wang et al. Citation1998), and chicken erythrocyte (Aydemir and Kuru Citation2003) with high purification folds.
In this study, we studied in vitro and in vivo alteration effects of some commonly used fungicides on catalase activity instead of inhibitory effects of the cefotaxime, ampicillin, and ceftriaxone antibiotics and various chemicals on purified enzyme from white-rot fungus Phanerochaete chrysosporium. According to our literature survey, there is no other paper concerning the purification of catalase from P. chrysosporium and alteration effects of agriculturally important fungicides on catalase activity.
Materials and methods
Materials
P. chrysosporium strains (DSM-1547 and DSM-6909) were purchased from German Collection of Microorganisms and Cell Culture (DSMZ). Fungicides and antibiotics were kindly provided from Izmir (Turkey) Provincial Directorate for Agriculture, Food Control Laboratory. All other reagents used in this study were analytically pure and purchased from Sigma and Merck companies.
Methods
The effects of the culture components and fermentation period on catalase production of P. chrysosporium strains
To optimize the culture components for the highest catalase production from two P. chrysosporium strains (DSM-1547 and DSM-6909), modified glucose yeast malt peptone agars (GYMP and GYMP + Asparagine), malt extract agar (MEA), potato dextrose agar (PDA) and malt yeast agar (MYA) and two different growth mediums () modified forms those from Tien and Kirk (Citation1988) together with their fermentation period were investigated. The strains were cultivated on agar mediums for 7 days at 28°C, and spores obtained from these agar plates were separately inoculated into the growth mediums. Incubation was carried out for 12 days at 28°C with 150-rpm agitation in the 250-mL Erlenmeyer flasks containing 90 mL growth medium and 10 mL spore suspension. The cells were harvested on 6th, 9th, and 12th days of the fermentation process and analyzed in terms of their catalase activities and total protein concentrations.
Table I. The components of the P. chrysosporium growth mediums.
Preparation of crude catalase extract from P. chrysosporium strains
The cells were washed several times with cold water, harvested by centrifugation (4000 rpm for 5 min), and then resuspended in pre-cold 20 mM potassium phosphate buffer, pH 7.4, in a volume (mL) equal to 3.0 times their weights (g). The homogenization procedure was performed at + 4°C for 3 min at 9000 rpm with 30-s time intervals. Cell debris was removed by centrifuge at 15000 rpm and 15 min at + 4°C. The supernatant was collected and used as catalase extract.
Catalase activity assay
Catalase activity was measured spectrophotometrically at 25°C by following the absorbance decrease at 240 nm caused by the disappearance of H2O2 according to the Aebi method (Aebi Citation1974). One unit of enzyme activity was defined as the amount of enzyme decomposing 1 μ of H2O2 under standard assay conditions.
Total protein assay
Total protein contents were measured with Bradford protein assay (Bradford Citation1976). Bovine serum albumin (BSA) was used as a standard.
Purification procedure of catalase from P. chrysosporium
All purification procedures were carried out at + 4°C. The supernatant obtained with the homogenization procedure as described above was concentrated with 10 kDa ultrafilter, saturated to 60% ammonium sulfate and then stirred for 3 h. The mixture was centrifuged at 15000 rpm for 15 min. The supernatant was discarded, and the pellet was dissolved in the least possible volume of 20 mM potassium phosphate buffer (pH, 7.4). This solution was then saturated to 60% ethanol and stirred for 1 h; the same procedure applied in the ammonium sulfate precipitation was followed. As a consequence, the obtained fraction is referred to as ammonium sulfate (60%)–ethanol (60%) precipitation fraction. For further purification, ammonium sulfate (60%)–ethanol (60%) precipitation fraction was ultrafiltrated (10 kDa) and applied to the DEAE cellulose anion exchange column (10 × 1 cm) which had been equilibrated with 10 mM potassium phosphate buffer (pH, 7.4). After the column was washed with the equilibration buffer, catalase was eluted by replacing the equilibration buffer in a linear gradient manner with an equal volume of 10 mM potassium phosphate buffer (pH, 7.4) containing 1.0 M NaCl. The flow rate was 0.75 mL/min. The catalase-active fractions were pooled (DEAE fraction), concentrated by ultrafiltration (10 kDa), and applied to the Sephacryl-S-200 gel filtration column (75 × 1.5 cm). Equilibration and elution were carried out with the 10 mM potassium phosphate buffer (pH, 7.4) containing 0.15 M NaCl with the flow rate of 0.2 mL/min. Catalase-active fractions were collected (sephacryl-S-200 fraction) and concentrated by ultrafiltration (10 kDa). The resultant preparation was stored as a purified enzyme at − 20°C for later analyses.
Native molecular weight determination of purified catalase from P. chrysosporium
In order to determine the native molecular weight of purified catalase from P. chrysosporium, standard protein mixture instead of sample was applied to the sephacryl-S-200 gel filtration column under same conditions; 2.0 mL standard protein mixture was prepared with 1 mg BSA, 1 mg alcohol dehydrogenase, 1 mg catalase, 1 mg L-glutamate dehydrogenase, and 1 mg apoferritin.
Electrophoretic procedures
SDS–PAGE was performed according to the method of Laemmli (Citation1970) using a vertical slab gel apparatus to control of enzyme purity. The following proteins were used as SDS–PAGE molecular weight standards: carbonic anhydrase (29 kDa), ovalbumin (45 kDa), bovine serum albumin (66 kDa), phosphorylase-b (97.4 kDa), β-galactosidase (116 kDa), and myosin (205 kDa).
Statistical analysis
All of the experiments were repeated thrice, and the values were the mean of three separate experiments. A two-tailed Student's t test was used for significance testing of the difference between means (control vs. test), and p < 0.05 was considered statically significant. GraphPad Prism was used to determine Ki and IC50 values.
Results
Alteration of catalase activity of P. chrysosporium strains depending on medium condition and incubation time
The catalase activities of the extracts prepared from DSM-1547 strain were substantially higher than those prepared from DSM-6909 for all of the mediums investigated. The highest catalase activities from P. chrysosporium (DSM-1547) were obtained in the GYMP + Asparagine-S2, GYMP-S2 and PDA-S2 combinations on 12th day with the values of 260.61, 254.98, and 238.56 U/mg, respectively. For further purification and characterization studies, PDA-S2 combination was chosen because of simple and cheap medium components instead of close specific activity compared to that of the others.
Purification of catalase from P. chrysosporium
Purification of the catalase from P. chrysosporium is summarized in , and the elution profiles of catalase from DEAE cellulose anion exchange and Sephacryl-S-200 gel filtration columns are shown in and , respectively. The ammonium sulfate-ethanol two-step precipitation step is simple and removed ˜90% of proteins with ˜50% retain of the catalase activity. The subsequent two runs of chromatography further purified catalase to nearly homogeneity. The procedure demonstrated approximately 129-fold purification with 20.63% recovery. The purified catalase showed a final specific activity of approximately 30770.31 U/mg of protein. The molecular weight of the native form of the catalase was found to be 290 ± 10 kDa using gel filtration chromatography (), and SDS–PAGE gave a single band at the molecular weight of 72.5 ± 2.5 kDa as the monomer subunit ().
Table II. Purification summary of catalase from P. chrysosporium.
Figure 1. Elution profile of catalase from DEAE-cellulose column; absorbance at 280 nm (-■-) and catalase activity (U/mg) (-▲-).
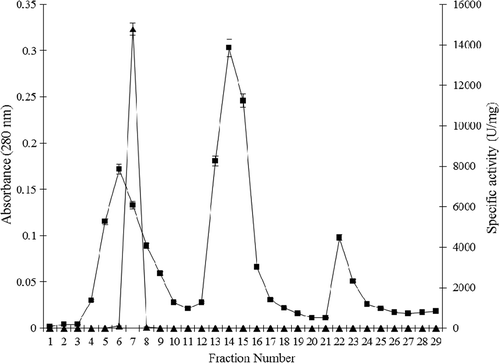
Figure 2. Elution profile of catalase from Sephacryl-S-200 column; (A) absorbance at 280 nm (-■-) and catalase activity (U/mg) (-▲-). (B) Molecular weight calibration curve; protein markers (-●-) and purified P. chrysosporium catalase (-○-).
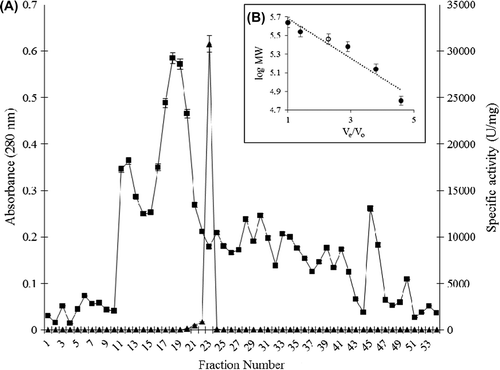
Figure 3. PAGE of P. chrysosporium catalase; lane 1: molecular weight markers, lane 2: crude extract of P. chrysosporium, and lane 3: purified P. chrysosporium catalase.
Characterization of purified catalase from P. chrysosporium
pH-dependent activity and stability changes
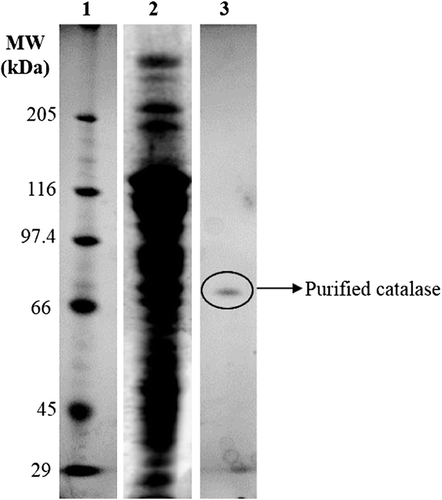
pH-dependent catalase activity variations were investigated under standard assay conditions but using 50 mM potassium phosphate buffer in the pH range of 5.0–8.0 and 20 mM Tris–HCl in pH range 8.0–9.0 instead of 50 mM potassium phosphate buffer, pH 7.0 in the assay. In addition, for the investigation of pH-dependent storage stability, purified catalase was incubated with 50 mM potassium phosphate buffer, pH 6.0, 7.0 and 8.5 for 10 h at 25°C, and the remaining activity values were determined every 2 h during incubation period under the standard activity assay conditions. As seen from , pH activity values showed almost linear increase up to pH 7.5. there was little decline in higher pH values. Results showed that the optimum pH of the P. chrysosporium catalase was 7.5. On the other hand, the stability of the purified catalase was the highest at pH 7.0 (). After incubation period at 25°C for 10 h in pH 7.0 buffer, the activity loss was nearly 17%.
Figure 4. pH activity (-X-) and stability profiles at pH 6.0 (-▲-), 7.0 (-●-), and 8.5 (-■-) depend on time at 25°C for purified P. chrysosporium catalase.
Temperature-dependent activity and stability changes
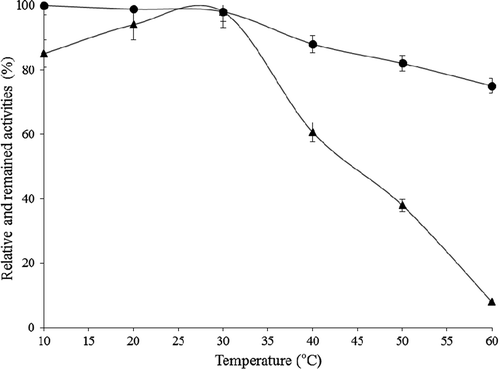
Temperature-dependent catalase activity variations were investigated under standard assay conditions but at 10, 20, 30, 40, 50, and 60°C rather than at 25°C. For the temperature-dependent storage stability assay, purified catalase was incubated between 10 and 80°C in 50 mM potassium phosphate buffer, pH 7.0, for 2 h. The remaining activity values were determined under the standard activity assay conditions. Optimum temperature for the purified catalase was found to be 30°C. Purified enzyme did not have activity at 70 and 80°C in practice (). As shown in , activity of purified catalase from P. chrysosporium was stable at temperatures between 10 and 30°C, but decreased with increasing temperatures and almost lost its activity of 25% at 60°C.
Figure 5. Temperature activity (-▲-) and stability after 2 h-incubation (-●-) profiles for purified P. chrysosporium catalase at pH 7.0.
Kinetic parameters of purified catalase from P. chrysosporium
To determine Km–Vmax values of purified catalase from P. chrysosporium, Lineweaver–Burke graph was drawn by measuring enzyme activity under standard assay conditions but with the different substrate concentrations (5–40 mM H2O2). According to the results, Km and Vmax values were found to be 289.86 mM and 250,000 U/mg for H2O2 at 25°C, respectively.
In vitro and in vivo inhibitory effects of some fungicides on catalase from P. chrysosporium
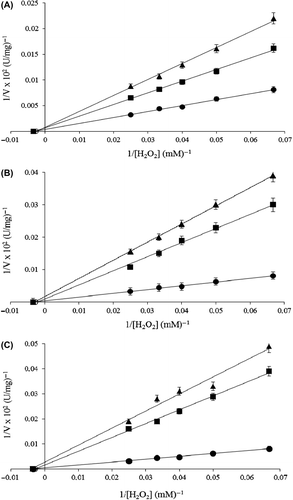
The inhibitory effects of several fungicides including azoxystrobin, benomyl, captan, carbendazim, chlorothalonil, imazalil, metalaxyl, procymidone, and triadimenol on purified catalase were investigated by measuring the remaining enzyme activity of the purified enzyme with the various concentrations of the so-called fungicides at 25°C under standard assay conditions after an incubation period of 1 h. These fungicides were chosen because they were commonly used in agriculture to prevent plants from harmful effects of fungi. It was determined that among the studied fungicides, benomyl, captan and chlorothalonil had inhibitory effects on purified catalase activity at in vitro conditions; that is, 45 ppm benomyl, 144 ppm captan, and 47.5 ppm chlorothalonil caused 14.57, 10.82, and 38.86% (p ˂ 0.05) inhibition compared to the control, respectively. To determine the inhibition types, fungicide solutions were added to the catalase activity assay medium, resulting in different fixed concentrations of fungicides in 1 ml of total reaction volume. In the assay medium, the substrate (H2O2) concentrations were in the range of 5–40 mM. From the Lineweaver–Burk graphs, it was found that the inhibition types of benomyl, captan, and chlorothalonil were compatible with non-competitive inhibition () with the Ki values of 1.158, 0.638, 0.145 mM and IC50 values of 0.573, 0.158, 0.010 mM, respectively.
Figure 6. Lineweaver–Burk graphs for purified P. chrysosporium catalase at five different substrate (H2O2) and two different fungicides concentration at pH 7.0 and 25°C; (A) control (-●-), 0.9 mM (-■-), 2.0 mM (-▲-) for benomyl; (B) control (-●-), 1.44 mM (-■-), 2.88 mM (-▲-) for captan and (C) control (-●-), 0.475 mM (-■-), 2 mM (-▲-) for chlorothalonil.
To determine whether benomyl, captan, and chlorothalonil had in vivo inhibitory effects on P. chrysosporium catalase as well as their in vitro effects, wet P. chrysosporium cells were treated with the so-called fungicides separately and catalase activity alterations were monitored for 24 h. After 24 h of incubation in the presence of the so-called fungicides, no significant changes were observed both in the OD values and in the protein levels of the samples. The time-dependent samples were sufficiently washed and homogenized. The crude extract was used for the evaluation of the activity alterations. The concentrations of the fungicides were the same as the ones adjusted in the in vitro experiments. The results showed that although there was no activity changes up to 10 h, benomyl, captan and chlorothalonil caused 15.25, 1.96 and 36.70% (p ˂ 0.05) activity decreases, respectively, after 24-h treatments compared to the control. This situation showed that the findings originated from in vivo inhibitory effects of the fungicides.
In vitro inhibitory effects of some chemicals and antibiotics on purified catalase from P. chrysosporium
To investigate and compare the response of purified catalase from P.chrysosporium to the several chemicals whose inhibitory effects have been studied on catalases purified from different sources, the purified enzyme was incubated with various concentrations of dithiothreitol (DTT), 2-mercaptoethanol, iodoacetamide, EDTA, urea, and SDS (0.3–30 mM) for 1 h. After incubation period, the remained catalase activity was measured under standard assay conditions and the results were summarized in . According to the findings, DTT, 2-mercaptoethanol, iodoacetamide, and EDTA caused 17, 76 (p < 0.05), 88 (p < 0.05), and 16% inhibition at 5 mM, respectively. On the other hand, the activity could not be observed at 15 and 10 mM of 2-mercaptoethanol and iodoacetamide, respectively. As seen from , urea did not influence the catalase activity even at 30 mM. In the case of sodium dodecyl sulfate (SDS), it was found that 16% (p ˂ 0.05) of catalase activity was protected after treatment with 30 mM anionic surfactant ().
Table III. The in vitro inhibitory effects of some chemicals on purified P. chrysosporium catalase after incubation for 1 h.
The studied antibiotics, cefotaxime, ampicillin, and ceftriaxone had no inhibitory effects on purified catalase activity up to 1000 ppm.
Discussion
Application of fungicides to soil to control plant diseases has become a common practice for crop production in many parts of the world. Understanding the possible impacts of fungicides on the beneficial activities of microorganisms is important to assess the hazards associated with fungicides used in agriculture. Recently, it has been known that fungicides show their negative effects on microorganisms by binding on DNA topoisomerases, inhibiting the synthesis of amino acids and nucleic acids, altering the membrane integrity, affecting signal transduction which involves the function of certain proteins, and inhibiting microbial respiration (CitationYang et al. 2011). However, action types of fungicide have never been well classified and the side effects of these important chemicals are not fully understood. In this paper, we primarily aimed to investigate the in vitro and in vivo effects of some widely used fungicides on catalase. As it is well known, catalase is one of the enzymatic antioxidants and has a significant role in the protection of the cells from oxidative stress. Therefore, it is important to determine the effects of fungicides on catalase in terms of elucidating the role of oxidative stress in killing mechanisms of the so-called fungicides.
In the presented study, easily producible white-rot fungus P. chrysosporium was used as the source of catalase. Since the agar and growth mediums used for the cultivation of microorganisms influence the beginning levels of target enzymes, various medium environments in terms of concentrations of carbon and nitrogen sources and mineral contents were investigated. Among the investigated mediums, nitrogen- and carbon-limited PDA-S2 with the specific catalase activity of 238.56 U/mg was chosen for the further studies. From the results, it can be interpreted as that the stress based on nutrient limitation triggered the catalase levels of P. chrysosporium. In addition, the wet biomass was greater in mineral-rich S2 medium compared to that of other growth medium. When initial catalase-specific activity of P. chrysosporium extract was compared with other sources, it can be absolutely stated that this fungus is one of the powerful sources for catalase instead of its well-known ligninolytic enzyme production property.
Enzyme was purified 129.10-fold using 60% ammonium sulfate and 60% ethanol precipitations, DEAE-cellulose anion-exchange and sephacryl-s-200 gel filtration chromatographies. The results of gel filtration chromatography and SDS-PAGE showed that the native molecular weight of purified catalase was 290 ± 10 kDa with four apparently identical subunits of 72.5 ± 2.5 kDa. It is known from other studies that the molecular weight of catalases isolated and purified from fungi are higher than the molecular weight of the ones from bacteria. Wang et al. (Citation1998) found the molecular weight of Thermoascus aurantiacus catalase to be 330 kDa, and Vainshtein et al. (Citation1986) determined the molecular weight of Penicillium vitale catalase to be 290 kDa. The optimum pH and temperature of the purified catalase from P. chrysosporium were found to be 7.5 and 30°C, respectively, and these results show similarities with catalases purified from other species such as Penicillium piceum (Eremin et al. Citation2004) and Bacillus sp. (CitationOgawa et al. 2004) with their optimum temperature of 30°C, and Escherichia coli (Claiborne and Fridovich Citation1979) with their optimum pH of 7.5. Km values of catalases for the substrate H2O2 change with the range of 0.025–1722 mM depending on the sources used for the purification of enzyme. We found Km value to be 289.86 mM and observed that this result was close to the Km values of the catalases obtained from Serratia marcescens and Bacteroides fragilis in particular (Switala and Loewen Citation2002). Inhibitory effects of some chemicals, DTT, 2-mercaptoethanol, iodoacetamide, EDTA, urea and SDS, on purified catalase from P. chrysosporium were investigated with the purpose of estimating the amino acids having significant impacts on the active center and activity of the enzyme. It was found that among the studied chemicals, DTT, 2-mercaptoethanol, iodoacetamide, and SDS inhibited activity in a considerable extent. According to these results, it may be said that methionine, cysteine, histidine, serine, and threonine residues are dominant residues in the active center of the enzyme. The obtained results are in accordance with the early inhibition studies except urea. From other characterization studies, it is well known that urea treatment causes a dissociation of the tetrameric native enzyme by the loss of catalytic function (Öztürk Ürek and Tarhan Citation2001). In contrast, we did not observed inhibition with urea treatment up to 30 mM.
In this research, we examined the inhibitory effects of the fungicides such as azoxystrobin, benomyl, captan, carbendazim, chlorothalonil, imazalil, metalaxyl, procymidone, and triadimenol on catalase activity. It was observed that benomyl, captan and chlorothalonil had both in vitro and in vivo inhibitory effects at the concentrations of 45, 144, and 47.5 ppm, respectively. Benomyl is a benzimidazole fungicide widely used on a variety of food crops. It is known from other studies that benomyl can interact with thiol groups and act as a potent glutathione-depleting agent. Further, benomyl is known for its ability to induce structural changes in the chromosomes and hepatocyte microtubule cytoskeleton, inhibit protein synthesis and lipid peroxidation in mammals (Min and Kang Citation2008). Since the results of the in vitro chemical inhibition experiments indicated that cysteine could be one of the important amino acids for the activity, it can be thought that benomyl showed its negative impact on activity over the thiol groups of this amino acid. Captan belongs to the phthalimide class of fungicides and the knowledge about its action mechanism is so limited. In our study, we found that 144 ppm captan caused 10.82 and 1.96% activity decreases in vitro and in vivo, respectively. The low inhibition percent for in vivo may result from the diffusion difficulty from membranes into the cell. In addition, the chlorine atoms of captan structure shows that the inhibitory effect of captan may originate from the interactions of chlorine atoms with amino groups. The fungicide chlorothalonil is considered to be non-selective and used commonly to control a broad spectrum of plant diseases. There is a lack of understanding of the fungi killing mechanism of chlorothalonil. We found that chlorothalonil significantly inhibited catalase activity about 35% both in vitro and in vivo. We also found that the inhibition types of all these three fungicides were non-competitive inhibition with the Ki values of 0.145, 0.638, 1.158 mM and IC50 values of 0.010, 0.158, 0.573 mM for chlorothalonil, captan, and benomyl, respectively. So we can conclude that chlorothalonil has most potent inhibitory effect on catalase from P. chrysosporium among the fungicides investigated. The results show that chlorothalonil generates oxidative stress condition in the cell by inhibiting one of the important antioxidant enzyme activities and that this stress condition may be among the significant negative impacts of the so-called fungicide.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Aebi H. 1974. Catalase. Methods of Enzymatic Analysis. New York: Academic Press.
- Akertek E, Tarhan L. 1995. Characterization of immobilized catalase and their application in pasteurization of milk with H2O2. Appl Biochem Biotechnol. 50:291–303.
- Al-Abrash AS, Al-Quobaili FA, Al-Akhras GN. 2000. Catalase evaluation in different human diseases associated with oxidative stress. Saudi Med J. 21:26–30.
- Aydemir T, Kuru K. 2003. Purification and partial characterization of catalase from chicken erythrocytes and the effect of various inhibitors on enzyme activity. Turk J Chem27:85–97.
- Bradford MM. 1976. A rapid and sensitive for the quantitation of microgram quantitites of protein utilizing the principle of protein-dye binding. Anal Biochem. 72:248–254.
- Ceyhun SB, Şentürk M, Erdoğan O, Küfrevioğlu Öİ. 2010. In vitro and in vivo effects of some pesticides on carbonic anhydrase enzyme from rainbow trout (Oncorhynchus mykiss) gills. Pestic Biochem Physiol. 97:177–181.
- Claiborne A, Fridovich I. 1979. Purification of the o-dianisidine peroxidase from Escherichia coli B. Physicochemical characterization and analysis of its dual catalatic and peroxidatic activities. J Biol Chem. 254:45–52.
- Costa SA, Tzanov T, Carneiro F, Gübitz GB, Cavaco-Paulo A. 2002. Recycling of textile bleaching effluents for dyeing using immobilized catalase. Biotechnol Lett. 24:173–176.
- Ebara S, Shigemori Y. 2008. Alkali-tolerant high-activity catalase from a thermophilic bacterium and its overexpression in Escherichia coli. Protein Expr Purif. 57:255–260.
- Eremin AN, Makarenko MV, Drozhdeniuk AP, Moroz IV, Mikhaǐlova RV. 2004. Precipitated, coprecipitated, and carbon-mineral sorbents for isolation of extracellular catalase from Penicillium piceum F-648. Prikl Biokhim Mikrobiol. 40:05–12.
- Haverkate F, Tempel A, Den Held AJ. 1969. Interaction of 2,4,5-trichlorophenyl sulphonyl methyl thiocyanate with fungal spores. Neth J Plant Pathol. 75:308–315.
- Horst F, Rueda EH, Ferreira ML. 2006. Activity of magnetite- immobilized catalase in hydrogen peroxide decomposition. Enzyme Microb Technol. 38:1005–1012.
- Kang YS, Lee DH, Yoon BJ, Oh DC. 2006. Purification and characterization of a catalase from photosynthetic bacterium Rhodospirillum rubrum S1 grown under anaerobic conditions. J Microbiol. 44:185–191.
- Kirk O, Borchert TV, Fuglsang CC. 2002. Industrial enzyme applications. Curr Opin Biotechnol. 13:345–351.
- Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227:680–683.
- Min EY, Kang JC. 2008. Effect of waterborne benomyl on the hematological and antioxidant parameters of the Nile tilapia, Oreochromis niloticus. Pestic Biochem Physiol. 92:138–143.
- Muri SD, van der Voet H, Boon PE, van Klaveren JD, Brüschweiler BJ. 2009. Comparison of human health risks resulting from exposure to fungicides and mycotoxins via food. Food Chem Toxicol. 12:63–74.
- Nakamura K, Watanabe M, Sawai-Tanimoto S, Ikeda T. 1998. A low catalase activity in dog erythrocytes is due to a very low content of catalase protein despite having a normal specific activity. Int J Biochem Cell Biol. 30:823–831.
- Ogawa J, Sulistyaningdyah WT, Li QS, Tanaka H, Xie SX, Kano K, et al. (2004). Two extracellular proteins with alkaline peroxidase activity, a novel cytochrome c and a catalase-peroxidase, from Bacillus sp. No.13. Biochim Biophys Acta. 1699:65–75.
- Ota Y, Ario T, Hayashi K, Nakagawa T, Hattori T, Maeshima M, Asahi T. 1992. Tissue-Specific Isoforms of Catalase Subunits in Castor Bean Seedlings. Plant Cell Physiol. 33:225–232.
- Öztürk Ürek R, Tarhan L. 2001. Purification and characterization of superoxide dismutase from chicken liver. Comp Biochem Physiol B. 128:205–212.
- Shi XL, Feng MQ, Shi J, Shi ZH, Zhong J, Zhou P. 2007. High-level expression and purification of recombinant humancatalase in Pichia pastoris. Protein Expr Purif. 54:24–29.
- Switala J, Loewen PC. 2002. Diversity of properties among catalases. Arch Biochem Biophys. 401:45–54.
- Şentürk M, Ceyhun SB, Erdoğan O, Küfrevioğlu Öİ. 2009. In vitro and in vivo effects of some pesticides on glucose-6-phosphate dehydrogenase enzyme activity from rainbow trout (Oncorhynchus mykiss) erythrocytes. Pestic Biochem Physiol. 95:95–99.
- Tarhan L. 1995. Use of immobilized catalase to remove H2O2 used in the sterilization of milk. Process Biochem. 70:623–628.
- Tien M, Kirk TK. 1988. Lignin peroxidase of Phanerochaete chrysosporium. Methods Enzymol. 161:238–249.
- Vainshtein BK, Melik-Adamyan WR, Barynin VV, Vagin AA, Grebenko AI, Borisov VV. 1986. Three-dimensional structure of catalase from Penicillium vitale at 2.0 Å resolution. J Mol Biol. 188:49–61.
- Wang H, Tokusige Y, Shinoyama H, Fujii T, Urakami T. 1998. Purification and characterization of a thermostable catalase from culture broth of Thermoascus aurantiacus. J Ferment Bioeng. 85: 169–173.
- Yang C, Hamel C, Vujanovic V, Gan Y. (2011)Fungicide: modes of action and possible impact on nontarget microorganisms, ISRN Ecology, doi:10.5402/2011/130289.
- Zámocký M, Godočíková J, Gašperík J, Koller F, Polek B. 2004. Expression, purification, and sequence analysis of catalase-1 from the soil bacterium Comamonas terrigena N3H. Protein Expr Purif. 36:115–123.