
Abstract
Induced pluripotent stem (iPS) cells are generated from differentiated adult somatic cells by reprogramming them. Unlimited self-renewal, and the potential to differentiate into any cell type, make iPS cells very promising candidates for basic and clinical research. Furthermore, iPS cells can be genetically manipulated for use as therapeutic tools. DNA can be introduced into iPS cells, using lentiviral vectors, which represent a helpful choice for efficient transduction and stable integration of transgenes. In this study, we compare two methods of lentiviral transduction of iPS cells, namely, the suspension method and the hanging drop method. In contrast to the conventional suspension method, in the hanging drop method, embryoid body (EB) formation and transduction occur concurrently. The iPS cells were cultured to form EBs, and then transduced with lentiviruses, using the conventional suspension method and the hanging drop method, to express miR-128 and green fluorescent protein (GFP). The number of transduced cells were assessed by fluorescent microscopy and flow cytometry. MTT assay and real-time PCR were performed to determine the cell viability and transgene expression, respectively. Morphologically, GFP+ cells were more detectable in the hanging drop method, and this finding was quantified by flow cytometric analysis. According to the results of the MTT assay, cell viability was considerably higher in the hanging drop method, and real-time PCR represented a higher relative expression of miR-128 in the iPS cells introduced with lentiviruses in drops. Altogether, it seems that lentiviral transduction of challenging iPS cells using the hanging drop method offers a suitable and sufficient strategy in their gene transfer, with less toxicity than the conventional suspension method.
Introduction
Human induced pluripotent stem (iPS) cells are generated from differentiated adult somatic cells, by reprogramming human somatic cells into iPS cells via ectopic expression of defined pluripotency-associated transcriptional factors, including Oct4, Sox2, Klf4, c-Myc, Lin28, and Nanog (CitationCitationOkita et al. 2007, CitationCitationYu et al. 2007, CitationCitationPark et al. 2007). They are unique in their ability to continuously self-renew, while retaining a normal karyotype, and the capacity to differentiate into all of the cell types (CitationCitationYu et al. 2007). Unlimited self-renewal, proliferation abilities, pluripotent gene expression, teratoma formation, and the potential to differentiate into any cell type, give iPS cells the potential to have far-reaching applications in many different aspects of basic and clinical research studies. They hold great promise for use as a unique tool in gene therapy, drug screening, and as an ex vivo source for cellular transplantation in regenerative medicine therapies (CitationCitationOkita et al. 2007, CitationCitationYu et al. 2007, CitationCitationLiao et al. 2009, CitationCitationTakahashi and Yamanaka 2006). They are also promising resources that can be easily genetically manipulated to generate disease-specific iPS cells for pathophysiological studies, and to produce transgenic breeding of chimeric, knock-out, and genetically modified domestic animals (CitationCitationOkita et al. 2007, CitationCitationYu et al. 2007, CitationCitationLiao et al. 2009). However, in order to use iPS derivatives for clinical applications, there is a need to optimize iPS cell-culture expansion, which is a labor-intensive procedure requiring the enrichment of the pluripotent cells from a heterogeneous population capable of spontaneous differentiation, as well as the optimization of differentiation protocols (CitationCitationYu et al. 2007). Furthermore, efficient genetic manipulation is one of the critical steps needed to establish human iPS cells. Stable gene transfer to these stem cells therefore has great potential to achieve appropriate advantages for therapeutic effects and basic research studies (CitationCitationNaldini et al. 1996).
DNA can be introduced into iPS cells using viral and non-viral methods, yielding both transient and stable transduction. The majority of successful genetic modifications of iPS cells have been done with lentiviral vectors, which represent a helpful choice for efficient transduction and stable integration of transgenes into iPS cells (CitationCitationNaldini et al. 1996). Lentiviral vectors are more advantageous than other members of the retrovirus family, in several ways. The most important is their potential to integrate into the cellular genome to generate stably, expressing cell lines and stable transgene expression at high efficiencies in a variety of cell types in vivo and in vitro (CitationCitationBlömer et al. 1997, CitationCitationKafri et al. 1997, CitationCitationNaldini et al. 1996). Second is their ability to promote efficient transduction of dividing and non-dividing cells, in contrast to most retroviral vectors, which can infect primary cells only (CitationCitationMa et al. 2003). Third, ‘transgene silencing’ was not observed in transduction by lentiviral vectors, in contrast to almost all the other retroviruses (CitationCitationHamaguchi et al. 2000). Transduction of iPS cells by lentiviral vectors facilitates transgene expression, to retain more than several passages all through the prolonged cultivation and differentiation, both in vitro and in vivo (CitationCitationPfeifer et al. 2002).
In this study, we compare two methods of lentiviral transduction of genetic elements into iPS cells, and discuss their differences.
Lentiviral transduction of iPS cells in suspension
iPS cells are clusters containing about 1000–2000 single cells, resulting in a diameter of 100–400 nm. Hence, only a small number of cells in the 2–3 marginal cell layers can be transfected with a transgene using lentiviral delivery systems, which leads to a low transfection efficiency (CitationCitationPfeifer et al. 2002).
Lentiviral transduction of iPS cells in hanging drops
The hanging drop – an ancient culture technique – has been employed to crystallize proteins (CitationCitationYoshizaki et al. 2004), to achieve embryoid body (EB) formation by stem cells (CitationCitationTeramoto et al. 2005), to designate the size-controlled islet spheroids (CitationCitationKim et al. 2013), to evaluate tumor invasion (CitationCitationTimmins et al. 2004), and for use in stem cell differentiation and cytotoxicity studies (CitationCitationBanerjee and Bhonde 2006). The technique enabled the first culturing of adult mammalian tissues and organs outside the body (CitationCitationCarrel and Burrows 1910). Cells are attached to a coverslip and bathed in a drop of partially coagulated serum or lymph (CitationCitationBanerjee and Bhonde 2006); the EBs are first cultured in “hanging drops” and then plated in media. Hanging drop cultivation depends on the force of microgravity in the droplet that enhances cell-cell interactions. In this method, colonies of iPS cells were dispersed into little particles, each containing approximately 150 cells, which may improve the efficiency of gene transduction due to the increased total surface of iPS cells (CitationCitationBanerjee and Bhonde 2006).
Therefore, the aim of our study was to explore the suitability of using the hanging drop method for stable lentiviral transduction, and to compare lentiviral transduction of iPS cells in the suspension and hanging drop methods.
Materials and methods
Cell culture
The iPS cells were cultured on previously mitomycin-inactivated SNL cells, in a 60mm culture dish coated with gelatin (0.1%). They were maintained in DMEM F12 supplemented with 15% ES (v/v) (Gibco), 1% nonessential amino acids (Sigma), 4 ng/mlb FGF (Peprotech), and 1mM L-glutamine (Gibco), and passaged every fifth day, with a daily change of media.
Lentivirus production and titration
To produce lentivirus particles, 21 μg of the miRNA-harboring plasmid Lenti-miR128 and the backbone were co-transfected to the Hek293 cells with 10.5 μg of the envelope plasmid pMD2G, and 21 μg of the packaging plasmid psPAX2, using calcium phosphate. After replacing the 16-h culture medium with fresh DMEM supplemented with 10% FBS, the supernatant containing viral particles was collected twice, with a 24-h interval. The viral supernatant was then filtered through 0.45 μm membranes and mixed overnight with 5% PEG-8000 and 0.15 M NaCl, with moderate shaking. The next day, the viruses were centrifuged at 4000 g and suspended in the EB medium. The titers of GFP-expressing lentiviral vector (TU/ml) were determined by infection of HEK 293T cells with serial dilutions and FACS analysis for each dilution, to determine transducing units (TU)/ml.
Lentiviral transduction of iPS cells in suspension
The iPS colonies were dissociated from the feeder layer by treating with collagenase IV1mg/ml (Gibco) for 20 min, 4 to 7 colonies were transferred to each well of the 6-well non-adherent petri dish; EBs formed after 2 days in iPS medium, except that bFGF was lacking for this step. The virus particles with the multiplicity of infection (MOI) of 30 were added to the wells, either concentrated or directly from the HEK-transfected viral supernatant, and incubated with 1 μg/ml polybrene for 3 h. After that, the viral supernatant was replaced with a new portion and incubated overnight with the cells. The next day, the viral supernatant was removed, and EBs attached to 60 mm petri dishes. Then, 1 μg/ml puromycin was added to the cells for antibiotic selection. The cells were analyzed after 10 days of culturing.
Lentiviral transduction of iPS cells in hanging drops
The iPS colonies were segmented to little particles, each containing nearly 150 cells, by treating with 0.05% trypsin for 5 min. Then, 25 μL drops, each containing approximately 150 cells and concentrated virus particles of pLenti miR128 GFP and the backbone (ABM) suspended in EB medium, were placed on the inside of a petri dish lid. The virus particles with the MOI of 30 were added to the iPS cells, and allowed to be transduced while forming EBs in the hanging drops for 2 days. The EBs were plated in 6-well non-adherent petri dishes, and cultured for 2 more days in suspension, to become larger. Subsequently, EBs were attached to 60 mm petri dishes in DMEMF12 supplemented with 10% FBS, and were analyzed after 10 days.
Flow cytometry
Single-cell suspensions of iPS cells, prepared by trypsinizing, were fixed with methanol at 4°C for 20 min. After labeling the cells with the antibodies, we detected them with the FACScan flow cytometer (Becton Dickinson, San Diego, CA, USA). At least 20,000 events were recorded for each sample, and then the results were analyzed using the FlowJo™ software.
Real-time RT-PCR
The reverse transcriptase reactions containing 1μg of RNA samples, including total RNA, were purified by the QIAZOL kit, 2pM stem–loop RT primer for miR128 (), 1x RT buffer, 0.25mM each of dNTPs, 1U/ml reverse transcriptase (Vivantis, Cat No: RTPL12). The reactions were incubated in a Bio-Rad thermocycler for 10 min at 25°C, 15 min at 37°C, 60 min at 42°C, and 10 min at 75°C, then held at 4°C. Relative miRNA expression levels were normalized to SNORD47 as an internal control. The RT products, reverse and forward primers (), were used for real-time PCR at 95°C for 10 min, followed by 35 cycles at 95°C for 15 s, and 60°C for 1 min, using the Takara SYBR Premix Ex Taq Master. Gene expression levels were quantified by Rotor-Gene.
Table I. The list of primers for miR128.
MTT assay
The viability assay was performed using the MTT reagent (3-[4, 5-dimethyl-2-thiazolyl]-2, 5 diphenyl tetrazolium bromide) according to the manufacturer's protocol. The iPS colonies were dissociated by 0.05% trypsin. The iPS cells were seeded, in triplicate, into 96-well microplates (at a density of 5000 cells/well) with DMEM F12 containing 15% FBS, and incubated for 24 h in a humidified atmosphere at 37°C and 5% CO2. On days 4 and 10, 50 μL of MTT reagent (2 mg/ml) were added to each well. Following incubation for 4 h at 37°C, the supernatants were removed carefully. Subsequently, 200 μL of DMSO and 25 μL of Sorenson glycine buffer were added, and the plates were incubated at 37°C in the dark for another half an hour to dissolve the formazan crystals. Finally, the absorbencies of wells were read at a wavelength of 570 nm, using an ELISA plate reader.
Results
Cell culture
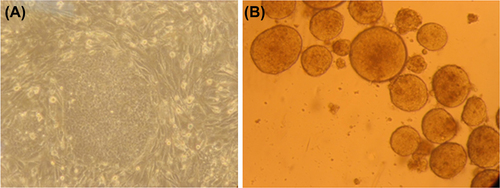
As shown in , the phase contrast image represents the growth of iPS cells in gelatin-coated culture dishes, using previously mitomycin-inactivated SNL cells. The iPS cells exhibited the morphology of an intact colony, with distinctive borders on the SNL feeder cells. After dissociating the iPS cells from the feeder layer by collagenase, EB formation in suspension confirmed the identity of the iPS cells.
Figure 1. (A) The iPS colony with distinctive borders on SNL feeder cells (100 × magnification). (B) EB formation in suspension culture.
Lentiviral transduction of iPS cells
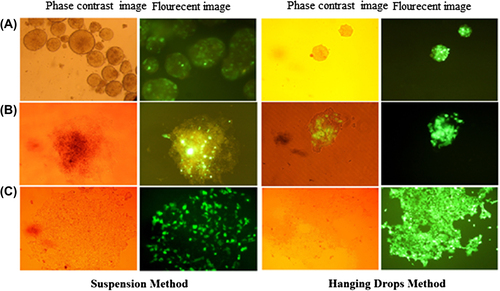
iPS cells were transduced with lentiviruses, to express miR-128 and GFP. On day-2, in the suspension method, just a few of the outer surface cells represent GFP; but in the hanging drop method, a comparison of the phase contrast image reveals a whole shiny green spherical shape, indicating that all cells were transduced. On the 4th day, after the iPS cells were flattened, the few GFP+ cells transduced in suspension became more remarkable; but in the hanging drop method, the iPS cells in the inner center of EBs were also transduced. On day- 10, in the suspension method, some untransduced cells were removed after puromycin selection; in contrast, in the hanging drop technique, there are a large number of transduced cells that are expanded after 10 days. Phase contrast and fluorescent images of the transduced cells on days 2, 4 and 10, are represented in .
Figure 2. Lentiviral transduction of iPS cells. (A) EBs harboring the miR128 gene and GFP after viral transduction on day-2; (B) Transduced iPS cells flattened on 60mm dishes on day-4; (C) iPS cells on day-10 (in the suspension method, the cells were selected by puromycin).
Flow cytometry
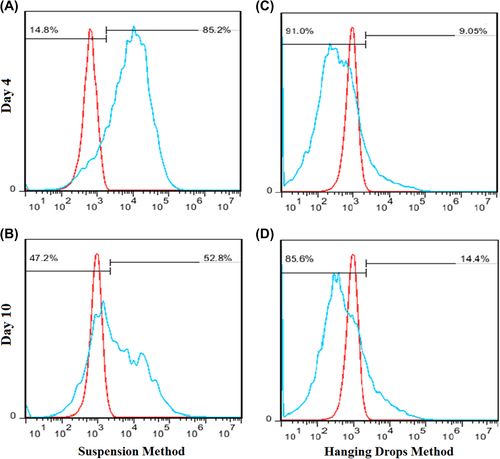
Transduction efficiencies were measured by the assessing GFP expression of transduced iPS cells, on days 4 and 10 (). In the cell suspension method, flow cytometry analysis performed after 4 days, showed 14.8% of GFP-positive cells. Then, puromycin selection was started on day-5, after the iPS cells were attached to 60mm dishes for 4–5 days. Subsequently, the iPS cells were split, and the GFP-positive cells were again assessed by the flow cytometry analysis, which demonstrated 47.2% of GFP-positive cells on day-10. In the hanging drop method, the flow cytometry analysis illustrated 91.0% and 85.6% of GFP-positive cells, respectively, on day-4 and day-10.
Figure 3. Flow cytometric analysis of lentiviral vector-mediated transduction of iPS cells, in comparison with untransduced cells. The red histograms represent untransduced controls, and the blue histograms represent cells transduced with miR-128 and the GFP-harboring vector. The percentages of GFP-positive cells are indicated. (A) The suspension cell method on day-4, before puromycin selection; (B) The suspension cell method on day-10, after puromycin selection; (C and D) The hanging drop method on days 4 and 10, respectively.
Real-time PCR
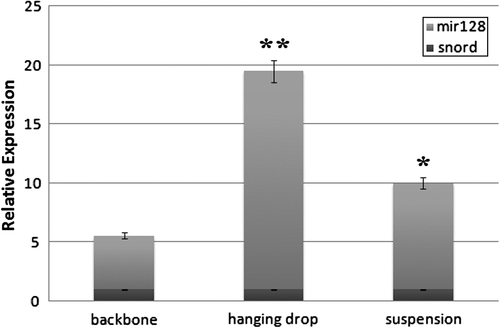
The total RNA of the iPS cells was extracted, cDNA was generated, and real- time PCR was performed on day-10. SNORD47 was evaluated as internal miRNA (control), and all data were normalized to day-1 of culture. As presented in , the transcription of miR-128 was elevated significantly in the hanging drop method, in comparison with iPS cells transduced using the suspension method, and with the backbone plasmid that just has the GFP marker. Although the transcription of miR-128 was increased in the suspension method (2 times more than in backbone transduction), its transcription was more significant in the hanging drop method (4 times more than in backbone transduction).
Figure 4. Relative expression of miR-128. The relative expression of miR-128 is significantly elevated in the hanging drop method (**P < 0.01) than in the suspension method (*P < 0.05), in comparison with the backbone. The error bars represent standard deviation.
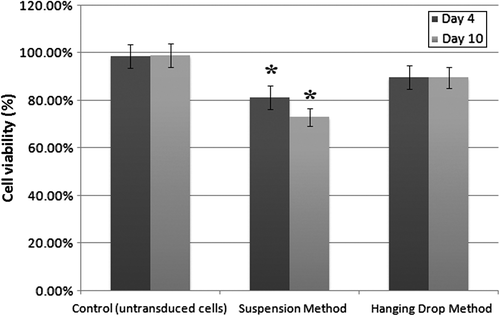
MTT assay
There was a significant difference between the survival of the transduced cells in the suspension method and in the hanging drop method. In the hanging drop method, cell viability was considerably higher than in the conventional suspension method. It seems that in the suspension method, some cells were missed due to the cytotoxic effects of polybrene on iPS cells; and some other cells were lost because of the sensitivity of the iPS cells to puromycin. Cell viability is represented in .
Figure 5. The viability of iPS cells transduced with miR-128 using the suspension method and the hanging drop method, in comparison with untransduced cells. Cell survival in the suspension method is significantly lower than that of untransduced cells (*P < 0.05). The error bar represents standard deviation.
Discussion
Genetically modified iPS cells provide unique tools in basic and clinical research by applying their features of self-renewal and pluripotency in a favorable manner. In contrast to murine iPS cells, human iPS cells are intractable in transgenesis using routine methods. This was also mentioned in previous studies on murine and human ESCs that may be due to their different stage with distinct developmental potentials (CitationCitationBuecker et al. 2010). The efficiency of transgene delivery and the cell survival are two considerably major points in gene modification systems. We choose lentiviral transduction in this study because of its highest efficiency (range: 22.4–25.3%), with more than 95% cell viability, in comparison to electroporation (range: 1.9–2.1%) and lipofection (range: 1.3–1.5%). Electroporation resulted in the lowest cell survival (range: 38–58%), while cell survival with lipofection is more than 90% (CitationCitationCao et al. 2010).
Obtaining a uniform clone of cells harboring the desired gene requires significant refinement and optimized protocols. Usually, transgene-expressing cells were enriched and purified to near homogeneity by cell sorting or antibiotic selection (CitationCitationCao et al. 2010, CitationCitationKita-Matsuo et al. 2009).
To determine a suitable strategy for high-content of lentiviral gene transfer to iPS cells, two methods of viral infection were assessed with iPS clones. In the conventional suspension method, after EBs formed in the suspension, the viral supernatant was added with polybrene to the cells; and antibiotic selection was subsequently carried out for purification. The spherical shape of iPS cells don't allow the inner cells’ contact with viral particles, and thus, transducing them as they aggregate to form spheroid shapes (transduction in hanging drops) may be a solution for this contact shortage. Usually, polybrene, at a concentration of 2–8 μgl/M, has its optimum effect on neutralizing the charge between the cell surface and viral particles. Since this concentration is toxic for iPS cells, polybrene is used at a concentration of 1 μg/Ml for the transduction of iPS cells, which may not have that much efficiency on better transduction; thus the small amount of cells that have been lost after transduction in the suspension method may be due to the toxic effects of polybrene. After viral transduction, puromycin selection removes some of untransduced cells, even though iPS cells are sensitive to high concentrations (more than 1 μgl/Ml) and long exposures to puromycin.
Using the hanging drop method, the single cells of the iPS clone will be transduced with lentiviruses while forming EBs. Because of the high efficiency (near 100%) of this viral transferring strategy, no selection is needed after transduction, which makes colonies with a high percentage of transduced cells advantageous, particularly because untransduced cells will affect the iPS colony niche, before their death by antibiotic.
In the hanging drop method, because the drop can just form in a volume of 25–30 μL, we have limitations in the volume of the viral suspension. We should have a high titer of concentrated viral particles, because iPS cells will be transduced with a high MOI of about 30. On the other hand, the cells should be viable in an adequate portion of EB medium in the drop; so it is crucial to use an appropriate ratio of cell numbers to viral particles. Too many cells will prevent EB formation; and virus particles exceeding the MOI will cause cellular death.
After 10 days, the ectopic over-expression of the miR128 gene is observed in both groups. However, the expression of miR128 in the backbone-transduced iPS cells may be intrinsically due to the existence of miR128 in the iPS cells. The expression analysis of miR128 and GFP suggests efficient and stable integration of these genes to the iPS cell genome, even after 10 days of culturing.
Collectively, it seems that conjugating the viral transduction of iPS cells with the hanging drop technique offers a promising strategy in gene transfer to these cells, in comparison with conventional methods refined by antibiotic selection. This substitute helps to design an appropriate protocol for transducing these rebellious cells, regarding their shape and high MOI. These findings have important implications for the possible future use of gene-modified human iPS cells in drug screening, cell modeling, transplantation, and tissue regeneration applications.
Authors’ contributions
AA and NZ conceived of the study and participated in its design and coordination. MZ, MS, MM, and PH participated in the sequence alignment and drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgments
The authors would like to thank the Neurosciences Research Center, Tabriz University of Medical Sciences, Tabriz, Iran, and the Department of Medical Biotechnology, Faculty of Advanced Medical Sciences, Tabriz University of Medical Sciences.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
References
- Banerjee M, Bhonde RR. 2006. Application of hanging drop technique for stem cell differentiation and cytotoxicity studies. Cytotechnology. 51:1–5.
- Blömer U, Naldini L, Kafri T, Trono D, Verma IM, Gage FH. 1997. Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. J Virol. 71:6641–6649.
- Buecker C, Chen H-H, Polo JM, Daheron L, Bu L, Barakat TS, et al. 2010. A murine ESC-like state facilitates transgenesis and homologous recombination in human pluripotent stem cells. Cell Stem Cell. 6:535–546.
- Cao F, Xie X, Gollan T, Zhao L, Narsinh K, Lee RJ, Wu JC. 2010. Comparison of gene-transfer efficiency in human embryonic stem cells. Mol Imaging Biol. 12:15–24.
- Carrel A, Burrows MT. 1910. Cultivation of adult tissues and organs outside of the body. J AMA. 55:1379–1381.
- Hamaguchi I, Woods N-B, Panagopoulos I, Andersson E, Mikkola H, Fahlman C, et al. 2000. Lentivirus vector gene expression during ES cell-derived hematopoietic development in vitro. J Virol. 74: 10778–10784.
- Kafri T, Blomer U, Peterson DA, Gage FH, Verma IM. 1997. Sustained expression of genes delivered directly into liver and muscle by lentiviral vectors. Nature Genet. 17:314–317.
- Kim H, Alam Z, Hwang J, Hwang Y, Kim M, Yoon S, et al. Eds. 2013. Optimal formation of genetically modified and functional pancreatic islet spheroids by using hanging-drop strategy. Transplantation Proc. 45:605–610.
- Kita-Matsuo H, Barcova M, Prigozhina N, Salomonis N, Wei K, Jacot JG, et al. 2009. Lentiviral vectors and protocols for creation of stable hESC lines for fluorescent tracking and drug resistance selection of cardiomyocytes. PloS One. 4:e5046.
- Liao J, Cui C, Chen S, Ren J, Chen J, Gao Y, et al. 2009. Generation of induced pluripotent stem cell lines from adult rat cells. Cell Stem Cell. 4:11–15.
- Ma Y, Ramezani A, Lewis R, Hawley RG, Thomson JA. 2003. High‐level sustained transgene expression in human embryonic stem cells using lentiviral vectors. Stem Cell. 21:111–117.
- Naldini L, Blömer U, Gage FH, Trono D, Verma IM. 1996. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci. 93:11382–11388.
- Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 272:263–267.
- Okita K, Ichisaka T, Yamanaka S. 2007. Generation of germline-competent induced pluripotent stem cells. Nature. 448:313–317.
- Park I-H, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, et al. 2007. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 451:141–146.
- Pfeifer A, Ikawa M, Dayn Y, Verma IM. 2002. Transgenesis by lentiviral vectors: lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proc Natl Acad Sci. 99:2140–2145.
- Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 126:663–676.
- Teramoto K, Hara Y, Kumashiro Y, Chinzei R, Tanaka Y, Shimizu-Saito K, et al. Eds. 2005. Teratoma formation and hepatocyte differentiation in mouse liver transplanted with mouse embryonic stem cell–derived embryoid bodies. Transplant Proc. 37:285–286.
- Timmins N, Dietmair S, Nielsen L. 2004. Hanging-drop multicellular spheroids as a model of tumour angiogenesis. Angiogenesis. 7: 97–103.
- Yoshizaki I, Nakamura H, Fukuyama S, Komatsu H, Yoda S. 2004. Scientific approach to the optimization of protein crystallization conditions for microgravity experiments. Ann N Y Acad Sci. 1027:28–47.
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. 2007. Induced pluripotent stem cell lines derived from human somatic cells. Science. 318:1917–1920.
- Kamata M, Liang M, Liu S, Nagaoka Y, Chen IS. 2010. Live cell monitoring of hiPSC generation and differentiation using differential expression of endogenous microRNAs. PloS One. 5:e11834.