
Abstract
The use of chimeric antigen receptor (CAR)-modified T cells is a promising approach for cancer immunotherapy. These genetically modified receptors contain an antigen-binding moiety, a hinge region, a transmembrane domain, and an intracellular costimulatory domain resulting in T-cell activation subsequent to antigen binding. Optimal tumor removal through CAR-modified T cells requires suitable target antigen selection, co-stimulatory signaling domain, and the ability of CAR T cells to traffic, persist, and retain antitumor function after adoptive transfer. There are several elements which can improve antitumor function of CAR T cells, including signaling, conditioning chemotherapy and irradiation, tumor burden of the disease, T-cell phenotype, and supplementary cytokine usage.
This review outlines four generations of CAR. The pre-clinical and clinical studies showed that this technique has a great potential for treatment of solid and hematological malignancies. The main purpose of the current review is to focus on the pre-clinical and clinical developments of CAR-based immunotherapy.
Introduction
Immunotherapy can be considered as a promising approach for the treatment of various diseases such as cancer, autoimmune disorder, and allergic and hypersensitivity disorders (CitationBurks et al. 2013, CitationPerosa et al. 2005, CitationTomihara et al. 2013, CitationZou et al. 2007). Cancer immunotherapy is generally classified into two main classes, including active and passive methods. Some interventions which promote the immune system of the patient, for example, by vaccination or adjuvant therapy, actively promote antitumor effector mechanisms to improve cancer elimination. On the other hand, administration of specific monoclonal antibodies (mAbs) against different tumor antigens and the adoptive transfer of genetically-modified specific T cells are currently the most rapidly developing approaches for targeted therapy of cancer (CitationAhmed and Cheung 2013, CitationBrayer and Pinilla-Ibarz 2013, CitationFenoglio et al. 2013, CitationKvistborg et al. 2012, CitationPranchevicius and Vieira 2013, CitationHosseini et al. 2015, CitationKazemi et al. 2015).
CD8+ T lymphocytes play a critical role in tumor eradication (CitationKvistborg et al. 2012). The main routes for T-cell therapy are described as follows: tumor-infiltrating lymphocyte (TIL) therapy, engineered TCR, and chimeric antigen receptor (CAR) (CitationHu et al. 2013, CitationJena et al. 2013, CitationKvistborg et al. 2012).
Recent efforts have been made to develop the recognition specificity of T cells, which redirects T cells to specific tumor-associated antigens (TAA). This technique uses both specific T and B cells by assembling an antigen-binding moiety, collected with an activating immune receptor (CitationHudecek et al. 2010).
The first chimeric TCR was created by joining a single-chain variable fragment (scFv) directly to the C region of either one of the α or β TCR chains, in 1989 (CitationGross et al. 1989).
CARs are proteins expressed on the surface of T and NK cells which contain extracellular binding domains, a hinge region which mediates the linkage of extracellular to transmembrane domains, a transmembrane domain, and an intracellular signaling domain (CitationChu et al. 2014, CitationTill et al. 2012).
The use of CARs is associated with several advantages, such as: CAR protein recognizes TAA in a major histocompatibility complex (MHC)-independent manner and therefore, this feature enables the T cells to overcome the tumor's capacity to escape immune recognition by downregulation of MHC molecules on the cell surface (CitationSadelain et al. 2003, CitationSchreiber et al. 2011, CitationSeliger 2008). The use of CARs can be applicable to a broad range of patients irrespective of MHC type, because tumor targeting is not restricted to MHC molecules. CAR proteins are able to recognize any cell surface antigen including proteins, carbohydrates, and glycolipids. These receptors enable genetically modified T cells to react to a broad range of molecules in comparison with the native TCR (CitationHombach et al. 1997, CitationKailayangiri et al. 2012, CitationKochenderfer et al. 2009). The T-cell engineering for expression of CAR protein can redirect the specificity for the most of the T-cell populations, including CD4, CD8, naive, and memory or T-cell effectors. This is essentially important since naive and antigen-experienced T cells have different functional capabilities which can make them more or less effective for adoptive cell therapy (CitationJameson and Masopust 2009). CAR modification can involve one or more T- cell-activating signals which will be associated with the higher potential of T cells to proliferate, persist, and lyse target cells (CitationPulè et al. 2005). Finally, the ability to produce a large number of tumor-specific T cells in a moderately short period of time makes this technique more attractive to use in the clinical background (CitationHollyman et al. 2009, CitationJune 2007).
CAR-modified T-cell production
To produce CAR-modified T cells, CD4+ and CD8+ T lymphocytes are separated from the patient's blood through leukapheresis. Isolated T cells are then transfected with a viral vector such as lentivirus, containing genes for CAR, directed against the tumor target antigen. The virus binds to the T-cell membrane and fuses with the cell membrane, and reverse transcription, DNA integration, expression of CAR genes, and insertion of CAR protein into the cell membrane of T cells then occur.
In the next step, the genetically modified T cells are cultured with a sufficient amount of IL-2 in the laboratory. To reduce the level of white blood cells, the patient receives lymphodepleting conditioning chemotherapy. Subsequently, autologous CAR T cells are injected, and patients monitor for the treatment response and for the function of modified T cells (CitationJacobson and Ritz 2011, CitationPorter et al. 2011).
CAR specificity
Antigen-targeting through CAR is a critical point in CAR design. Generally, the recognition of a target by CAR includes the usage of scFv that has been assembled from monoclonal antibodies (CitationBridgeman et al. 2010, CitationSadelain et al. 2003). Eradication of tumor by CAR requires recognition of antigen that only expresses on malignant cells and is pivotal for tumor cell survival. Since this antigen is critical for tumor survival, it cannot escape from immune response (CitationGoldberger et al. 2008). Unfortunately, the expression of most antigens is not limited to tumor cells, resulting in undesirable effects on the normal tissue. CAR-modification of T cells can only recognize antigens expressed on the target cell surface (CitationStewart-Jones et al. 2009, CitationTassev et al. 2012). Moreover, tumor targeting by scFv is similar to antigen–antibody interaction. Various factors, including the affinity of binding domains, structure of epitopes, and amount of antigen which expresses on the tumor cells can affect the response of the modified T cells to target antigen (CitationHudecek et al. 2013).
Several studies have addressed various anchored ligands to the extracellular domain which contain scFv against certain molecules like CD19, CD20, HER2, FITC, etc., on tumor cell surface, or some agents like avidin that are designed, at the N-terminal of CAR, to recognize soluble biotin targets which are conjugated with monoclonal antibodies (CitationBrentjens et al. 2011, CitationPinthus et al. 2003, CitationTamada et al. 2012, CitationUrbanska et al. 2012, CitationWang et al. 2004).
Until now, the genetically engineered T cells have successfully been developed against various tumor antigens such as CD19 and CD20 for the treatment of B-cell malignancies (CitationBrentjens et al. 2011, CitationWang et al. 2004), ERRB2 (estrogen-related receptor beta type 2) for the treatment of prostate and breast cancer (CitationPinthus et al. 2003, CitationTeng et al. 2004), Carbonic anhydrase IX (CAIX) for the treatment of renal cell carcinoma (CitationLamers et al. 2006), prostate-specific membrane antigen (PSMA) for the treatment of prostate cancer (CitationGong et al. 1999, CitationMaher et al. 2002), Lewis Y for the treatment of lung cancer (CitationMezzanzanica et al. 1997, CitationWestwood et al. 2005), carcinoembryonic antigen for the treatment of colon cancer (CitationDarcy et al. 1998, CitationHaynes et al. 2002), TAG72 for the treatment of breast, colorectal, pancreatic cancer, etc. (CitationSharifzadeh et al. 2013), folate-binding protein and MUC16 for the treatment of ovarian cancer (CitationChekmasova et al. 2010, CitationHwu et al. 1993), and the diasialoganglioside GD2 for the treatment of neuroblastoma (CitationRossig et al. 2002). Studies have shown various CARs containing receptor polypeptide ligands linked to intracellular domains such as an integrin-binding peptide (anti-avb6), a polypeptide against vascular endothelial growth factor receptor (anti-VEGFR2), heregulin (anti-Her3/4 receptor), or interleukin (IL)-13 mutein (anti-IL13 receptor-a) (CitationKahlon et al. 2004, CitationMuniappan et al. 2000, CitationNiederman et al. 2002, CitationPameijer et al. 2007).
Recently, two general CAR systems have been described which include anti-biotin–CAR and an anti-fluorescein isothiocyanate (anti-FITC)–CAR. In the anti-biotin–CAR system, the extracellular domain of CAR attaches to avidin, which can bind to soluble biotin-conjugated therapeutic molecules (monoclonal antibodies, nucleic acid-based aptamers, or ligands). In other CAR systems that have been developed by Tamada et al., the binding region anchored to an anti-FITC scFv recognized tumor cells via binding to the FITC monoclonal antibody. Patients received these conjugated therapeutic molecules before infusion of CAR T cells, which labeled the tumor target. This technique allows the recognition of various tumor antigens which restrict the capacity of tumors to escape targeting by downregulating a single target antigen. Furthermore, genetically modified T cells can be useful for several types of tumors with only one CAR (CitationTamada et al. 2012, CitationUrbanska et al. 2012). In another experiment, Ahmed et al. introduced a dual-specific CAR T cell—TanCAR—which contains two scFvs associated with a flexible linker on the surface of T cells against HER2 and CD19. This approach could enhance the anti-tumor function of CAR T cells, in vivo (CitationGrada et al. 2013). One disadvantage of the CAR strategies using scFvs derived from murine antibodies is the appearance of both antibody- and cell- mediated responses against these foreign molecules. In order to avoid this adverse effect, we can use the humanized scFv domains or human antibodies to produce CARs (CitationHombach et al. 2000, CitationLamers et al. 1995) ().
Table I. Targets for CAR-modified T cells.
CAR T-cell Signaling
The optimal activation of T cells requires at least two distinct signals including interaction between the TCR–CD3 complexes with peptide presented by MHC molecules (signal 1), and recruitment of CD28 on the T-cell surface with co -stimulatory molecules CD80 (B7.1) or CD86 (B7.2) on the antigen-presenting cells such as dendritic cells (signal 2). Signal 1 and signal 2 are necessary for the optimal activation, effector function, proliferation, and survival of T cells. Delivery of signal 1 without signal 2 leads to the anergy and apoptosis of T cells and consequently leads to their hyporesponsiveness (CitationChen 2004, CitationChen and Flies 2013, CitationCroft 2005, CitationLadygina et al. 2013, CitationNäslund et al. 2013). The first generation of CARs contain a scFv joined to the intracellular signaling domain of CD3ζ (CitationEshhar et al. 1993) (). The CD3ζ-derivedsignal provides the required ‘signal 1’ resulting in T-cell activation, modest IL-2 secretion, target cell lysis, and in vivo antitumor function (CitationEshhar et al. 1993, CitationHaynes et al. 2001, CitationHeuser et al. 2003). The activation of the first-generation CARs with only signal 1 results in T-cell anergy, unfavorable cytokine secretion, weak proliferation, and apoptosis of CAR T cells (CitationGrossi et al. 1992, CitationLenschow et al. 1996). The second-generation CAR T cells are composed of scFv linked to co-stimulatory domains such as CD28 and CD3ζ which were generated to improve CAR activation signals and increase the proliferation and cytokine secretion (). Interaction between CD28 and B7 family molecules such as B7.1 and B7.2, which are located on the surface of target cells, provides a second activation signal that enhances ‘signal 1’ from the TCR–CD3 complex (CitationBretscher 1999, CitationChambers and Allison 1997, CitationSmith-Garvin et al. 2009). Delivery of signal 1 and signal 2 via CAR molecules in the second-generation CARs leads to T-cell proliferation, lysis of target cells, IL-2 secretion, and expression of some proteins such as anti-apoptotic protein Bcl-xL (CitationChambers and Allison 1997). Studies have shown that the second-generation CAR T cells against PSMA are much more effective than the first-generation in IL-2 production and proliferation (CitationMaher et al. 2002).
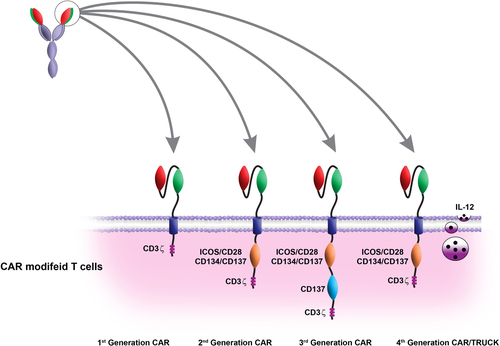
Another study indicated that the treatment of established tumor using the second-generation CAR which contains CD28 increased the eradication efficacy in comparison with the first-generation CAR for the CD19 antigen (CitationBrentjens et al. 2007). The use of CD28-containing second-generation CARs was also associated with their increased persistence (CitationSavoldo et al. 2011). Generally, the enhanced cytokine production will increase the cell proliferation and upregulation of anti-apoptotic proteins such as Bcl-xL in the second- generation CARs containing CD28, in comparison with the first-generation CARs (CitationBeecham et al. 2000, CitationHaynes et al. 2002, CitationHombach et al. 2001a, Citation2001b, CitationKalos et al. 2011, CitationKowolik et al. 2006, CitationVera et al. 2006). Other B7 family members such as inducible co-stimulation (ICOS) and tumor necrosis factor receptor consisting of CD137–4-1BB or CD134–OX-40 can be replaced with CD28-containing second-generation CARs (CitationFinney et al. 2004).
To improve CAR design and enhance the activation of the second-generation CARs, the ‘third-generation’ CARs were developed containing 3 co-stimulatory domains (). The third-generation CARs provide signal 1, signal 2, and a supplementary co-stimulatory signal to boost T-cell activation signals, and include CD28, 4-1BB, and CD3ζ. Several studies have demonstrated the improvement of T-cell survival, cytokine production, and anti-tumor potential for genetically modified T cells that express the third-generation CAR (CitationCarpenito et al. 2009, CitationChang et al. 2007, CitationWang et al. 2007, CitationZhao et al. 2009, CitationZhong et al. 2010). In another study, Zhong et al. (CitationZhong et al. 2010) have reported that the use of PMSA targeting CAR T cells was associated with the improved in vivo T-cell survival, enhanced cytokine production, improved PI3K/AKT pathway activation, enhanced tumor elimination, reduced T-cell apoptosis, and enhanced Bcl-xL expression. In recent studies, scientists have developed T cells redirected for universal cytokine-mediated killing or TRUCKs (fourth-generation CARs) (). This type of CAR is able to secrete a pro-inflammatory cytokine such as IL-12 into the tumor microenvironment (CitationChmielewski et al. 2011, CitationPegram et al. 2012). Interaction between the fourth-generation CARs and their target leads to the activation of T cells and secretion of IL-12 by the CAR T cells in the target tissue. The accumulation of IL-12 is an important strategy for attraction of innate immune cells, mainly NK cells and macrophages, in a local tumor lesion (CitationKerkar et al. 2011, CitationKerkar et al. 2010). IL-12 is a heterodimeric protein which is physiologically secreted by macrophages and dendritic cells in response to intracellular pathogens (CitationTrinchieri et al. 2003). Secretion of IL-12 by the fourth-generation CARs, leads to the infiltration of NK cells and macrophages to the tumor site, which consequently improve tumor eradication (CitationPritchard et al. 2005, CitationZhang et al. 2011). The optimal tumor eradication with CARs requires incorporation of different signaling domains which leads to complete activation of T cells to promote the secretion of certain cytokines.
CAR T-cell Trafficking
In order to achieve effective tumor eradication, genetically modified T cells should traffic to the tumor site(s) where CAR T cells will be able to target tumor cells. In fact, poor T-cell homing to tumor sites is the main reason for the reduced efficiency of adoptive immunotherapy based on CAR-engineered T cells (CitationParente-Pereira et al. 2011). Migration of genetically-modified T cells to the tumor site has been demonstrated through a dual bioluminescent imaging of CAR T cells in a murine tumor model (CitationSantos et al. 2009). Accumulation of CAR T cells at the most site(s) of tumor, and persistence of CAR T cells, are the major principles for effective tumor eradication (CitationParente-Pereira et al. 2011). Studies have shown that genetically-modified T cells which express the second-generation CAR will accumulate in the disease sites (CitationBrentjens et al. 2011, CitationKalos et al. 2011, CitationSavoldo et al. 2011). Trafficking of CAR T cells to several sites including liver, lymph nodes, and bone marrow have been demonstrated by Brentjens et al. through the second-generation CAR against CD19 (CitationBrentjens et al. 2011). In another investigation, Savoldo et al. (CitationSavoldo et al. 2011) has reported the homing of genetically-modified T cells to a skin lesion after adoptive cell therapy in patients with non-Hodgkin's lymphoma (NHL). The second-generation CAR containing CD28 has shown effective trafficking to the tumor site compared to the first-generation CAR against CD19 (CitationSavoldo et al. 2011). Overproduction of several molecules such as integrins and chemokines was associated with T-cell trafficking to tumor sites. Chemokines and their receptors play a critical role in the homing of CAR T cells to tumor sites (CitationDi Stasi et al. 2009). Kershaw et al. (CitationKershaw et al. 2002) have demonstrated that the expression of CXCR2 and CCR4 in CAR-modified T cells will enhance T-cell trafficking to the disease site(s).
Persistence of CAR T cells
Poor survival of the infused T cells limits the efficiency of adoptive T-cell immunotherapy. Genetically modified T cells need to survive for a sufficient time period to achieve complete and successful tumor elimination. Robbins et al. have indicated that the efficacy of anti-cancer response is correlated with the persistence of infused T cells (CitationRobbins et al. 2004). Brocker et al. have also reported poor survival of the first-generation CAR T cells containing CD3ε against lymphoma (CitationBrocker 2000). In another study, Lamers et al. demonstrated poor persistence of the first-generation CAR T cells against carbonic anhydrase IX (CAIX) in metastatic renal cell carcinoma (CitationLamers et al. 2006). In order to overcome the limitations of the first-generation CARs, the second- and third-generation CARs have been developed to increase persistence of genetically modified T cells. Activation of CAR T cells with two or more co-stimulatory domains can enhance the persistence of infused T cells (CitationCarpenito et al. 2009, CitationWang et al. 2007).
Moreover, lymphodepleting chemotherapy can also improve the persistence of CAR T cells through the modification of the host environment prior to the infusion of CAR T cells (CitationMuranski et al. 2006, CitationRosenberg and Dudley 2009). Conditioning chemotherapy can remove suppressive cells such as regulatory T (Treg) cells and myeloid cells and enhance the persistence of CAR T cells.
Conditioning chemotherapy and low-dose irradiation can improve the survival of adoptively transferred T cells through a number of mechanisms, as demonstrated in preclinical models (CitationCuriel et al. 2004, CitationDurrant and Metzger 2010, CitationGattinoni et al. 2005). Brentjens et al. reported their observations for 10 patients with chronic lymphocytic leukemia (CLL) or acute lymphoblastic leukemia (ALL). In the first group, patients had been treated with the second- generation CAR containing CD28 anti-CD19 without conditioning chemotherapy, and showed undetectable persistence. Controversially, observations in the next group of patients that received prior lymphodepleting conditioning chemotherapy (cyclophosphamide), showed that genetically modified T cells can persist for 6 weeks after adoptive transfer in the bone marrow (CitationBrentjens et al. 2011).
A chemotherapy regimen can reduce the patient's disease burden and enhance the persistence of the infused T cells. Correlation between patient's tumor burden and persistence of CAR T cells was demonstrated by Park et al. in patients with metastatic neuroblastoma (CitationPark et al. 2007).
One approach to improve the persistence of genetically modified T cells can be the administration of IL-2. An enhanced persistence of the first-generation CAR anti- CD20 was also observed in patients with indolent NHL who were administered low dosages of subcutaneous IL-2. Moreover, production of some cytokines such as IL-2 and IL-15, as well as the overexpression of their receptors, improved the persistence of CAR T cells (CitationKershaw et al. 2006, CitationTill et al. 2008).
Another approach to enhance in vivo T-cell persistence involves the application of virus-specific T cells which are genetically modified to express CAR. Murphy et al. have demonstrated the increased persistence of CAR-specific influenza virus T cells in mice with breast cancer (CitationMurphy et al. 2007). In another experiment, improved persistence of CAR-specific Epstein Barr virus (EBV) T cells against GD2 was observed in patients with neuroblastoma (CitationPule et al. 2008). Increased persistence of CAR T cells in the circulation for up to 6 weeks was also observed in patients that were treated with EBV-specific T cells engineered with a CAR recognizing the diasialoganglioside GD2, representing an antigen expressed by neuroblastoma cells (CitationLouis et al. 2011).
In conclusion, conditioning chemotherapy, cytokine supplementation, the patient's tumor burden, T-cell phenotype, co-stimulatory signaling, and possibly the use of viral-specific T cells are several factors which are able to improve the persistence of infused T cells.
CAR T cells and immunosuppressive tumor microenvironment
The maintenance of cytolytic function is an important element for successful tumor eradication by CAR-genetically modified T cells subsequent to an adoptive transfer. In addition, several mechanisms may inhibit the function of adoptively transferred CAR T cells. These mechanisms include suppressor cells such as Tregs cells, myeloid-derived suppressor cells (MDSCs), and immunosuppressive molecules/cytokines (e.g., TGF-β) (CitationRabinovich et al. 2007, CitationSchreiber et al. 2011). Moreover, some genetic modifications mentioned above can also help genetically modified T cells to resist immunosuppressive factors in the tumor microenvironment. For instance, the incorporation of co-stimulatory molecules such as CD28 into CARs can assist T cells in being more resistant to Treg cells and TGFβ (CitationKoehler et al. 2007, CitationLee et al. 2011, CitationLoskog et al. 2006). Overexpression of AKT can also increase the resistance of CAR (CitationSun et al. 2010). A promising approach to inhibit the effects of TGFβ is expression of a decoy form of the TGFβ receptor, which can be achieved through genetic strategies in CAR T-cell design (CitationBollard et al. 2002, CitationGorelik and Flavell 2002). Activation of T cells can be suppressed by programmed cell death protein 1 (PD1) which is expressed on the surface of T cells. Interaction between PD1 and their ligands that are expressed in surface of cancerous cells can suppress the activation of CAR T cells in the tumor microenvironment. PD1-blocking by small interfering RNA (siRNA) or mAbs is a promising approach to reduce this form of immunosuppression (CitationBorkner et al. 2010, CitationTopalian et al. 2012). Expression of IL-12 by genetically modified CAR T cells is another strategy to enhance the resistance and may also change the tumor microenvironment. Genetic engineering of T cells to express IL12 can also enhance antitumor function and will improve the capacity to resist the immunosuppression exerted by Treg cells (CitationKerkar et al. 2010, CitationSpear et al. 2012). Secretion of IL-12 by T cells leads to the formation of immunosupportive myeloid cells in tumors (CitationChmielewski et al. 2011, CitationPegram et al. 2012). Genetically modified T cells expressing a chimeric NKG2D were found to improve cytokine secretion, leading to a change in myeloid cells from an immunosuppressive to an immunostimulatory phenotype (CitationKerkar et al. 2011). Lee et al. have demonstrated that conditioning chemotherapy can also reduce the number of Tregs and improve the antitumor function of T cells (CitationLee et al. 2011). Other immunosuppressive factors such as galectins and amino acid metabolites may also suppress the function of CAR T cells, and future approaches that aim to block these factors may generate more effective tumor-specific T cells.
Toxicity of CAR T cells
Unwanted toxicity is a major problem that can limit CAR T-cell-based immunotherapy. Genetically modified T cells have to recognize antigens that only express on the surface of tumors. Morgan et al. treated a colon cancer patient with the third-generation CAR against ERBB2. The patient that received lymphocyte-depleting chemotherapy died 5 days after infusion of CAR T cells. Interaction between genetically modified CAR T cells and ERBB2 led to the release of pro-inflammatory cytokines. Hence, pulmonary toxicity, multi-organ failure, and death of the patient were observed after infusion of T cells (CitationMorgan et al. 2010). In another clinical study, 11 renal-cell carcinoma patients were treated with the first-generation CAR-engineered T cells recognizing the CAIX, representing an antigen expressed by renal cells. Targeting CAIX in bile duct epithelial cells led to serious liver toxicity in 5 patients after the infusion of CAR T cells (CitationLamers et al. 2006, CitationLamers et al. 2011). In another clinical trial, neurotoxicity was reported in 2 of 8 patients treated with genetically modified T cells to express a MAGE-A3-specific CAR. It was concluded that neurotoxicity was the result of cross-reactivity between MAGE-12, which is expressed on some neurons, and MAGE-A3 that is only expressed on tumors (CitationMorgan et al. 2013). These studies demonstrated the need for tumor-targeting antigens that are only expressed on cancer cells. Some important characteristics of the fourth-generation of CAR T cells have been summarized in .
Table II. Different characteristics of the four generations of CAR T cells.
Clinical trials with different generations of CAR T cells
Several studies have evaluated the antitumor effectiveness of the first-generation CAR-modified T cells. No significant results were found in the early clinical trials using the first-generation CAR T cells. Furthermore, there are certain differences between these clinical trials, including the targeted antigen selection, gene transfer method, the technique of ex vivo expansion, injection of exogenous IL-2, cell dosage, and the use of conditioning therapy.
Lamers and colleagues (CitationLamers et al. 2006, CitationLamers et al. 2011) described their experience on 11 patients with metastatic cell renal carcinoma treated with the first-generation anti-CAIX CAR which contained a CD3ζ endodomain with or without subcutaneous IL-2. Liver toxicity was observed after infusion of CAR T cells in 5 out of 8 patients. This side-effect is due to the low-level expression of CAIX on bile duct epithelial cells which are targeted through CAR T cells. Moreover, no liver toxicity was observed in 3 patients who received anti-CAIX mAb (to block CAIX on normal liver tissue) prior to the infusion of CAR T cells. The persistence of infused T cells was limited for all patients, and no objective clinical responses were observed. Importantly, humoral and cellular anti-CAR responses were developed in all patients, which can describe the limited persistence of infused T cells.
Kershaw et al. (CitationKershaw et al. 2006) reported their studies on 14 patients with ovarian cancer treated with CAR-modified T cell anti-α-folate receptor in the presence and absence of subcutaneous IL-2. Infused T cells had limited persistence, and no objective clinical responses were observed.
Park et al. (CitationPark et al. 2007) tried to treat 6 neuroblastoma patients with CAR-modified T cells against L1-cell adhesion molecule (L1-CAM; CD171); however, no objective clinical responses were observed. However, increased T-cell survival was observed in a patient with a low disease burden in comparison with the patients with larger tumor burdens.
Till et al. (CitationTill et al. 2008) treated 7 patients with relapsed or refractory indolent NHL with T-cell targeting CD20. In the first group, no objective clinical responses were observed, and the persistence of infused CAR T cells was also limited. However, patients in the second group who received subcutaneous IL-2, the survival of modified CAR T cells was improved significantly. One patient in the second group demonstrated a limited remission, and one patient showed a reduction in the metabolic activity of tumor.
Moreover, Jensen et al. (CitationJensen et al. 2010) investigated the efficacy of CAR-modified T cells targeting CD19 and CD20 in the treatment of four patients with recurrent NHL. Two patients with B-cell lymphoma were treated with modified T cells expressing a CD20-specific CAR. Treatment of two follicular lymphoma patients with CAR-modified T cells expressing a CD19-specific CAR and low dosage of subcutaneous IL-2 showed the limited persistence of CAR T cells, which was associated with no clinical responses.
The initial clinical trial based on the application of the second-generation CAR-modified T cells developed in 2010, also focused on B-cell malignancy.
Kochenderfer et al. (CitationKochenderfer et al. 2010) described their experience regarding the 1 patient with NHL treated with a CD28-containing second-generation CAR anti-CD19 antigen. The patient received lymphodepleting conditioning chemotherapy that included cyclophosphamide and fludarabine prior to the infusion of CAR T cells and IL-2. A partial remission of the patient's lymphoma was also observed for up to 32 weeks. The persistence of CAR T cells was shown by quantitative polymerase chain reaction (Q-PCR) in the peripheral blood for 27 weeks, and no significant toxicity was found.
Savoldo et al. (CitationSavoldo et al. 2011) tried to treat 6 relapsed or refractory NHL patients with genetically modified anti-CD19CAR T cells. Their patients were treated with a mixture of engineered T cells included CD3ζ-containing first- generation, and CD28-, CD3ζ-containing second-generation CAR anti-CD19, without lymphodepletion. They showed that the second-generation CARs had higher persistence, expansion, and traffic in comparison to the first-generation CARs.
Brentjens and colleagues (CitationBrentjens et al. 2011) treated 9 leukemia patients with CD28-containing second-generation CAR T cells against CD19 antigen. In this study, 8 CLL patients and 1 ALL patient were treated with CAR T cells with and without conditioning chemotherapy. Enhanced persistence of the modified CAR T cells was observed in 6 patients who received lymphodepleting conditioning chemotherapy with cyclophosphamide before T-cell infusion. One patient died 2 days after modified CAR T-cell infusion. Out of 4 patients with bulky CLL, 3 demonstrated a response to treatment with conditioning chemotherapy and cyclophosphamide followed by infusion of modified CAR T cells. The one ALL patient treated with CAR T cells showed a B-cell aplasia. Modified CAR T cells showed efficient traffic to the sites of disease and retained ex vivo antitumor function after adoptive transfer.
Kalos et al. (CitationKalos et al. 2011) described the adoptive transfer of 4-1BB-containing second-generation CAR-modified T cells directed against CD19 antigen in 3 patients with CLL who received conditioning chemotherapy prior to the T-cell infusion. Modified T cells showed traffic to site of the disease and were detected at high levels for up to 6 months post-infusion in peripheral blood and bone marrow. Responses to treatment and tumor lysis syndrome were observed in 3 patients (2 complete remissions and 1 partial remission) which correlated with increased CAR T-cell number and pro-inflammatory cytokine levels.
Kochenderfer et al. (CitationKochenderfer et al. 2012) treated 8 advanced B-cell malignancy patients with second-generation CAR directed against CD19 with low dosage of IL-2. Six treated patients (6 of 7 evaluable) had a clinical remission. Trafficking of modified T cells to bone marrow and complete remission was demonstrated in few patients.
Saar Gill et al. (CitationGill et al. 2014) have introduced CD123, a transmembrane chain of IL-3R, as a worthy target for AML-directed CAR T-cell therapy. As it is over-expressed in most of the AML cells in vivo, eradication of primary AML with the help of human CD123-CD28-CD3ζ T cells (CAR T123) in immunodeficient mice suggested that CAR T123 is one of the best choices for AML therapy, in this study.
Kochenderfer and colleagues (CitationKochenderfer et al. 2014) evaluated the efficacy of CD28- and CD3ζ-containing second-generation CAR-engineered T cell anti-CD19 in 15 patients with advanced B-cell malignancies following conditioning chemotherapy. In this study, 9 patients with diffuse large B-cell lymphoma (DLBCL) and 6 patients with indolent B-cell malignancies were treated. Of the 7 patients with DLBCL, 4 achieved complete remissions (CRs), 2 achieved partial remissions (PRs), and 1 had stable disease (SD) after T-cell therapy. Of the 6 evaluable patients with indolent B-cell malignancies, 4 achieved CR and 2 achieved PR. One patient died after infusion of CAR T cells, and one patient was withdrawn to follow-up. Several manifestations including hypotension, fever, delirium, and other neurologic toxicities were observed in some patients after treatment; these toxicities were eliminated within 3 weeks after T-cell infusion.
Katz et al. (CitationKatz et al. 2015) treated 6 patients with liver metastases (LM) with second-generation of CAR-T cell (CD28 and CD3ζ) anti-CEA with and without IL-2 injection. Of the 6 evaluable patients, 5 died due to disease progression. One patient had stable disease at 23 months after treatment. Elevated serum IFN-γ levels and improved CEA responses were demonstrated in second-generation groups who received systemic IL-2.
Morgan et al. treated 1 colon cancer patient with CD28- and 4-1BB-containing third-generation CAR targeting ERBB2 with lymphocyte-depleting chemotherapy including cyclophosphamide and fludarabine (CitationMorgan et al. 2013). The patient died 5 days after the infusion of modified T cells. Tumor-targeting T cells recognized ERBB2 on the lung epithelium, which ultimately leads to death of the patient.
Till et al. reported their experience regarding 4 patients with mantle cell or follicular non-Hodgkin's lymphoma. In their investigation, patients were treated with CD28- and 4-1BB-containing third-generation CAR targeting CD20. Two patients remained progression-free for 12 and 24 months. An objective partial remission was found in the third patient 12 months after infusions (CitationTill et al. 2012).
Recently, fourth-generation CARs have been introduced. Some studies have revealed the anti-tumor function of CAR-modified T cells in vitro and in several animal models.
Koneru et al. (CitationKoneru et al. 2015) have designed the fourth-generation of CAR (4H11-28z/IL-12) against MUC-16ecto antigen that overexpresses on the surface of ovarian tumor cells and is the residue of MUC-16 after CA-125 cleavage. This study indicates that co-expression of IL-12 and 4H11-28z CAR T cells’ increased proliferation and IFN-γ secretion when compared with 4H11-28z CAR T cells in the SKOV3 cell line. The antitumor efficacy of IL-12-secreting 4H11-28z CAR T cells is also examined in SCID-Beige mice with human ovarian cancer xenografts, through enhanced survival, prolonged persistence of T cells, higher systemic IFN-γ level, and in turn, complete eradication of tumors.
Although these experiments have shown the potential of T cells engineered with the fourth-generation CARs for the treatment of patients, further investigations in various clinical studies (see ) (CitationChmielewski et al. 2011, CitationKerkar et al. 2011, CitationKerkar et al. 2010, CitationPegram et al. 2012) are needed.
Table III. Clinical trials with different types of CAR-modified T cells.
Conclusion& Future perspective
CD4+and CD8+ T lymphocytes play an important role in the treatment of cancer. CD8+ CTLs are able to eradicate tumor cells efficiently, whereas CD4+ T lymphocytes can improve the capacity of antigen-presenting cells (APC) and antitumor function of CTLs through the enhanced secretion of some cytokines. CAR-based immunotherapy was developed for use both in cellular and humoral arms of the immune system for effective tumor eradication. The first generation of CARs includes a binding domain which specifically targets a tumor antigen, and a lymphocyte-activating intracellular domain. Clinical trials utilizing the first-generation CAR-engineered T cells have failed to exhibit significant clinical benefits. The next generations of CARs were developed to improve the persistence and anti-tumor function of the first generation of CARs. Genetically modified T cells which express CARs are capable of eradicating malignant cells in vitro, and the infusion of engineered T cells results in remission of tumors in various animal models. Recent clinical trials which are based on the infusion of CAR-engineered T cells into tumor patients have indicated that this approach is feasible and safe. Despite some promising results which were derived from clinical studies, some side-effects were reported which can limit the efficacy of CAR T cells. Additionally, unwanted toxicity is a major problem which was observed in several clinical studies. Another problem in immunotherapeutic approaches that are based on CAR-modified T cells is their restricted availability of proper surface tumor-associated antigen (TAAs). Finally, the poor persistence of infused CAR-genetically modified T cells results in restricted antitumor responses. Second, the third and fourth generations of CAR-engrafted T cells were also developed to overcome this obstacle. These generations of CARs exhibited increased proliferation rates and increased cytokine secretion, and are more persistent in the immunosuppressive mechanism of the tumor microenvironment. It is expected that in the near future, many efforts will focus particularly on the development of the third and fourth generations of CARs to acquire more successful results for the treatment of cancer.
Disclosures of financial & competing interests
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
References
- Ahmed M, Cheung N-KV. 2013. Engineering anti-GD2 monoclonal antibodies for cancer immunotherapy. FEBS Lett. 588:288–297.
- Beecham EJ, Ma Q, Ripley R, Junghans RP. 2000. Coupling CD28 co-stimulation to immunoglobulin T-cell receptor molecules: the dynamics of T-cell proliferation and death. J Immunother. 23:631–642.
- Bollard CM, Rössig C, Calonge MJ, Huls MH, Wagner H-J, Massague J, et al. 2002. Adapting a transforming growth factor β–related tumor protection strategy to enhance antitumor immunity. Blood. 99:3179–3187.
- Borkner L, Kaiser A, Van De Kasteele W, Andreesen R, Mackensen A, Haanen JB, et al. 2010. RNA interference targeting programmed death receptor-1 improves immune functions of tumor-specific T cells. Cancer Immunol Immunother. 59:1173–1183.
- Brayer JB, Pinilla-Ibarz J. 2013. Developing strategies in the immunotherapy of leukemias. Cancer Control. 20:49–59.
- Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. 2011. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 118:4817–4828.
- Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, et al. 2007. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 13:5426–5435.
- Bretscher PA. 1999. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc Natl Acad Sci USA. 96: 185–190.
- Brocker T. 2000. Chimeric Fv-ζ or Fv-ε receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood. 96:1999–2001.
- Burks A, Calderon MA, Casale T, Cox L, Demoly P, Jutel M, et al. 2013. Update on allergy immunotherapy: American Academy of Allergy, Asthma & Immunology/European Academy of Allergy and Clinical Immunology/PRACTALL consensus report. J Allergy Clin Immunol. 131:1288–1296.e3.
- Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. 2009. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. 106:3360–3365.
- Chambers CA, Allison JP. 1997. Co-stimulation in T cell responses. Curr Opin Immunol. 9:396–404.
- Chang L, Chang W, Mcnamara G, Aguilar B, Ostberg J, Jensen M. 2007. Transgene-enforced co-stimulation of CD4+ T cells leads to enhanced and sustained anti-tumor effector functioning. Cytotherapy. 9:771–784.
- Chekmasova AA, Rao TD, Nikhamin Y, Park KJ, Levine DA, Spriggs DR, Brentjens RJ. 2010. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin Cancer Res. 16:3594–3606.
- Chen L. 2004. Co-inhibitory molecules of the B7–CD28 family in the control of T-cell immunity. Nat Rev Immunol. 4:336–347.
- Chen L, Flies DB. 2013. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 13:227–242.
- Chmielewski M, Kopecky C, Hombach AA, Abken H. 2011. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 71:5697–5706.
- Chu J, Deng Y, Benson D, He S, Hughes T, Zhang J, et al. 2014. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 28:917–927.
- Croft M. 2005. The evolving crosstalk between co-stimulatory and co-inhibitory receptors: HVEM–BTLA. Trends Immunol. 26:292–294.
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. 2004. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 10:942–949.
- Darcy PK, Kershaw MH, Trapani JA, Smyth MJ. 1998. Expression in cytotoxic T lymphocytes of a single-chain anti-carcinoembryonic antigen antibody. Redirected Fas ligand-mediated lysis of colon carcinoma. Eur J Immunol. 28:1663–1672.
- Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. 2009. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 113:6392–6402.
- Durrant DM, Metzger DW. 2010. IL-12 can alleviate Th17-mediated allergic lung inflammation through induction of pulmonary IL-10 expression. Mucosal Immunol. 3:301–311.
- Eshhar Z, Waks T, Gross G, Schindler DG. 1993. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. 90:720–724.
- Fenoglio D, Traverso P, Parodi A, Kalli F, Zanetti M, Filaci G. 2013. Generation of more effective cancer vaccines. Hum Vaccin Immunother. 9:2543–2547.
- Finney HM, Akbar AN, Lawson AD. 2004. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCRζ chain. J Immunol. 172:104–113.
- Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. 2005. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8 + T cells. J Exp Med. 202:907–912.
- Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, et al. 2014. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor–modified T cells. Blood. 123:2343–2354.
- Goldberger O, Volovitz I, Machlenkin A, Vadai E, Tzehoval E, Eisenbach L. 2008. Exuberated numbers of tumor-specific T cells result in tumor escape. Cancer Res. 68:3450–3457.
- Gong MC, Latouche J-B, Krause A, Heston WD, Bander NH, Sadelain M. 1999. Cancer patient T cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen. Neoplasia. 1:123.
- Gorelik L, Flavell RA. 2002. Transforming growth factor-β in T-cell biology. Nat Rev Immunol. 2:46–53.
- Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, et al. 2013. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids. 2:e105.
- Gross G, Waks T, Eshhar Z. 1989. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA. 86:10024–10028.
- Grossi JA, Raulet DH, Allison JP. 1992. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature. 356:607–609.
- Haynes NM, Snook MB, Trapani JA, Cerruti L, Jane SM, Smyth MJ, Darcy PK. 2001. Redirecting mouse CTL against colon carcinoma: superior signaling efficacy of single-chain variable domain chimeras containing TCR-ζ vs FcεRI-γ. J Immunol. 166:182–187.
- Haynes NM, Trapani JA, Teng MW, Jackson JT, Cerruti L, Jane SM, et al. 2002. Rejection of syngeneic colon carcinoma by CTLs expressing single-chain antibody receptors codelivering CD28 costimulation. J Immunol. 169:5780–5786.
- Heuser C, Hombach A, Lösch C, Manista K, Abken H. 2003. T-cell activation by recombinant immunoreceptors: impact of the intracellular signalling domain on the stability of receptor expression and antigen-specific activation of grafted T cells. Gene Ther. 10:1408–1419.
- Hollyman D, Stefanski J, Przybylowski M, Bartido S, Borquez-Ojeda O, Taylor C, et al. 2009. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother. 32:169.
- Hombach A, Heuser C, Sircar R, Tillmann T, Diehl V, Kruis W, et al. 1997. T cell targeting of TAG72 + tumor cells by a chimeric receptor with antibody-like specificity for a carbohydrate epitope. Gastroenterology. 113:1163–1170.
- Hombach A, Schneider C, Sent D, Koch D, Willemsen RA, Diehl V, et al. 2000. An entirely humanized CD3 zeta-chain signaling receptor that directs peripheral blood t cells to specific lysis of carcinoembryonic antigen-positive tumor cells. Int J Cancer. 88:115–120.
- Hombach A, Sent D, Schneider C, Heuser C, Koch D, Pohl C, et al. 2001a. T-Cell activation by recombinant receptors cd28 costimulation is required for interleukin 2 secretion and receptor-mediated T-cell proliferation but does not affect receptor-mediated target cell lysis. Cancer Res. 61:1976–1982.
- Hombach A, Wieczarkowiecz A, Marquardt T, Heuser C, Usai L, Pohl C, et al. 2001b. Tumor-specific T cell activation by recombinant immunoreceptors: CD3ζ signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3ζ signaling receptor molecule. J Immunol. 167:6123–6131.
- Hosseini M, Haji-Fatahaliha M, Jadidi-Niaragh F, Majidi J, Yousefi M. 2015. The use of nanoparticles as a promising therapeutic approach in cancer immunotherapy. Artif Cells Nanomed Biotechnol. 1–11.
- Hu Z, Xia J, Wargo J, Yang Y-G. 2013. Generation of human T cells expressing only the engineered tumor antigen specific TCR in humanized mice for preclinical research and anticancer therapy (P4364). J Immunol. 190:177.15.
- Hudecek M, Lupo-Stanghellini M-T, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, Riddell SR. 2013. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 19:3153–3164.
- Hudecek M, Schmitt TM, Baskar S, Lupo-Stanghellini MT, Nishida T, Yamamoto TN, et al. 2010. The B-cell tumor–associated antigen ROR1 can be targeted with T cells modified to express a ROR1- specific chimeric antigen receptor. Blood. 116:4532–4541.
- Hwu P, Shafer G, Treisman J, Schindler D, Gross G, Cowherd R, et al. 1993. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor gamma chain. J Exp Med. 178:361–366.
- Jacobson CA, Ritz J. 2011. Time to put the CAR-T before the horse. Blood. 118:4761–4762.
- Jameson SC, Masopust D. 2009. Diversity in T cell memory: an embarrassment of riches. Immunity. 31:859–871.
- Jena B, Maiti S, Huls H, Singh H, Lee DA, Champlin RE, Cooper LJ. 2013. Chimeric antigen receptor (CAR)-specific monoclonal antibody to detect CD19-specific T cells in clinical trials. PloS one. 8:e57838.
- Jensen MC, Popplewell L, Cooper LJ, Digiusto D, Kalos M, Ostberg JR, Forman SJ. 2010. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19- specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 16:1245–1256.
- June CH. 2007. Adoptive T cell therapy for cancer in the clinic. J Clin Invest. 117:1466–1476.
- Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. 2004. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 64:9160–9166.
- Kailayangiri S, Altvater B, Meltzer J, Pscherer S, Luecke A, Dierkes C, et al. 2012. The ganglioside antigen GD2 is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br J Cancer. 106:1123–1133.
- Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. 2011. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 3:95ra73.
- Katz SC, Burga RA, Mccormack E, Wang LJ, Mooring JW, Point G, et al. 2015. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor modified T cell therapy for CEA+ liver metastases. Clin Cancer Res. 1421.2014.
- Kazemi T, Younesi V, Jadidi-Niaragh F, Yousefi M. 2015. Immunotherapeutic approaches for cancer therapy: an updated review. Artif Cells Nanomed Biotechnol. 1–11.
- Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, et al. 2011. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest. 121:4746.
- Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, Yu Z, et al. 2010. Tumor-specific CD8 + T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 70:6725–6734.
- Kershaw MH, Wang G, Westwood JA, Pachynski RK, Tiffany HL, Marincola FM, et al. 2002. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther. 13:1971–1980.
- Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. 2006. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 12:6106–6115.
- Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. 2012. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood. 119:2709–2720.
- Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. 2014. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 33:540–549.
- Kochenderfer JN, Feldman SA, Zhao Y, Xu H, Black MA, Morgan RA, et al. 2009. Construction and Pre-clinical Evaluation of an Anti-CD19 Chimeric Antigen Receptor. J Immunother. 32:689.
- Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler- Stevenson M, Feldman SA, et al. 2010. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 116: 4099–4102.
- Koehler H, Kofler D, Hombach A, Abken H. 2007. CD28 Costimulation Overcomes Transforming Growth Factor-β–Mediated Repression of Proliferation of Redirected Human CD4+ and CD8+ T Cells in an Antitumor Cell Attack. Cancer Res. 67:2265–2273.
- Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. 2015. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. 4:e994446.
- Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, et al. 2006. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 66:10995–11004.
- Kvistborg P, Shu CJ, Heemskerk B, Fankhauser M, Thrue CA, Toebes M, et al. 2012. TIL therapy broadens the tumor-reactive CD8 + T cell compartment in melanoma patients. Oncoimmunology. 1:409–418.
- Ladygina N, Gottipati S, Ngo K, Castro G, Ma JY, Banie H, et al. 2013. PI3Kγ kinase activity is required for optimal T-cell activation and differentiation. Eur J Immunol. 43:3183–3196.
- Lamers CH, Gratama JW, Warnaar SO, Stoter G, Bolhuis RL. 1995. Inhibition of bispecific monoclonal antibody (bsAb)-targeted cytolysis by human anti-mouse antibodies in ovarian carcinoma patients treated with bsAb-targeted activated T-lymphocytes. Int J Cancer. 60:450–457.
- Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. 2006. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 24:e20–e22.
- Lamers CH, Willemsen R, Van Elzakker P, Van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J, et al. 2011. Immune responses to transgene and retroviral vector in patients treated with ex vivo–engineered T cells. Blood. 117:72–82.
- Lee JC, Hayman E, Pegram HJ, Santos E, Heller G, Sadelain M, Brentjens R. 2011. In vivo inhibition of human CD19-targeted effector T cells by natural T regulatory cells in a xenotransplant murine model of B cell malignancy. Cancer Res. 71:2871–2881.
- Lenschow DJ, Walunas TL, Bluestone JA. 1996. CD28/B7 system of T cell costimulation. Ann Rev Immunol. 14:233–258.
- Loskog A, Giandomenico V, Rossig C, Pule M, Dotti G, Brenner M. 2006. Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia. 20:1819–1828.
- Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. 2011. Antitumor activity and long-term fate of chimeric antigen receptor– positive T cells in patients with neuroblastoma. Blood. 118:6050–6056.
- Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. 2002. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nat Biotechnol. 20:70–75.
- Mezzanzanica D, Canevari S, Mazzoni A, Figini M, Colnaghi MI, Waks T, et al. 1997. Transfer of chimeric receptor gene made of variable regions of tumor-specific antibody confers anticarbohydrate specificity on T cells. Cancer Gene Ther. 5:401–407.
- Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. 2013. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 36:133–151.
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. 2010. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 18:843–851.
- Muniappan A, Banapour B, Lebkowski J, Talib S. 2000. Ligand- mediated cytolysis of tumor cells: use of heregulin-ζ chimeras to redirect cytotoxic T lymphocytes. Cancer Gene Ther. 7:128–134.
- Muranski P, Boni A, Wrzesinski C, Citrin DE, Rosenberg SA, Childs R, Restifo NP. 2006. Increased intensity lymphodepletion and adoptive immunotherapy—how far can we go? Nat Clin Pract Oncol. 3:668–681.
- Murphy A, Westwood J, Brown L, Teng M, Moeller M, Xu Y, et al. 2007. Antitumor activity of dual-specific T cells and influenza virus. Cancer Gene Ther. 14:499–508.
- Näslund TI, Gehrmann U, Qazi KR, Karlsson MC, Gabrielsson S. 2013. Dendritic cell–derived exosomes need to activate both T and B cells to induce antitumor immunity. J Immunol. 190:2712–2719.
- Niederman TM, Ghogawala Z, Carter BS, Tompkins HS, Russell MM, Mulligan RC. 2002. Antitumor activity of cytotoxic T lymphocytes engineered to target vascular endothelial growth factor receptors. Proc Natl Acad Sci USA. 99:7009–7014.
- Pameijer C, Navanjo A, Meechoovet B, Wagner J, Aguilar B, Wright C, et al. 2007. Conversion of a tumor-binding peptide identified by phage display to a functional chimeric T cell antigen receptor. Cancer Gene Ther. 14:91–97.
- Parente-Pereira AC, Burnet J, Ellison D, Foster J, Davies DM, Van Der Stegen S, et al. 2011. Trafficking of CAR-engineered human T cells following regional or systemic adoptive transfer in SCID beige mice. J Clin Immunol. 31:710–718.
- Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. 2007. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 15:825–833.
- Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ. 2012. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 119:4133–4141.
- Perosa F, Favoino E, Caragnano MA, Prete M, Dammacco F. 2005. CD20: a target antigen for immunotherapy of autoimmune diseases. Autoimmun Rev. 4:526–531.
- Pinthus JH, Waks T, Kaufman-Francis K, Schindler DG, Harmelin A, Kanety H, et al 2003. Immuno-gene therapy of established prostate tumors using chimeric receptor-redirected human lymphocytes. Cancer Res. 63:2470–2476.
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. 2011. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. N Engl J Med. 365:725–733.
- Pranchevicius M-CS, Vieira TR. 2013. Production of recombinant immunotherapeutics for anticancer treatment: The role of bioengineering. Bioengineered. 4:305–312.
- Pritchard M, Wolf S, Kraybill W, Repasky EA. 2005. The anti-tumor effect of interleukin-12 is enhanced by mild (fever-range) thermal therapy. Immunol Invest. 34:361–380.
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. 2008. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 14:1264–1270.
- Pulè MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. 2005. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 12:933–941.
- Rabinovich GA, Gabrilovich D, Sotomayor EM. 2007. Immunosuppressive strategies that are mediated by tumor cells. Ann Rev Immunol. 25:267.
- Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, et al. 2004. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 173:7125–7130.
- Rosenberg SA, Dudley ME. 2009. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol. 21:233–240.
- Rossig C, Bollard CM, Nuchtern JG, Rooney CM, Brenner MK. 2002. Epstein-Barr virus–specific human T lymphocytes expressing antitumor chimeric T-cell receptors: potential for improved immunotherapy. Blood. 99:2009–2016.
- Bridgeman JS, Hawkins RE, Hombach AA, Abken H, Gilham DE. 2010. Building better chimeric antigen receptors for adoptive T cell therapy. Curr Gene Ther. 10:77–90.
- Sadelain M, Rivière I, Brentjens R. 2003. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 3:35–45.
- Santos EB, Yeh R, Lee J, Nikhamin Y, Punzalan B, Punzalan B, et al. 2009. Sensitive in vivo imaging of T cells using a membrane-bound Gaussia princeps luciferase. Nat Med. 15:338–344.
- Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. 2011. CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. J Clin Invest. 121:1822.
- Schreiber RD, Old LJ, Smyth MJ. 2011. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 331:1565–1570.
- Seliger B. 2008. Different regulation of MHC class I antigen processing components in human tumors. J Immunotoxicol. 5:361–367.
- Sharifzadeh Z, Rahbarizadeh F, Shokrgozar MA, Ahmadvand D, Mahboudi F, Jamnani FR, Moghimi SM. 2013. Genetically engineered T cells bearing chimeric nanoconstructed receptors harboring TAG-72-specific camelid single domain antibodies as targeting agents. Cancer Lett. 334:237–244.
- Smith-Garvin JE, Koretzky GA, Jordan MS. 2009. T cell activation. Ann Rev Immunol. 27:591–619.
- Spear P, Barber A, Rynda-Apple A, Sentman CL. 2012. Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-γ and GM-CSF. J Immunol. 188:6389–6398.
- Stewart-Jones G, Wadle A, Hombach A, Shenderov E, Held G, Fischer E, et al. 2009. Rational development of high-affinity T-cell receptor-like antibodies. Proc Natl Acad Sci USA. 106:5784–5788.
- Sun J, Dotti G, Huye LE, Foster AE, Savoldo B, Gramatges MM, et al. 2010. T cells expressing constitutively active Akt resist multiple tumor-associated inhibitory mechanisms. Mol Ther. 18:2006–2017.
- Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, Davila E. 2012. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res. 18:6436–6445.
- Tassev D, Cheng M, Cheung N-K. 2012. Retargeting NK92 cells using an HLA-A2-restricted, EBNA3C-specific chimeric antigen receptor. Cancer Gene Ther. 19:84–100.
- Teng MW, Kershaw MH, Moeller M, Smyth MJ, Darcy PK. 2004. Immunotherapy of cancer using systemically delivered gene-modified human T lymphocytes. Hum Gene Ther. 15:699–708.
- Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, et al. 2008. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 112:2261–2271.
- Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. 2012. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4–1BB domains: pilot clinical trial results. Blood. 119:3940–3950.
- Tomihara K, Curiel TJ, Zhang B. 2013. Optimization of immunotherapy in elderly cancer patients. Critical Reviews™ in Oncogenesis. 18.
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, Mcdermott DF, et al. 2012. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med. 366:2443–2454.
- Trinchieri G, Pflanz S, Kastelein RA. 2003. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity. 19:641–644.
- Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, et al. 2012. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 72:1844–1852.
- Vera J, Savoldo B, Vigouroux S, Biagi E, Pule M, Rossig C, et al. 2006. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood. 108:3890–3897.
- Wang J, Jensen M, Lin Y, Sui X, Chen E, Lindgren CG, et al. 2007. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther. 18:712–725.
- Wang J, Press OW, Lindgren CG, Greenberg P, Riddell S, Qian X, et al. 2004. Cellular immunotherapy for follicular lymphoma using genetically modified CD20-specific CD8 + cytotoxic T lymphocytes. Mol Ther. 9:577–586.
- Westwood JA, Smyth MJ, Teng MW, Moeller M, Trapani JA, Scott AM, et al. 2005. Adoptive transfer of T cells modified with a humanized chimeric receptor gene inhibits growth of Lewis-Y-expressing tumors in mice. Proc Natl Acad Sci USA. 102:19051–19056.
- Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, et al. 2011. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther. 19:751–759.
- Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, et al. 2009. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol. 183:5563–5574.
- Zhong X-S, Matsushita M, Plotkin J, Riviere I, Sadelain M. 2010. Chimeric Antigen Receptors Combining 4–1BB and CD28 Signaling Domains Augment PI3kinase/AKT/Bcl-XL Activation and CD8 & plus; T Cell–mediated Tumor Eradication. Mol Ther. 18:413–420.
- Zou Y, Stastny P, Süsal C, Döhler B, Opelz G. 2007. Antibodies against MICA antigens and kidney-transplant rejection. N Engl J Med. 357: 1293–1300.