
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Context
Vesicular transdermal delivery can enhance the bioavailability of a drug especially affected by first-pass metabolism, e.g. nitrendipine. However effective transdermal delivery employs permeation enhancer, e.g oleic acid (OA) with ceramide 2, stearic acid, behenic acid, and cholesteryl sulfate lipid complex.
Objective
This study investigated the preparation, characterization of physicochemical properties, ex vivo permeation using human skin, pharmacokinetic parameters and antihypertensive potential in rats, of nitrendipine-loaded nanovesicles of ceramide 2, stearic acid, behenic acid and cholesteryl sulfate containing oleic acid gel (NOVG).
Materials and methods
The nanovesicles were made using film hydration method and characterized for physicochemical properties, ex vivo permeation using human skin, pharmacokinetic parameters and antihypertensive potential.
Results
Nitrendipine-loaded nanovesicles of ceramide-2 containing oleic acid (NOV-5) have shown fluxes in the range of 4.88–24.72 μg/cm2/h nitrendipine oral suspension (NOS) at equal dose. NOVG-5 has shown almost 33% reduction in blood pressure in the first hour and a further decrease of 25% in the second hour to restore the normal pressure.
Discussion
The permeation increases with increase in OA content. OA gets integrated in vesicle wall and enhances its permeability, whereas ceramide content makes sure that skin does not become damaged even after permeation.
Conclusion
NOVG-5 has shown the most favorable physicochemical properties and good permeation through skin providing good management of hypertension during crucial initial hours.
Introduction
Nitrendipine is a dihydropyridine calcium-channel blocker used in the management of hypertension. It is reported to be well absorbed following oral administration but it undergoes extensive first-pass metabolism having an absolute oral bioavailability of 10–20%. After an oral dose of nitrendipine 20 mg, peak plasma concentrations (between 5 and 40 μg/L) are achieved within 1–2 h. The commonly reported side effects are headache, flushing, edema and palpitations which are the usual extension of vasodilating property of the drug. Along with their relatively frequent occurrence, these adverse reactions are generally pronounced in oral drug delivery system, having a variable absorption.
The formulation development for nitrendipine has been focused on enhancing absorption and bioavailability using controlled/sustained formulations or by using permeation enhancers in transdermal formulations. Some of the studies are summarized here.
Giannakou et al. examined combination of N-methyl-2-pyrrolidone and glyceryl monooleate and obtained higher flux values at enhancer concentration >4.5% w/w (Giannakou et al. Citation1995). Tipre and Vavia (Citation2003) fabricated transdermal formulation using d-limonene and obtained high input rate. Cui et al. (Citation2003) prepared sustained-release microspheres and obtained higher bioavailability with respect to Baypress™ tablets.
Yang et al. (Citation2004) formulated solid dispersion and a novel pH-dependent gradient release delivery system and obtained increased bioavailabilities in comparison with Baypress™ tablets. Manjunath et al. assessed the improvement in bioavailability using solid lipid nanoparticles and attained several folds higher effective bioavailability with the suspension (Manjunath and Venkateswarlu Citation2006). Gannu et al. (Citation2007) developed transdermal formulation using 6% v/w of carvone as penetration enhancer.
Kumar et al. (Citation2007) prepared nitrendipine-loaded solid lipid nanoparticles and attained several folds higher bioavailability after intraduodenal administration in comparison to suspension. Bhaskar et al. (Citation2008, Citation2009) formulated nitrendipine-loaded solid lipid nanoparticles and nanostructured lipid carriers and obtained maximum enhancement for nitrendipine flux.
Mittal et al. (Citation2009) prepared matrix-type patches of nitrendipine and attained higher drug flux. Xia et al. (Citation2010) prepared nitrendipine nanosuspensions and obtained higher Cmax (peak plasma concentration) and AUC0→12 (area under the curve) values with respect to commercial tablets. Wang et al. (Citation2010) prepared solid self-emulsifying pellets of nitrendipine and obtained higher AUC compared to the conventional tablets.
Transdermal delivery became popular due to several notable advantages over conventional drug delivery system, e.g. avoidance of gastrointestinal transit and hepatic first-pass metabolism. It also offers simultaneous control of the input. But skin is a barrier which is designed to resist the passage of foreign materials. The main component in this barrier is stratum corneum (10–20 μm). In SC, corneocytes are surrounded by lipid regions, oriented parallel to the corneocytes surfaces. The substances move mainly along the tortuous pathways in the intercellular lipid regions which have been reported to be arranged in lamellar form. SC lipids are composed of free fatty acids (FFA), cholesterol, ceramides and cholesteryl sulfate representing approximately 25%, 25%, 40% and 10% (w/w) of the SC lipids, respectively (Gaur et al. Citation2014c). These SC lipids play an essential role in maintaining and structuring the lipid barrier which affords protection against external insults and water loss through the skin (Gaur et al. Citation2014d).
Several methods had been devised to enhance the transdermal permeation. Here, we will discuss only those which are directly related to our work. The use of nanosized formulations and formulation using chemical enhancer are the main approaches for enhancing the transdermal permeation. Cooper et al. reported enhancement of transport of nonpolar materials by the addition of oleic acid (OA) (Cooper Citation1984). Several workers have reported mechanistic permeation studies on OA. It was reported that OA disrupts the lipid structure and increases the fluidity of the lipid region. It can exist in a fluid phase at temperatures below the Tm and can fluidize the lipid bilayer at temperatures above the Tm (Barry Citation1987). On the other hand, it also reduces Tm for porcine skin from 63.2 to 53.6 °C (Francoeur et al. Citation1990). The synergistic exponent was also observed for OA when incorporated as a vesicular component (Maghraby et al. Citation2000). OA was reported to increase the permeation of 5-fluorouracil (Yamane et al. Citation1995), fluconazole (Zakir et al. Citation2010) and methotrexate (Srisuk et al. Citation2012). Stratum corneum lipid liposomes were first reported by Wertz et al. (Citation1986); however, it was used as stratum corneum substitute for experimental studies. Ceramide has earlier been employed for dermal delivery of 5-amino laevulinic acid for skin cancer (Pierre et al. Citation2001) and retinoic acid (Contreras et al. Citation2005). Also it has been used in transdermal delivery of diclofenac and ibuprofen (Gaur et al. Citation2014a, Citation2014d).
Nitrendipine suffers from low bioavailability due to high hepatic first pass and till date no study has reported on stable nanovesicular formulation of nitrendipine; so in this study, a nanovesicle formulation was developed for transdermal delivery of nitrendipine using ceramide 2 (biomaterial), stearic acid, behenic acid, cholesteryl sulfate and OA. The nanovesicles were formulated and characterized for different physicochemical parameters, transdermal absorption, pharmacokinetics and antihypertensive activity.
Materials and methods
Materials
Nitrendipine was the gift sample from Concept Pharmaceuticals Ltd, Mumbai, Maharashtra, India and ceramide-2 was gift sample from Sederma (France), supplied by Croda Chemicals (India) Pvt. Ltd, Thane, Mumbai, India. Stearic acid (SA), behenic acid (BA), cis-9-octadecenoic acid (oleic acid ≥99%, OA) and cholestryl-3-sulphate (CS) were purchased from Sigma–Aldrich (New Delhi, India). All other chemicals were of analytical grade.
Methods
Vesicle preparation
The nanovesicles were prepared using thin lipid film method using the composition given in . Suitable amount of drug (1 g) was dissolved in organic solvent (chloroform) and then lipid components were mixed into it. This lipid drug mixture was evaporated using rotary evaporator to deposit a thin film in a round-bottom flask (Rotavapor® R II, BUCHI India Private Ltd.). The resultant lipid film was hydrated by mixing with buffer (pH 6.8) at 200 rpm and 25 °C for 1 h. Further a probe sonicator was employed for 15 min at 100 W for obtaining nanodispersion.
Table I. Formulation of lipid components (lipid drug ratio = 3:1).
This nanodispersion was extruded through a membrane (Immobilon-P Membrane, 0.22 μm pore size, Millipore Pvt. Ltd., New Delhi, India) and finally unentrapped drug was removed using a sephadex G-20 minicolumn (Gaur et al. Citation2014c, Citation2014d, Sinico et al. Citation2005).
Surface morphology and size distribution analysis
Surface morphology was analyzed by SEM. Sample was prepared by lightly sprinkling vesicles on a double adhesive carbon tape with aluminum stub followed by coating with gold. The sample was analyzed using Zetasizer.
Size distribution was analyzed by TEM. TEM analysis was done after staining the vesicles with negative stain (phosphotungstic acid). An aliquot of the formulation was placed above a copper grid and allowed to soak. The excess was removed by a filter paper. Then 1% solution of phosphotungstic acid was added and allowed to absorb. The grid was dried at room temperature and then analyzed using Philips CM-10 (acceleration voltage: 100 kV; magnification: upto 450,000×; cryo-attachment).
Zeta potential and polydispersity index (PDI)
Zeta potential and PDI were determined by means of Zetasizer (NanoZS, Malvern Instruments, UK) which was fitted with a 4 mW He–Ne laser. Appropriate volume was taken in microcentrifuge tube and lyophilized. Then this sample was resuspended in phosphate buffer, pH 6.8 and analyzed for zeta potential and PDI at room temperature.
Entrapment efficiency
Entrapment efficiency was estimated by ultracentrifugation method. Appropriate volume of formulation was taken and centrifuged at 12,000 rpm (Remi C-24 BL with angular R-241 rotor, Remi House, Mumbai, India). The supernatant and pellets were individually analyzed for drug content. The entrapment efficiency was computed by using Eq. (1) (Gaur et al. Citation2014d):
Assay
Drug content was determined using HPLC method. The instrument consisted of an LC-10AT VP pump, an SIL-10AF auto injector, an SPD-10A UV–VIS detector and an SCL-10A VP system controller (Shimadzu, Japan). The method was followed as per Gaur et al. (Citation2013) and Bhaskar et al. (Citation2009).
The HPLC column was Shim-pack VP-ODS having internal diameter 4.6 mm and length 150 mm, packed with 5 μm particles. The system was injected with 20 μL sample, eluted isocratically with double-distilled water, glacial acetic acid and pH 7.4 phosphate buffer (50:40:10, v/v/v) at a flow rate of 1.0 mL/min and detected at 237 nm.
A calibration curve was prepared between concentration and peak area using various concentrations and a linear relation was attained within the concentration range of 1–50 ng/mL. The calibration curve equation was y = 32,360x − 59,222 (R2 = 0.9995), where x is the concentration and y is the peak area.
In vitro drug release
In vitro drug release was estimated on a semipermeable cellulose acetate membrane with a pore size of 12 KDa (molecular cutoff). To make it suitable for permeation experiment, the membrane was stabilized in saline solution and placed over Franz diffusion cell having a nominal area of 1 cm2. The receiver chamber was filled with pH 5.5 buffer. A sample of 200 mg formulation containing 2 mg drug was applied onto the donor side. The cell was operated for 24 h and suitable volume of aliquot was taken out at predecided time intervals. The aliquots were diluted and analyzed using HPLC assay (Gaur et al. Citation2014d).
Stability studies
The stability was analyzed under two different conditions, viz. 25 ± 2 °C; 60 ± 5% relative humidity (RH) and 40 ± 2 °C; 75 ± 5% RH for 90 days. The formulations were stored in above-mentioned conditions and periodically (30, 60 and 90 days) analyzed for physical parameters and in vitro release (Ishida et al. Citation2002, Gaur et al. Citation2014d).
Preparation of gel
The vesicle formulations were dispersed in the carbopol gel for optimizing transdermal administration. For preparation of gel, carbopol 934 was dispersed in water to make 1% gel and shaken by employing mechanical stirrer. The gel was neutralized by using 0.5% w/w aqueous triethanolamine solution. The gel was kept aside for 24 h for exclusion of entrapped air bubbles. Then, the gel was homogeneously mixed with drug-loaded nanovesicles to formulate nanovesicle gels. CG (carbopol gel containing unentrapped nitrendipine) was formulated for comparison purpose and was prepared by adding plain unentrapped drug in carbopol gel (Gaur et al. Citation2014d).
Viscosity
The viscosity determination was performed by using Brookfield rheometer (Brookfield DV III ultra V6.0 RV cone and plate rheometer; Brookfield Engineering Laboratories, Inc., Middleboro, MA). The spindle # CPE40 was employed at 25 ± 0.5 °C for the estimation of viscosity.
Ex vivo skin permeation studies
Ex vivo permeation study protocol was prepared and presented to Institutional Ethical committee for approval. After getting necessary permissions, full thickness human skin obtained from plastic surgery patients was taken. The skin sample was washed with ringers’ solution and then subcutaneous fatty tissues were removed by means of scalpel and forceps. The skin sample was again washed with ringers’ solution and packed in aluminum foil. These packets were placed inside polyethylene bags and stored at −20 °C till further use; however, no more than 3 days were allowed for storage before any ex vivo permeation studies.
For the ex vivo studies, skin was brought to 37 °C and washed with saline solution. It was then fixed onto the donor side of Franz diffusion cell with epidermal side toward donor chamber. The receiver chamber was packed with pH 5.5 buffer and the set-up was kept overnight for saturation. The formulation was applied on the epidermal surface and the set-up was further operated for 24 h. Suitable aliquots were removed from the receiver chamber and analyzed by HPLC assay after appropriate dilutions. The volume was restored in receiver chamber by adding the fresh buffer solution after removal of each aliquot.
After finishing the ex vivo permeation study, the skin sample was separated in various layers by means of tape stripping. The skin sample used in permeation study was removed and mounted with pins on a flat surface; then it was stripped by means of scotch tape (10 times). After every strip, the tape was weighed and stored in a vial [strip 1 (vial 1), strips 2 and 3 (vial 2), strips 4–6 (vial 3) and strips 7–10 (vial 4)]. After stripping, epidermis was scratched by means of scalpel and placed in a separate vial. Remaining skin sample containing some epidermis and dermis was kept separately. All the skin samples (tape strips, scratched epidermal portions and dermis) were homogenized in methanol and analyzed for drug content (Gaur et al. Citation2014b, Citation2014d, Manconi et al. Citation2011).
All the nanovesicle gel (NOVG) formulations and nitrendipine plain gel (NCG) were evaluated at a dose of 200 mg (containing 2 mg nitrendipine).
In vivo skin permeation
In vivo skin permeation study was performed on Wistar albino rats. The experimental protocol was prepared and approved by Institutional Animal Ethical committee.
Twenty-four male albino rats (6–8 weeks) were kept for 1 week in polypropylene cages in standard laboratory conditions, with continuous access to normal laboratory diet (Lipton feed, Mumbai, India) and water ad libitum. Seven- to nine-week-old (average weight 200–210 g) animals were used in the study. The animals were divided in 4 groups:
Group I = no treatment
Group II = oral suspension (5 mg/kg)
Group III = NCG (200 mg formulation containing 2 mg drug)
Group IV = NOVG-5 (100 mg formulation containing 1 mg drug)
For administration of transdermal formulations, animals were sedated by administering a combination of ketamine hydrochloride (75 mg/kg) and xylazine (5 mg/kg) intravenously. After anesthetizing, the abdominal portion was cleaned and hairs were shaved.
The transdermal gels were applied to the skin surface by a gentle rub in a circular motion covering an area of 1 cm2. The blood sample was taken through the tail vein at predecided time interval and stored in vacutainer tubes. The blood sample was centrifuged at 5000 rpm for 20 min for plasma separation. Drug content was analyzed using HPLC assay soon after plasma isolation (Gaur et al. Citation2014c).
Antihypertensive activity
The protocol for antihypertensive activity determination on experimentally induced hypertension was prepared and reviewed by Institutional Animal Ethical committee. The antihypertensive activity was determined by noninvasive BP measurement tail cuff method. Methyl prednisolone acetate at a dose of 20 mg/kg/week was administered subcutaneously for 2 weeks to induce hypertension. Thirty-six Wistar albino rats (average age 6–7 weeks, average weight 200–220 g) were housed in polypropylene cages with adequate access to standard feed (Lipton, Mumbai India) and water ad libitum. Before the study, the animals were acclimatized with the restrainer by keeping them inside restrainer for 10 min every day till 30 days. For the blood pressure measurement, the animal was held in a restrainer and its tail was put inside the cuff. Then the blood pressure was recorded using the instrument. The readings were taken initially and then at predecided time interval after treatment administration till 24 h (Bhaskar et al. Citation2008, Citation2009).
Group I = normotensive (negative control)
Group II = hypertensive (toxic control)
Group III = hypertensive + blank carbopol gel (placebo)
Group IV = hypertensive + NOS (treatment)
Group V = hypertensive + NCG (treatment)
Group VI = hypertensive + NOVG-5
Skin irritation studies
The study protocol was approved by Institutional Ethical committee. All the participants were demonstrated the procedure and consent forms were signed. The study was done on 6 male volunteers. The upper arm area was carefully checked and an area was marked for application. 5% w/v solution of sodium lauryl sulfate (SLS) was applied in the left arm, whereas the treatment (NOVG-5) was administered to the right arm. Untreated skin served as a negative control. SLS or NOVG-5 formulation was held with a cotton bandage. The study was done for 7 days and every day the bandage was removed and observations were made before fresh application till 7 days.
Irritation potential was assessed by visual observations and scores were given as follows:
0 = no reaction;
1 = weak spotty or diffuse erythema;
2 = weak but well perceptible erythema covering the total exposure area;
3 = moderate erythema;
4 = severe erythema with edema;
5 = very severe erythema with epidermal defects (blisters, erosions, etc.) (Patel et al. Citation2009).
Data and statistical analysis
Percentage cumulative amount of drug released was plotted with time to construct the in vitro drug release profile.
The cumulative amount of drug permeated was plotted versus time to generate ex vivo permeation profile. The slope of the profile was taken as steady-state drug flux (Jss) and x-intercept of the linear region of the curve gave lag time (Tlag). Further, determination of permeability coefficient through the membrane (Kp) and diffusion constant within the membrane (D) were done by using below mentioned formulae (Eqs. 2 and 3).
Where D is the diffusion coefficient within the skin (cm2 h−1), h is the diffusional path length and Cd is the initial drug concentration in donor compartment.
Plasma concentration time profiles were constructed by plotting plasma drug concentration (μg) with time (h). Cmax and tmax (time for peak plasma concentration) were read directly from the profiles. AUC0→24 was estimated employing linear trapezoidal method in Graph pad Prism Version 4.
Triplicate values were used for data analysis in physicochemical parameters, drug release and stability study. In ex vivo/in vivo permeation study and antihypertensive activity determination, mean of 6 values ± S.D. was used for statistical comparison. Statistical analyses were executed using the Graph pad Prism Version 4 software by employing analysis of variance (ANOVA) or the paired t-test, where appropriate with statistical significance at P < 0.05.
Results
Physicochemical parameters
Physicochemical characteristics such as size, charge and drug content have significant effect on the performance of the vesicular formulations inside the physiological system so much, so that sometimes the effectiveness of a formulation can be significantly altered only by altering the physicochemical properties such as size.
The vesicle size and its distribution were determined by SEM. The vesicle size was between 89.9 and 113.3 nm giving rise to nanovesicle term. The vesicle shape was spherical ( and
Figure 1. (A) SEM photograph of NOV-5. (B) TEM photograph of NOV-5. (C) Particle size distribution analysis report of NOV-5.

Table II. Nanovesicles and corresponding gels: physicochemical parameters.
In vitro drug release
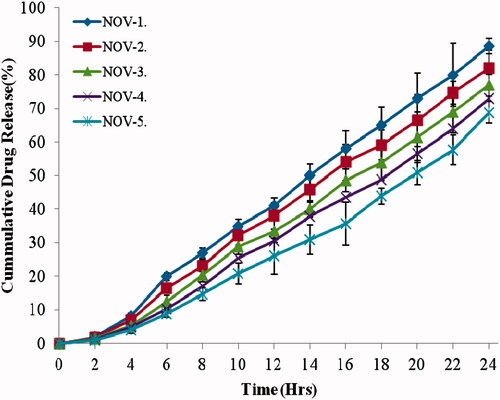
In vitro drug release was done on a 12 KDa molecular cutoff semisynthetic membrane to exclude any hindrance from the barrier. Drug release was controlled in all the vesicular formulations with highest drug release in NOV-1 since it contained the lowest lipid content (). NOV-5 contained 25% more lipid content than NOV-1 reflecting in 17% higher entrapment efficiency than NOV-1.
Figure 2. In vitro drug release profile for nanovesicle formulations in 24 h. Data presented as mean ± SD; (n = 3); p ≤ 0.01.
Stability studies
Stability evaluation was done at two different storage conditions for NOV-5 which was selected on the basis of physicochemical parameters. NOV-5 has shown good stability at even 40 °C after a period of 90 days ().
Table III. Physicochemical characterization of nitrendipine ceramide oleic vesicles (NOV-5) after stability studies.
Ex vivo drug permeation
The permeation profiles of the formulations were studied using full thickness human skin mounted in Franz diffusion cell and various permeation parameters were calculated using drug permeation across human skin. depicts the amount of drug permeated during 24 h to the receiver chamber via unit area of skin. The drug content permeated ranged from 97.15 to 606.55 μg/cm2 for NOVG formulations, whereas NCG was able to permeate only 0.27 μg/cm2. The formulation NOVG-5 permeated the highest amount of drug in receiver chamber. Based on ex vivo permeation data, flux, lag time, permeability coefficient and diffusion parameter were computed for all NOVG formulations (). The flux values were in the range of 4.88–24.72 μg/cm2/h with respective lag time values of 2.5–0.4 h, whereas NCG has shown flux and lag time value of 0.0236 μg/cm2/h and 3.4 h, respectively.
Figure 3. Cumulative amount of drug permeated per unit area through human skin in 24 h. Data presented as mean ± SD; (n = 6); p ≤ 0.05.
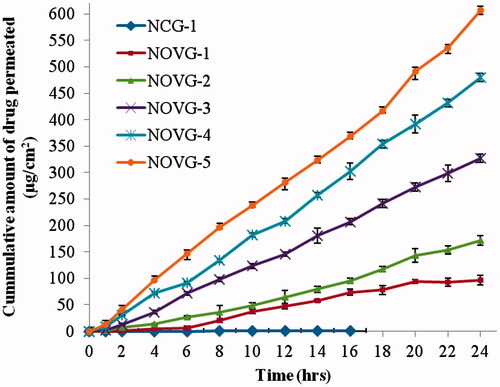
Table IV. Flux, permeability coefficient and diffusion parameters of NOVG formulations.
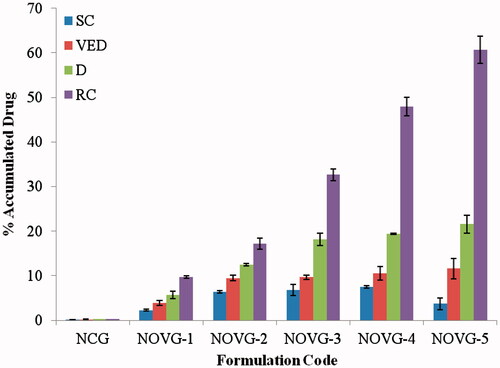
shows the drug permeation levels attained in various layers of skin. The data was obtained by stripping the skin used in ex vivo experiment after completion of 24 h resulting in drug content in SC, viable epidermis, dermis and receiver chamber. The drug content in receiver chamber represents the approximate drug concentration which has crossed all the transdermal barriers. NOVG-5 has shown highest drug content in receiver chamber which means highest drug permeation through transdermal route.
Figure 4. Drug concentration in various strata of skin; SC, stratum corneum; VED, viable epidermis; D, dermis; RC, receiver chamber. Data presented as mean ± SD; (n = 6); p ≤ 0.05.
In vivo drug permeation
The optimum formulation NOVG-5 was evaluated against nitrendipine oral suspension and nitrendipine carbopol gel. Plasma concentration time profiles have shown 5.6 times higher Cmax for NOVG-5 than NOS at equivalent dosage. It also increases tmax from 2 (NOS) to 12 h (NOVG-5) (). It is visible from plasma concentration profile that NOVG-5 maintains the drug content above the Cmax of oral suspension since the start of 2nd hour upto 24th hour. Consequently, the AUC (bioavailability) of the NOVG-5 stands at 78.34% in comparison to the oral suspension (10%) ().
Figure 5. Plasma drug concentration profiles of NOVG-5, NOS and NCG. Data presented as mean ± SD; (n = 6); p ≤ 0.05.
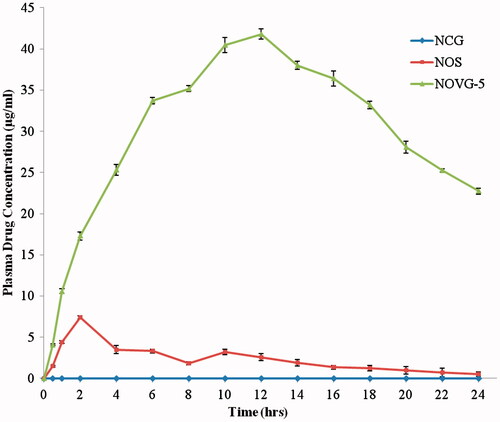
Table V. Cmax, tmax and AUC0→24 values for NOS, NCG and NOVG formulations.
Antihypertensive activity
shows the reductive potential of NOVG-5 formulation in experimentally hypertensive rats. NOVG-5 has shown the reduction of 60 mm Hg with the first hour of treatment which must be the confirmation of the smallest lag time (0.4 h = 24 min) shown by it in ex vivo permeation study. By the second hour of treatment, it restores the normal blood pressure.
Table VI. Antihypertensive effect of NOVG-5 and NCG in comparison to oral route.
Skin irritation potential
NOVG-5 was compared with SLS for skin irritation potential since OA has irritation potential. None of the participants reported any kind of skin injury (erythema or edema). The OA has been reportedly successfully used as a permeation enhancer upto 5% v/v and in our study, the maximum content of OA was 2.5. Also ceramide 2 has regenerative potential which further supports normal skin structure and functions.
Discussion
Lipid composition decides the physicochemical parameters which affects the behavior of nanovesicles in a physiological system. Nanosized vesicles will be more able to permeate upto deeper layers. The thickness of SC is usually 15 μm and there is a chain of pores which are smaller than 4 nm size. The formulated vesicle population was relatively homogenous.
All the NOVG formulations possess negative charge owing to the presence of CS and OA. OA has a pka value of 5.02, so it will be 75.1% ionized at skin pH (5.5) imparting a net negative charge to the vesicle membrane. Zeta potential is the product of electrostatic charge in the vesicle membrane and effective surface area. Same electrical charge in vesicle membrane provides stability against vesicle aggregation since vesicular formulations are thermodynamically unstable. Zeta potential values were within the range of −35 to −41 mV which is a product of increasing content of OA (6.25–25%).
In this study, zeta potential increased with increase in the content of oleic acid in formulation composition. Oleic acid is negatively charged in ionized form so it is mainly responsible for negative values of zeta potential. However, the increase in value of charge is counter balanced with decreased surface area owing to increase in vesicle size.
Entrapment efficiency was highest in NOV formulation since the total lipid content has been increased by 25%. Nitrendipine is a basic drug which has its pKa at 5.43 which makes it 46% ionized at skin pH. Since ionic fraction is more hydrophilic than nonionized fraction, it remains inside the vesicle core, maintaining a reservoir, whereas nonionized fraction goes through the vesicle wall and exerts therapeutic effect.
Drug release study shows that as the content of oleic acid increases, the drug release increases. OA has a bent structure due to presence of unsaturation in the mid of hydrocarbon chain which makes the vesicle membrane permeable for drug release.
NOVG-5 was selected for stability evaluation since it has shown the most optimum physicochemical parameters. The selected formulation was proven to be stable at elevated conditions.
Ex vivo permeation study was done using human skin because it can give the realistic observations about drug permeation in clinical settings. The concentration in receiver chamber represents the drug in systemic circulation and NOVG-5 has shown the highest drug content in receiver chamber. It contains the highest content of OA which acts as a permeation enhancer by means of permeating into SC cells and introducing kink in lamellar SC lipids. Initially the drug release was slow; however, after 3–5 h, all the oleic acid formulations showed exponential drug release. This could be attributed to saturation of skin layers with OA and opening up of various paths for drug permeation through skin. Various permeation parameters have been calculated on the basis of permeation data. NOVG-5 has shown the flux value 24.72 (μg/cm2/h) and a lag time value of 0.4 h, which is excellent and will prove beneficial in effective management of hypertension.
Since in vivo study on human subjects is not a viable option during formulation development, albino rats were used for in vivo study and determination of pharmacokinetic parameters along with the measurement of antihypertensive activity.
In vivo drug permeation study of NOVG-5 was done in comparison of oral suspension (5 mg/kg dose; app 1 mg nitrendipine), and NOVG-5 was administered in the identical dosage. The in vivo study replicates the results of ex vivo drug permeation study. Rat skin is thinner than human skin, so drug content increases further. Antihypertensive study shows the clinical implication of NOVG-5 formulation. It restores the normal blood pressure in 2 h of treatment.
NOVG-5 has shown the highest drug content permeated to the receiver chamber. All the NOVG formulations have similar compositions except the OA content. During assembly of vesicles, all lipid components equally distribute according to their steric requirements. Ceramide 2 has a hairpin structure (Polar head group and two hydrocarbon chains), whereas stearic acid, behenic acid and cholesteryl sulfate are planar molecules. OA is an unsaturated 16-carbon fatty acid with an unsaturation at Δ9. Once OA gets integrated in vesicle wall, it introduces a kink in the vesicle wall due to cis-configuration. Further it modifies the configuration of the intercellular lipids causing increase in permeability of SC which have similar arrangement, permitting molecules to migrate across the SC. OA is a known permeation enhancer which enhances the drug permeation across the skin by a selective perturbation of the intracellular lipid bilayer present in the SC below the Tm (Francoeur et al. Citation1990, Ongpipattanakul et al. Citation1991, Torchilin et al. Citation1992, Yamane et al. Citation1995, Gaur et al. Citation2014c). OA was reported to increase the permeation of 5-fluorouracil (Yamane et al. Citation1995), fluconazole (Zakir et al. Citation2010) and methotrexate (Srisuk et al. Citation2012). It has also been reported that OA dramatically change the morphology and the density of epidermal Langerhans cells and generates pores on the surface of epidermal corneocytes at a concentration of 10% (Touitou et al. Citation2002). The concentration used in our study was well below this irritant concentration and ceramide-2 in vesicle composition counterbalances its effects since it is a skin protectant.
Conclusion
Nitrendipine is an important antihypertensive drug for primary hypertension; however, its bioavailability is low. Nanovesicle formulation of nitrendipine can provide controlled delivery through the transdermal route. Vesicles – composed of ceramide, stearic acid, behenic acid, cholesterol sulfate and fatty acid – have been reported for dermal drug delivery, however, with the addition of OA as a fatty acid component permeation enhancement can be introduced for effective transdermal drug delivery. OA is a permeation enhancer with own hazards however formulating it with ceramide 2 will prove to be effective and safe for transdermal drug delivery. On the basis of this study, we propose OA-containing ceramide nanovesicles as promising carrier for systemic transdermal delivery.
Further, we acknowledge the Advanced Analytical Instruments facility (AIRF), JNU, New Delhi for SEM analysis and Advanced Centre for Nanotechnology (ACN), National Institute of Pharmaceutical Education & Research (NIPER) for providing facility of Zetasizer nano ZS.
Acknowledgements
The authors want to acknowledge Sederma, France (supplier Croda India) for genuine supply of Ceramide-2 and Concept Pharmaceuticals Ltd for providing nitrendipine.
Declaration of interest
The authors declare that there is no conflict of interests regarding the publication of this article.
References
- Barry BW. 1987. Mode of action of permeation enhancers in human skin. J Control Release. 6:85–97.
- Bhaskar K, Anbu J, Ravichandiran V, Venkateswarlu V, Rao YM. 2009. Lipid nanoparticles for transdermal delivery of flurbiprofen: formulation, in vitro, ex vivo and in vivo studies. Lipids Health Dis. 8:6.
- Bhaskar K, Krishna Mohan C, Lingam M, Prabhakar Reddy V, Venkateswarlu V, Madhusudan Rao Y. 2008. Development of nitrendipine controlled release formulations based on SLN and NLC for topical delivery: in vitro and ex vivo characterization. Drug Dev Ind Pharm. 34:719–725.
- Contreras MJF, Jim, enez Soriano MM, Di, eguez ARı. 2005. In vitro percutaneous absorption of all-trans retinoic acid applied in free form or encapsulated in stratum corneum lipid liposomes. Int J Pharma. 297:134–145.
- Cooper ER. 1984. Increased skin permeability for lipophilic molecules. J Pharm Sci. 73:1153–1156.
- Cui F, Yang M, Jiang Y, Cun D, Lin W, Fan Y, Kawashima Y. 2003. Design of sustained-release nitrendipine microspheres having solid dispersion structure by quasi-emulsion solvent diffusion method. J Control Release. 91:375–384.
- Francoeur ML, Gloden GM, Potts PO. 1990. Oleic acid: its effects on stratum corneum in relation to (trans) dermal drug delivery. Pharm Res. 7:621–627.
- Gannu R, Vishnu YV, Kishan V, Rao YM. 2007. Development of nitrendipine transdermal patches: in vitro and ex vivo characterization. Curr Drug Deliv. 4:69–76.
- Gaur PK, Purohit S, Mishra S. 2013. Solid lipid nanoparticles of guggul lipid as drug carrier for transdermal drug delivery. Biomed Res Int. 2013:10.
- Gaur PK, Bajpai M, Mishra S, Verma A. 2014a. Development of ibuprofen nanoliposome for transdermal delivery: physical characterization, in vitro/in vivo studies, and anti-inflammatory activity. Artif Cells Nanomed Biotechnol. 1–6. DOI: https://doi.org/10.3109/21691401.2014.953631.
- Gaur PK, Mishra S, Kumar A, Panda BP. 2014b. Development and optimization of gastroretentive mucoadhesive microspheres of gabapentin by Box–Behnken design. Artif Cells Nanomed Biotechnol. 42:167–177.
- Gaur PK, Purohit S, Kumar Y, Mishra S, Bhandari A. 2014c. Ceramide-2 nanovesicles for effective transdermal delivery: development, characterization and pharmacokinetic evaluation. Drug Dev Ind Pharm. 40:568–576.
- Gaur PK, Purohit S, Kumar Y, Mishra S, Bhandari A. 2014d. Preparation, characterization and permeation studies of a nanovesicular system containing diclofenac for transdermal delivery. Pharma Dev Technol. 19:48–54.
- Giannakou SA, Dallas PP, Rekkas DM, Choulis NH. 1995. Development and in vitro evaluation of nitrendipine transdermal formulations using experimental design techniques. Int J Pharm. 125:7–15.
- Ishida T, Takanashi Y, Doi H, Yamamoto I, Kiwada H. 2002. Encapsulation of an antivasospastic drug, fasudil, into liposomes, and in vitro stability of the fasudil-loaded liposomes. Int J Pharm. 232:59–67.
- Kumar VV, Chandrasekar D, Ramakrishna S, Kishan V, Rao YM, Diwan PV. 2007. Development and evaluation of nitrendipine loaded solid lipid nanoparticles: influence of wax and glyceride lipids on plasma pharmacokinetics. Int J Pharm. 335:167–175.
- Maghraby GMME, Williams AC, Barry BW. 2000. Skin delivery of oestradiol from lipid vesicles: importance of liposome structure. Int J Pharm. 204:159–169.
- Manconi M, Caddeo C, Sinico C, Valenti D, Mostallino MC, Biggio G, Fadda AM. 2011. Ex vivo skin delivery of diclofenac by transcutol containing liposomes and suggested mechanism of vesicle-skin interaction. Eur J Pharm Biopharm. 78:27–35.
- Manjunath K, Venkateswarlu V. 2006. Pharmacokinetics, tissue distribution and bioavailability of nitrendipine solid lipid nanoparticles after intravenous and intraduodenal administration. J Drug Target. 14:632–645.
- Mittal A, Sara US, Ali A, Mohammed A. 2009. Design, development, physicochemical, in vitro and in vivo evaluation of monolithic matrix type transdermal patches containing nitrendipine. Pharm Dev Technol. 14:422–434.
- Ongpipattanakul B, Burnette R, Potts PO. 1991. Evidence that oleic acid exists as a separate phase within stratum corneum. Pharm Res. 8:350–354.
- Patel NA, Patel NJ, Patel RP. 2009. Formulation and evaluation of curcumin gel for topical application. Pharm Dev Technol. 14:80–89.
- Pierre MBR, Tedesco AC, Marchett JM, Vitória M, Bentley L. 2001. Stratum corneum lipids liposomes for the topical delivery of 5- aminolevulinic acid in photodynamic therapy of skin cancer: preparation and in vitro permeation study. BMC Dermatol 1:5.
- Sinico C, Manconi M, Valenti M, Fadda AM. 2005. Liposomes as carriers for dermal delivery of tretinoin: in vitro evaluation of drug permeation and vesicle-skin interaction. J Control Release. 103:123–136.
- Srisuk P, Thongnopnua P, Raktanonchai U, Kanokpanont. S. 2012. Physico-chemical characteristics of methotrexate-entrapped oleic acid-containing deformable liposomes for in vitro transepidermal delivery targeting psoriasis treatment. Int J Pharm. 427:426–434.
- Tipre DN, Vavia PR. 2003. Acrylate-based transdermal therapeutic system of nitrendipine. Drug Dev Ind Pharm. 29:71–78.
- Torchilin VP, Lukyanov AN, Klibanov AL, Omelyancnkod VG. 1992. Interaction between oleic acid-containing pH-sensitive and plain liposomes. Fluorescent spectroscopy studies. FEBS Lett. 305:185–188.
- Touitou E, Godin B, Karl Y, Bujanover S, Becker Y. 2002. Oleic acid, a skin permeation enhancer, affects Langerhans cells and corneocytes. J Control Release. 80:1–7.
- Wang Z, Sun J, Wang Y, Liu X, Liu Y, Fu Q, Meng P, He Z. 2010. Solid self-emulsifying nitrendipine pellets: preparation and in vitro/in vivo evaluation. Int J Pharm. 383:1–6.
- Wertz PW, Abraham W, Landmann L, Downing DT. 1986. Preparation of liposomes from stratum corneum lipids. J Invest Dermatol. 87:582–584.
- Xia D, Quan P, Piao H, Piao H, Sun S, Yin Y, Cui F. 2010. Preparation of stable nitrendipine nanosuspensions using the precipitation—ultrasonication method for enhancement of dissolution and oral bioavailability. Eur J Pharm. Sci 40:325–334.
- Yamane MA, Williams AC, Barry BW. 1995. Effects of terpenes and oleic acid as skin permeation enhancers towards 5-fluorouracil as assessed with time; permeation, partitioning and differential scanning calorimetry. Int J Pharm. 116:237–251.
- Yang M, Cui F, You B, Wang L, Yue P, Kawashima Y. 2004. A novel pH-dependent gradient-release delivery system for nitrendipine: II. Investigations of the factors affecting the release behaviors of the system. Int J Pharm. 286:99–109.
- Zakir F, Vaidya B, Goyal AK, Malik B, Vyas SP. 2010. Development and characterization of oleic acid vesicles for the topical delivery of fluconazole. Drug Deliv. 17:238–248.