
Abstract
Ginsenosides are triterpenoid saponins, which is an active compound responsible for most of the pharmacological effects of ginseng (Panax ginseng Meyer). It is known to have numerous structural and pharmacological properties. However, aqueous solubility and delivery of ginsenosides in targeted region by avoiding undesirable toxicity to normal cell is also of prime importance. The aim of this study was to obtain amphiphilic ginsenoside derivatives in which hydrophilic polymers were conjugated to ginsenosides to enhance the water solubility and targeted delivery. To this end, the hydrophobic protopanaxadiol ginsenoside aglycone (aPPD) was covalently conjugated to the backbone of hydrophilic polyethylene glycol (PEG) through a pH sensitive ester linkage, which was confirmed by 1H NMR and FTIR. The resultant PPD is covalently conjugated to hydrophilic PEG through esterification (PEG-PPD) forming self-assembled spherical nanoparticles, whose average particle diameter was 189 nm as observed by FE-TEM and particle size analyzer respectively. In vitro release experiments revealed that the release rate of PPD was rapidly increased from the self-assembled nanoparticles under acidic conditions (pH 5.0) than in a physiological buffer (pH 7.4) condition. Furthermore, in vitro cytotoxicity assays revealed that PEG-PPD conjugates exhibited lower cytotoxicity in HT-29 cancer cells compared with PPD alone. Since the slow release of PPD from conjugates is triggered only by acidic environmental conditions, such as those found in extracellular solid tumor tissues, intracellular endosomes, and intracellular lysosomes, the conjugation of PPD may aid its selective delivery to these targets. Overall, results suggest that pH-dependent release of PPD, which expected in reduced cytotoxicity to non-targeted regions, may enhance the overall efficacy of PPD.
Introduction
Korean Ginseng (Panax ginseng Meyer) is a plant traditionally used in Asian medicine that is believed to cure all diseases. Ginsenosides are triterpenoid steroidal saponins (Subramaniyam et al. Citation2014), these active compounds have also been reported to mediate the various pharmacological effects of ginseng. Ginsenosides are classified as either protopanaxadiols (PPD) or protopanaxatriols (PPT), according to the manner in which their sugar chains are covalently linked with their triterpenoid aglycone by glycosidic bonding. Ginsenosides and ginseng extracts have been reported to exert various pharmacological activities, including antitumor activity, anti-inflammatory activity, and protective effects against Alzheimer’s disease and diabetes (Choi Citation2008, Sathishkumar et al. Citation2012).
However, orally-administered PPD-type ginsenosides and crude extracts of these major ginsenosides are predominantly hydrolyzed into minor ginsenosides, such as 20-O-β-(D-glucopyranosyl)-20(S)-protopanaxadiol (also called compound K or CK) and further aglycone protopanaxadiol (aglycone PPD or aPPD) via stepwise cleavage of their sugar moieties by intestinal gut microbiota. Both CK and PPD are major metabolites that reach systemic circulation (Ren et al. Citation2008). Compared with other ginsenosides, these metabolites have been reported to exert potent antitumor activities in various cell lines (Park et al. Citation2013). Moreover, the highest uptake ratios of aglycone PPD and CK have been reported in lysates from red ginseng (RG) extract-treated (10 and 50 μg/mL) MCF-7 cancer cells (Ha et al. Citation2010). Furthermore, the bioavailability of PPD has been shown to be very high compared with other ginsenosides, such as Rg3 and Rh2, which are currently used to treat cancer in China (Ren et al. Citation2008). In addition, PPD has undergone phase I clinical trials as an anticancer drug candidate (Saklani and Kutty Citation2008).
The utilization of multiple therapeutic drugs for the treatment of cancer and other related diseases appears to be a clinically valuable strategy. The aqueous solubility of a drug is one of the most important physiochemical factors regulating its absorption and distribution, and thus can determine how effective the drug is in treating these diseases. In addition, targeting the delivery of ginsenosides to specific sites of interest, thereby avoiding any unwanted toxicity to normal cells, is also of prime importance. Thus, polymeric nanoparticle-based approaches are needed for the ginsenosides PPD and CK to enhance their solubility and to achieve a more targeted delivery.
Various hydrophilic polymeric carriers (Cai et al. Citation2014, Deepagan et al. Citation2013, Li et al. Citation2013, Sivasubramanian et al. Citation2013a, Thambi et al. Citation2014a) have been utilized for efficient drug delivery. Polyethylene glycol (PEG) is a hydrophilic, non-toxic polymer, and non-ionic that is widely used as a polymeric nano carrier in drug delivery (Li et al. Citation2013). PEG is classified to be Generally Regarded as Safe (GRAS) by the US Food and Drug Administration (FDA). In addition, PEG also exhibits a low polydispersity index (PDI), which is a prerequisite for any polymer to have pharmaceutical applications. The conjugation of small molecular hydrophobic drugs to hydrophilic PEG molecules can result in the following benefits: increases water solubility of the hydrophobic drugs, protection from proteolytic digestion, prolonged circulation time of drugs in in vivo, and a reduction of non-targeted toxicity (Cai et al. Citation2014, Li et al. Citation2013). Owing to these unique features, a variety of FDA-approved PEG-based nanocarriers or drug conjugates are currently in different phases of clinical trials; some of these are even available in the market for treatment (Chao Deng, 2012, Cheng et al. Citation2012, Li et al. Citation2013). Another pertinent aspect of these PEGylated macromolecular conjugates is their ability to effectively reach tumors through passive targeting, since the tumors accumulate more conjugates due to their leaky vasculature. After reaching tumor sites via leaky endothelium tissue, the target drug is expected to be released at the tumor site following exposure to intracellular stimuli. Variation of the intracellular normal tissues and tumor tissues pH, have garnered increased pH-responsive drug conjugates to improve the therapeutic efficacy of parent drugs (Min et al. Citation2012, Sivasubramanian et al. Citation2013b, Thambi et al. Citation2014b). We previously described the conjugation of Glycol chitosan and PEG with ginsenosides CK and its properties (Mathiyalagan et al. Citation2014a, Mathiyalagan et al. Citation2014b).
Here, we report a method by which the solubility of PPD can be readily enhanced. In this method, PPD is covalently conjugated to hydrophilic PEG through esterification (PEG-PPD). Successful conjugation was confirmed by 1H NMR and FTIR. The physicochemical characteristics of PEG-PPD, such as solubility (data not shown), particle morphology and particle size in aqueous solution were determined using FE-TEM and a particle size analyzer respectively. The cytotoxicity of PEG-PPD was also determined using HT-29 cells. Cumulatively, these results indicate that PEG-PPD facilitated the release of PPD into cancer cells.
Materials and methods
Materials
The ginsenoside PPD was purchased from the Ginseng Genetic Resource Bank (Kyung Hee University, Yongin, South Korea). Polyethylene glycol monomethyl ether (mPEG, MW = 2 kDa); dimethyl amino pyridine (DMAP); N, Ǹ-dicyclohexyl carbodiimide (DCC); triethylamine (TEA); anhydrous 1,4-dioxane; dimethylformamide (DMF); and succinic anhydride were purchased from Sigma Aldrich Co. (St. Louis, MO). All other chemicals were of analytical grade.
Preparation of PEG-PPD conjugates
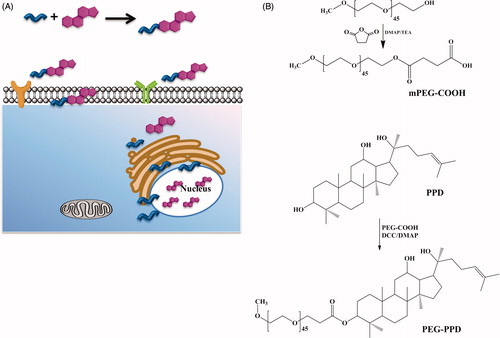
PEG-PPD conjugates were synthesized as described by Mathiyalagan et al. (Citation2014b). The synthesis involved a two-step process, as shown in . Briefly, the active carboxyl-terminus of PEG (α-carboxy-ω-methoxy polyethylene glycol - mPEG-COOH) was first prepared using succinic anhydride and then conjugated to PPD. mPEG-COOH was synthesized as follows: mPEG (MW 2000 g/mol; 1 g, 0.5 mmol), succinic anhydride (0.06 g, 0.6 mmol), DMAP (0.061 g, 0.5 mmol), and TEA (0.05 g, 0.5 mmol) were dissolved in 10 mL of anhydrous dioxane. The resultant mixture was stirred for 24 h at room temperature. Next, mPEG-COOH was precipitated with diethyl ether, filtered, and then dried under a vacuum. The resultant mPEG-COOH (0.044 g, 0.02 mmol), DCC (0.06 g, 0.03 mmol), and DMAP were added to a stirred solution, in which DMAP was used as a catalyst and DCC was used as a condensation agent. The condensation agent was added to remove the water formed during esterification (Yang et al. Citation2008).
Figure 1. Schematic illustrations of PEG-PPD conjugate synthesis. (A) Graphical illustration of PPD release from PEG-PPD conjugate at the intracellular environment. (B) mPEG-COOH was initially prepared using succinic anhydride, DMAP, and TEA. Then mPEG-COOH, DCC, and DMAP with PPD were added to obtain PEG-PPD conjugate.
After 15 min, the PPD was dissolved in DMF, added to the mixture, and stirred overnight. Next, the reaction mixture was transferred to dialysis membrane (MWCO = 3500) and dialyzed against distilled water for 24 h. Finally, the dialyzed mixture was filtered through a syringe (0.45 μM) and lyophilized to obtain the PEG-PPD conjugates.
Characterization of the PEG-PPD conjugates
The structures of the PEG-PPD conjugates and their intermediates were confirmed by 1H NMR and FTIR. 1H NMR spectra were recorded at 300 MHz on a spectroscopy (JEOL, Tokyo, Japan); samples were dissolved in deuterated dimethylsulfoxide (DMSO-d6). FTIR spectra were recorded using a Perkin-Elmer Spectrum One FTIR spectrophotometer (Boston, U.S.A); samples were prepared using the KBr pellet method. The morphology of the conjugates was observed on a field emission transmission electron microscope (FE-TEM) (JEM-2000F; JEOL Tokyo, Japan), operated at an acceleration voltage of 200 kV. For FE-TEM observations, one drop of sample solution was placed onto a 200-mesh carbon-coated copper grid and allowed to air dry. A drop of phosphotunstic acid (PTA) solution was used for negative staining and air dried. The particle size distributions of the PEG-PPD conjugates were determined by a particle analyzer, after dispersing the particles in phosphate-buffered saline (PBS, pH 7.4).
In vitro pH-dependent release of PPD from the PEG-PPD conjugates
The rates of hydrolysis and release of PPD from the PEG-PPD conjugates were determined at different pHs, including pH 5.0 (10 mM acetate buffer) and pH 7.4 (0.1X PBS). The PEG-PPD conjugates were dissolved in different buffers and gently stirred at 37°C for 24, 48, or 72 h. At predetermined intervals, samples were withdrawn and extracted with water-saturated n-BuOH. The amount of PPD hydrolyzed from the conjugate was monitored by HPLC [Agilent, C18 column (3.0 × 50 mm, particle size 2.7 μm)] with acetonitrile (solvent A) and distilled water (solvent B) as the mobile phases and a flow rate of 1.0 mL/min. UV detection was performed at 203 nm.
In vitro cytotoxicity assay
The human colon cancer (HT-29) cell line purchased from the Korean Cell Line Bank (Seoul, Korea) was cultured in RPMI 1640 medium (Welgene, Inc., Daegu, South Korea) containing 10% (v/v) fetal bovine serum (Welgene, Inc.) and 1% (w/v) penicillin-streptomycin (Welgene, Inc.). Cells were maintained in an atmosphere of 5% CO2 and 95% air at 37°C. HT-29 cells were seeded at a density of 2 × 104 cells/well in 96-well flat-bottomed plates (SPL Life Sciences, Gyeonggi-do, Korea). After one day of growth, cells were deprived of serum for 12 h; then, the culture medium was replaced with 100 μL of various concentrations of either free PPD or PEG-PPD. Cells were then incubated for 24 h at 37°C. Next, 10 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tertazolium bromide (MTT) was added, and the plates were incubated for an additional 3 h at 37°C. The culture medium was then discarded, and the resultant blue formazan products were dissolved in 100 μL of DMSO. The optical densities of the products were then measured at 570 nm using a Synergy HT multidetection microplate reader (Molecular Devices, BioTek Instruments, Winooski, VT). Three biological replicates were measured for each sample, and each experiment was performed in triplicate.
Statistical analysis
The statistical significances of differences between the control versus PPD and PEG-PPD groups were determined using Student’s t-test. p Values <0.01 were considered to be significant, and are indicated in the figures with asterisks (*).
Results and discussion
To enhance the solubility and more efficiently target ginsenosides to tumor sites, we endeavored to prepare polymeric ginsenosides as self-assembled nanoparticles. To this end, acid-labile ester linkages were used to conjugate PEG to the surface of the PPD. The selective hydrolysis of these ester bonds is predicted to result in the triggered release of PPD in pathophysiological environments, such as tumor environments, as illustrated in .
Synthesis and characterization of PEG-PPD conjugates
Active carboxylated PEG (mPEG-COOH) was prepared with succinic anhydride and then conjugated onto the surface of hydrophobic PPD molecules, thereby yielding the PEG-PPD conjugates (). Successful esterification of mPEG was confirmed by 1H NMR and FTIR. The 1H NMR spectra of mPEG-COOH revealed the characteristic succinic acid peak at 2.6 ppm (). In addition, the FTIR spectra of mPEG-COOH revealed the presence of an intense carboxylic C = O bond at 1735 cm−1, which confirmed the successful esterification of mPEG ().
Figure 2. Characterization of PEG-PPD conjugates and its intermediates by 1H NMR spectra. (A) PEG-COOH, (B) PPD, and (C) PEG-PPD.
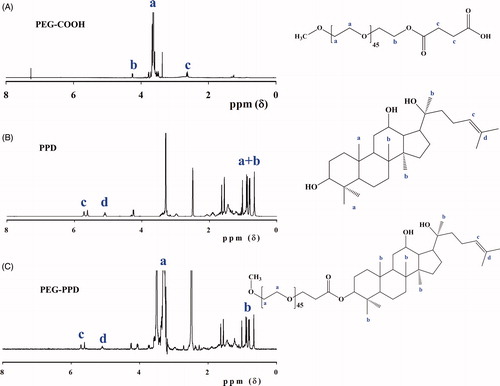
Figure 3. Characterization of PEG-PPD conjugates and its intermediates by FTIR spectra. (A) PEG-COOH, (B) PPD, and (C) PEG-PPD.
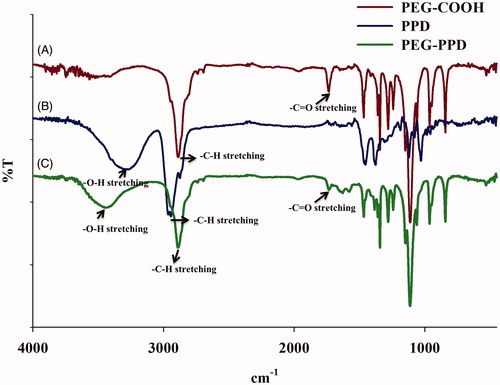
In the next step, mPEG-COOH was conjugated to PPD via the formation of ester bonds; this step was also confirmed by 1H NMR and FTIR. shows the 1H NMR spectra of PEG-PPD, which exhibited the characteristic peaks of PEG and PPD. The 1H NMR results indicated that the C-3 hydroxyl group of PPD aglycone underwent esterification. Compared with PPD alone (), the FTIR spectra of the PEG-PPD conjugates () exhibited an increased intensity of the peak at 1733 cm−1; this peak indicates the presence of a carboxylic bond (C = O) and the formation of an ester bond between mPEG-COOH and PPD. Covalent conjugation to PEG with ester bonds of several different anticancer drugs has been reported to confer improved antitumor efficacy and decreased toxicity, compared with their parent drugs (Cai et al. Citation2014, Lee et al. Citation2008, Li et al. Citation2009, Yang et al. Citation2008).
Size and morphology of the PEG-PPD conjugates
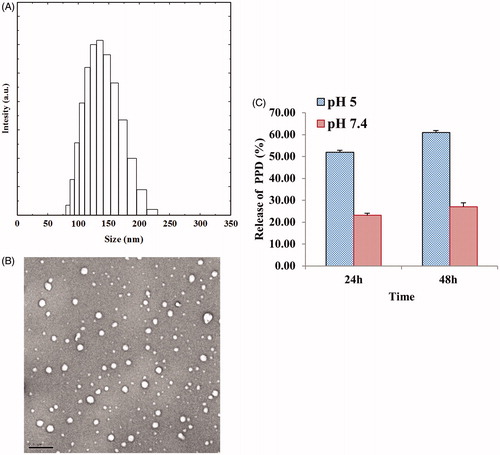
Owing to the amphiphilic nature of drug conjugates, i.e. the hydrophobic nature of the inner drug and the hydrophilic nature of the polymers, these conjugates spontaneously form self-assembled nanoparticles in an aqueous environment. Thus, since the PEG hydrophilic shell surrounds the inner core of the hydrophobic PPD, this arrangement could enhance the solubility of the drug and also protect it from proteolytic digestion, thereby enabling more prolonged half-life in vivo (Li et al. Citation2009). Accordingly, the solubility of the PEG-PPD conjugates in aqueous medium was greater than the solubility of PPD alone (data not shown). Examination of the morphology of the PEG-PPD conjugates by FE-TEM revealed that the conjugates were spherical in aqueous medium (). Moreover, a particle size analyzer determined that the average diameter of the PEG-PPD conjugates in aqueous medium was 189 ± 15.69 nm (). Other polymer drug conjugates have been reported to exhibit similar sizes (Lee et al. Citation2008). Due to differences in sample preparation, the average diameter of the conjugates differed depending on whether FE-TEM or the particle size analyzer was used for analysis. The samples were dried before the FE-TEM evaluation, whereas the particle size analyzer evaluated hydrated samples (Yu et al. Citation2008).
Figure 4. Physiochemical characterization of PEG-PPD conjugates. (A) Particle size of PEG-PPD conjugate. (B) The spherical shaped PEG-PPD conjugates observed in TEM images. The scale bar represents 200 nm. (C) In vitro pH-dependent release of PPD from PEG-PPD conjugates at different pH values. The error bar represent standard deviation (n=3).
In vitro pH-dependent release of PPD from the PEG-PPD conjugates
PEG conjugated macromolecular drugs are able to efficiently reach tumors, due to the leaky tumor vasculature; moreover, these conjugated drugs are expected to be released in the extracellular tumor tissue due to its mildly acidic environment. After entering cells by endocytosis, these spherical pegylated particles release their internal drugs upon exposure to the acidic conditions of intracellular endosomes and lysosomes (pH 4-6). The observation that extracellular and intracellular tumor tissues generally exhibit lower than physiological pH conditions (i.e. lower than pH 7.4), encouraged researchers to develop pH-responsive polymeric drug conjugates with the aim of improving the therapeutic efficacy of these drugs (Lee et al. Citation2008, Min et al. Citation2012, Thambi et al. Citation2011). Accordingly, the release of PPD from the PEG-PPD conjugates over time was monitored by incubating the samples at pH 7.4 (physiological) and pH 5.0 (pathophysiological) conditions. Although the ester bond conjugates did not completely block the release of PPD from the PEG-PPD conjugates, these ester bond conjugates inhibited the release of PPD at pH 7.4; however, increased rate of PPD release occurred at pH 5 (). The increased release under acidic pH conditions (pH 5.0) is representative of what would be expected in extracellular solid tumor tissues, intracellular endosomes, and intracellular lysosomes. Therefore, the pH-responsive release of PPD in conjugated form may be an effective mechanism by which PPD can be accurately targeted to pathophysiological sites, such as tumors.
In vitro cytotoxicity of the PEG-PPD conjugates
The in vitro anticancer activities of the PEG-PPD conjugates and their intermediates were examined in HT-29 cancer cells by the MTT assay. Due to the high biocompatibility of the hydrophilic PEG polymer, most of the cells were viable, and the polymer did not exhibit any significant cytotoxicity in cancer cells at 2, 20, or even 200 μM PEG and PEG-COOH (). On the other hand, PPD and PEG-PPD exhibited dose-dependent cytotoxicity in HT-29 cancer cells at 24 h (). The slightly lower cytotoxicity of PEG-PPD was presumably due to the slow discharge of PPD upon induction of hydrolysis by acidic conditions, which would be expected to occur in intracellular endosomes and lysosomes. This slow discharge might explain the slight delay in cytotoxicity of the PEG-PPD conjugates. In addition, free PPD might accumulate in cell nuclei after its addition to the culture medium. In contrast, PEG-PPD conjugates presumably enter HT-29 cells by endocytosis, and only activate the release of PPD upon acidic hydrolysis. Similarly, slower cytotoxicity of conjugates in in vitro and better antitumor activity in vivo was reported (Min et al. Citation2012).
Figure 5. In vitro cytotoxicity of (A) PEG, PEG-COOH and (B) free PPD and PEG-PPD on HT-29 cancer cells. (A) Different concentration of PEG and PEG-COOH did not show any significant cytotoxicity in cancer cells. (B) PEG-PPD exhibited lower toxicity than free PPD, which may be due to slow release of PPD upon hydrolysis ester linkage. Error bars represent the standard deviation (n=3). ***p<0.001 versusss control (untreated group).
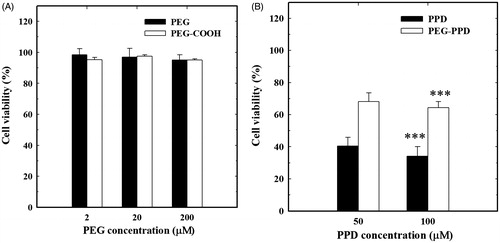
These results indicate that while unconjugated PPD is unable to distinguish between normal cells and cancer cells, the macromolecular PEG-PPD conjugates may selectively accumulate in cancer cells. Thus, the non-targeted cytotoxicity of the conjugates in normal cells may be greatly decreased.
In conclusion, nanoparticle-based approaches enabling the efficient utilization of multiple pharmacologically active herbal drugs for antitumor purposes are more advanced than those for chemotherapeutic purposes. Therefore, we successfully synthesized PEG-PPD conjugates resulted in enhanced solubility, which is most important physiochemical factor, for drug absorption and proper distribution. The release of PPD from the PEG-PPD conjugates was increased under acidic pH (pathophysiological) conditions compared with normal (physiological) conditions. The high drug solubility and pH-selectivity of the PEG-PPD conjugates might enhance their accumulation at pathophysiological sites. Thus, the conjugation of PPD to PEG may increase the overall efficacy of PPD, and make this ginsenoside a more promising drug candidate.
Acknowledgments
This research was fully supported by the Korea Institute of Planning & Evaluation for Technology in Food, Agriculture, Forestry & Fisheries (KIPET no: 313038-03-2-SB010) and also by a grant from the Next-Generation BioGreen 21 Program (SSAC, grant no: PJ00952903), Rural Development Administration, Republic of Korea.
Declaration of interest
There is no conflict of interest.
References
- Cai L, Qiu N, Xiang M, Tong R, Yan J, He L, et al. 2014. Improving aqueous solubility and antitumor effects by nanosized gambogic acid-mPEG2000 micelles. Int J Nanomedicine. 9:243–255.
- Chao Deng YJ, Ru Cheng, Fenghua Meng, Zhiyuan Zhong. 2012. Biodegradable polymeric micelles for targeted and controlled anticancer drug delivery: promises, progress and prospects. Nano Today. 7:467–480.
- Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. 2012. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 338:903–910.
- Choi KT. 2008. Botanical characteristics, pharmacological effects and medicinal components of Korean Panax ginseng C A Meyer. Acta Pharmacol Sin. 29:1109–1118.
- Deepagan VG, Thambi T, Ko H, Kang YM, Park JH. 2013. Amphiphilic polysialic acid derivatives: synthesis, characterization, and in-vitro cytotoxicity. J Nanosci Nanotechnol. 13:7312–7318.
- Ha YW, Ahn KS, Lee JC, Kim SH, Chung BC, Choi MH. 2010. Validated quantification for selective cellular uptake of ginsenosides on MCF-7 human breast cancer cells by liquid chromatography-mass spectrometry. Anal Bioanal Chem. 396:3017–3025.
- Lee H, Lee K, Park TG. 2008. Hyaluronic acid-paclitaxel conjugate micelles: synthesis, characterization, and antitumor activity. Bioconjug Chem. 19:1319–1325.
- Li J, Wang Y, Yang C, Wang P, Oelschlager DK, Zheng Y, et al. 2009. Polyethylene glycosylated curcumin conjugate inhibits pancreatic cancer cell growth through inactivation of Jab1. Mol Pharmacol. 76:81–90.
- Li W, Zhan P, De Clercq E, Lou H, Liu X. 2013. Current drug research on PEGylation with small molecular agents. Prog Poly Sci. 38:421–444.
- Mathiyalagan R, Subramaniyam S, Kim YJ, Kim YC, Yang DC. 2014a. Ginsenoside compound K-bearing glycol chitosan conjugates: synthesis, physicochemical characterization, and in vitro biological studies. Carbohydr Polym. 112:359–366.
- Mathiyalagan R, Subramaniyam S, Kim YJ, Natarajan S, Min JW, Kim SY, Yang DC. 2014b. Synthesis and pharmacokinetic characterization of a pH-sensitive polyethylene glycol ginsenoside CK (PEG-CK) conjugate. Biosci Biotechnol Biochem. 78:466–468.
- Min KH, Lee HJ, Kim K, Kwon IC, Jeong SY, Lee SC. 2012. The tumor accumulation and therapeutic efficacy of doxorubicin carried in calcium phosphate-reinforced polymer nanoparticles. Biomaterials. 33:5788–5797.
- Park B, Lee YM, Kim JS, Her Y, Kang JH, Oh SH, Kim HM. 2013. Neutral sphingomyelinase 2 modulates cytotoxic effects of protopanaxadiol on different human cancer cells. BMC Complement Altern Med. 13:194.
- Ren HC, Sun JG, Wang GJ, A JY, Xie HT, Zha WB, et al. 2008. Sensitive determination of 20(S)-protopanaxadiol in rat plasma using HPLC-APCI-MS: application of pharmacokinetic study in rats. J Pharm Biomed Anal. 48:1476–1480.
- Saklani A, Kutty SK. 2008. Plant-derived compounds in clinical trials. Drug Discov Today. 13:161–171.
- Sathishkumar N, Sathiyamoorthy S, Ramya M, Yang DU, Lee HN, Yang DC. 2012. Molecular docking studies of anti-apoptotic BCL-2, BCL-XL, and MCL-1 proteins with ginsenosides from Panax ginseng. J Enzyme Inhib Med Chem. 27:685–692.
- Sivasubramanian M, Thambi T, Deepagan VG, Saravanakumar G, Ko H, Kang YM, Park JH. 2013a. Carboxymethyl dextran-cyclodextrin conjugate as the carrier of doxorubicin. J Nanosci Nanotechnol. 13:7271–7278.
- Sivasubramanian M, Thambi T, Park JH. 2013b. Mineralized cyclodextrin nanoparticles for sustained protein delivery. Carbohydr Polym. 97:643–649.
- Subramaniyam S, Mathiyalagan R, Natarajan S, Kim YJ, Jang MG, Park JH, Yang DC. 2014. Transcript expression profiling for adventitious roots of Panax ginseng Meyer. Gene. 546:89–96.
- Thambi T, Deepagan VG, Yoo CK, Park JH. 2011. Synthesis and physicochemical characterization of amphiphilic block copolymers bearing acid-sensitive orthoester linkage as the drug carrier. Polymer. 52:4753–4759.
- Thambi T, You DG, Han HS, Deepagan VG, Jeon SM, Suh YD, et al. 2014a. Bioreducible carboxymethyl dextran nanoparticles for tumor-targeted drug delivery. Adv Healthcare Mater. 3:1829–1838.
- Thambi T, Park JH. 2014b. Recent advances in shell-sheddable nanoparticles for cancer therapy. J Biomed Nanotechnol. 10:1841–1862.
- Yang DB, Zhu JB, Huang ZJ, Ren HX, Zheng ZJ. 2008. Synthesis and application of poly(ethylene glycol)-cholesterol (Chol-PEGm) conjugates in physicochemical characterization of nonionic surfactant vesicles. Colloids Surf B Biointerfaces. 63:192–199.
- Yu JM, Li YJ, Qiu LY, Jin Y. 2008. Self-aggregated nanoparticles of cholesterol-modified glycol chitosan conjugate: preparation, characterization, and preliminary assessment as a new drug delivery carrier. Eur Pol J. 44:555–565.