
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The aim of present study was to develop VIN-loaded mPEG-PLA nanoparticle systems. The VIN mPEG-PLA nanoparticles were prepared using an emulsion solvent evaporation method, and studied their particle size, morphology, encapsulation efficiency and drug-loading coefficient. Moreover, the nanoparticles were evaluated on the drug release behaviors in vitro and bioavailability in vivo. The results show that the spherical nanoparticles obtained were negatively charged with a zeta potential of about −23.4 mV and characterized ∼110 nm with a narrow size distribution. The encapsulation efficiency and drug loading of prepared NPs were 76.4 ± 6.3 and 9.2 ± 2.2% (n=5), respectively. The in vitro release showed that the percent of accumulated dissolution of VIN NPs in phosphate-buffered saline 6.8 over 24 h was <80%, which was almost 100% of VIN in commercial injections. The in vivo study indicated that systemic absorption of VIN was significantly enhanced by incorporating into mPEG–PLA NPs compared with VIN injection (2.87-fold in AUC0–t). The results suggested that the form of VIN in mPEG–PLA NPs could enter the body circulation to perform sustained release in vitro and in vivo.
Introduction
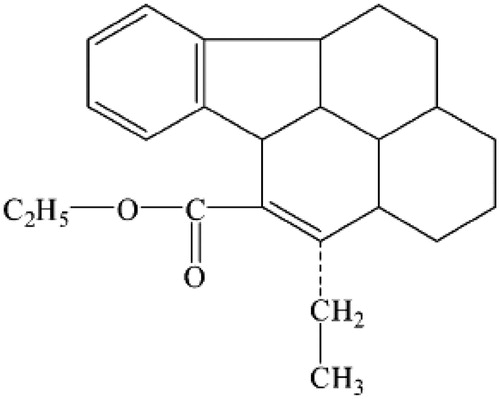
Vinpocetine (VIN), the chemical structure of which is shown in , is a vincamine derivative used for the treatment of chronic cerebral vascular ischemia, acute stroke, senile cerebral dysfunction and Alzheimer’s disease (Bönöczk et al. Citation2000, Chiu et al. Citation1988, Subhan and Hindmarch Citation1985). Over last several decades, it has been found high clinical value and little adversary effects (Hindmarch et al. Citation1991). However, due to its poor aqueous solubility (water solubility ∼5 μg/mL) (Miskolczi et al. Citation1990) and a short half-life time (∼2 h) (Polgár et al. Citation1985) and extensively metabolized during first pass, its clinical use is greatly restricted by the low bioavailability after oral administration (the oral bioavailability of VIN in human is as low as 7%) (Pudleiner and Vereczkey Citation1993, Szakács et al. Citation2001). Therefore, a kind of controlled release dosage form for parenteral administration is highly desirable because it may provide enhanced clinical value over conventional oral formulations as a result of possibly promoted bioavailability (Qiu et al. Citation2003).
Figure 1. The structure of VIN.
Nanoscale vehicles have also been used as a versatile carrier for the delivery of therapeutic drug (Fabregat et al. Citation2014). Polymeric nanoparticles have recently received considerable attention as a promising nanoscale delivery system (Rancan et al. Citation2014, Sada Tabatabaei Mirakabad et al. Citation2014, Xiong et al. Citation2014). Polymeric nanoparticles consist of a hydrophobic inner core surrounded by a hydrophilic outer shell, which are easy to prepare and can incorporate various drugs into their inner cores with relatively high stability by chemical conjugation or physical entrapment (Lavasanifar et al. Citation2002, Kataoka et al. Citation2001, Nishiyama and Kataoka Citation2006).
Some characteristics can be the prerequisites for a nano drug delivery system: (i) a low cytotoxicity and the possibility of biodegradability of the vector itself, (ii) versatile surface functionalization, (iii) a high drug-loaded content related with elevated therapeutic efficacy, (iv) a good dispersibility and colloidal stability of the vector in physiological conditions (Adair et al. Citation2010, Elsabahy and Wooley Citation2012, Kievit and Zhang Citation2011).
As one of the most prominent biodegradable polymers, polylactic acid (PLA) is widely used in the fields of drug delivery and tissue engineering due to its non-toxic, biocompatible and biodegradable characteristics (Chen et al. Citation2006, Garlotta Citation2001, Jain et al. Citation2009, Nagarwal et al. Citation2009). Polyethylene glycol (PEG), a FDA-approved polymer highly soluble in water, is the most popular hydrophilic outer shell, has been used as a source of hydrophilicity due to its outstanding physicochemical and biological properties, including solubility in water and organic solvents, non-toxicity, and providing a stabilizing interface between the nanoparticle core and the aqueous phase (Chen et al. Citation2006, Discher and Ahmed Citation2006, Layre et al. Citation2006, Shah et al. Citation2012, Veronese and Pasut Citation2005, Walkey et al. Citation2012). PEG–PLA nanoparticles have been used as carriers of hydrophobic compounds to increase solubility and loading capability (Nishiyama and Kataoka, Citation2006). The further development of PEG–PLA, monomethyl poly(ethylene glycol)-poly(d,l-lactide) (mPEG-PLA) diblock copolymers have been of great interest as a completely biocompatible material for drug delivery (Dong and Feng Citation2004, Zhang et al. Citation2005). Moreover, mPEG-PLA could make long circulation possible for pharmaceutical uses and opened new perspectives for controlled drug delivery in particular.
In this study, the preparation of VIN-loaded mPEG-PLA nanoparticle (VIN-NPs) systems was optimized by an emulsion solvent evaporation method and characterized the nanoparticles in terms of particle size, morphology, encapsulation efficiency and drug-loading coefficient. Moreover, the nanoparticles were evaluated on the drug release behaviors in vitro and bioavailability in vivo.
Materials and methods
Materials
mPEG (Mn = 2000; Fluka, MO), PLA was purchased from Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, China. mPEG-PLA were synthesized according to our previous work and molecular weight of mPEG-PLA is 2000 (mPEG)–30,000 (PLA) (determined by 1H-NMR). VIN was obtained from Dongbei Pharmaceutical Co. (Shenyang, China). Diazepam (internal standard) was obtained from National Institute for Food and Drug Control (purity >98%; Beijing, China). Sodium cholate and Tween-80 were supplied by Shanghai Chemical Reagent Company (Shanghai, China). All other chemicals used were at least of analytical grade.
Preparation of VIN-NPs
VIN-NPs were prepared by an emulsion solvent evaporation method. The desired amounts of VIN (1 mg) and mPEG-PLA (22 mg) were mixed with a sprinkle of dichloromethane (1.5 mL). Emulsifiers [sodium cholate (1%)/Tween-80(0.5%)] were dispersed in distilled water with magnetic stirring at the indoor temperature. After completely dissolved, the organic phase was then slowly added to water phase (20 mL) and then the coarse emulsion was subjected to 400 W of ultrasonic treatment for 2 min using a high-intensity probe ultrasonicator with water bath (0 °C). After the dichloromethane had completely evaporated (40 °C rotary evaporation), 1 mL of aqueous mannitol (20%, w/v) was added to prevent the aggregation of microparticles, the NPs were freeze dried.
Morphology
The morphology of VIN-NPs solution was observed by transmission electron microscopy (TEM, Hitachi H-100; Japan). The sample demanded was prepared by placing a drop of VIN-NPs which was diluted 50-fold with double-distilled water onto a 400-mesh copper grid coated with carbon film and followed by negative staining with 1.5% (w/v) phosphotungstic acid. Then, the sample was dried in the air before TEM observation.
Particle size and zeta potential
The average particle size, PDI and zeta potential of VIN-NPs were determined under room temperature by a dynamic light scattering (DLS) instrument (Nicomp 380 ZLS; PSS, Santa Barbara, CA) operating at 633 nm. The light scattering angle was set at 90 °, Sartorius Stedim Biotech, Goettingen, Germany. The particle size analysis data obtained were evaluated using the volume distribution. The samples were allowed to dilute with double-distilled water prior to zeta potential determination.
Encapsulation efficiency and drug loading
Encapsulation efficiency (EE) of VIN-NPs was calculated by determining the amount of free drug using hypothermy ultrafiltration technique. One milliliter of VIN-NPs solution was placed in the upper chamber of a centrifuge tube matched with an ultrafilter [Amicon Ultra, Millipore Co., MA; molecular weight cut-off (MWCO) 10 kDa] and centrifuged for 10 min at 4000 rpm. The ultrafiltrate containing the unencapsulated drug was determined by high-performance liquid chromatography (HPLC). The drug entrapment efficiency and drug loading was calculated by the following equations:
WVIN represents the amount of VIN-loaded in the NPs, WTotal represents the total VIN amount added during preparation of the NPs and WVIN–NPs represents the weight of the VIN-NPs.
In vitro release studies
The in vitro release studies of VIN from NPs were carried out by dialysis method (Levy and Benita Citation1990, Washington Citation1990). The release medium was phosphate-buffered saline (PBS) pH 6.8 containing 0.5% Tween-80 at 37 °C. And the stirring speed was set at 50 rpm. As previously reported, the MWCO of dialysis bags were 8–10 kDa. Aliquots of 2 mL of VIN-NPs were directly placed into dialysis bags and then released in 200 mL tubes of release medium (200 mL). Samples of 2 mL were removed from the tubes at sampling times of 1, 2, 4, 6, 8, 10, 12, 14, 20 and 24 h after centrifugation at 4000 rpm for 5 min. Then the VIN was analyzed by HPLC. Meanwhile, the release of VIN solution (2 mL, 50 μg/mL) was monitored as control. The accumulative release was calculated, and the results were shown as mean ± standard deviation (SD) (n=3).
Stability studies
A sufficient quantity of VIN-NPs was stored in a desiccator containing saturated solution of sodium chloride, which gave a relative humidity of 75 ± 5%. The desiccator was placed in a hot air oven maintained at 25 ± 2 °C, and samples were withdrawn at 0, 1, 2, 3 and 6 months. The drug content remaining and physical stability of formulation were measured at predetermined time interval.
Pharmacokinetic study
All animal experiments were carried out according to the Principles of Laboratory Animal Care (People’s Republic of China), and the protocols for the animal studies were approved by the Department of Laboratory Animal Research. Male Wistar rats weighing 220–250 g were obtained from Animal experimental center of China Medical University and housed at temperature of 22 ± 2 °C and 45–50% relative humidity. All the rats were divided randomly into two groups comprising six animals in each and fasted overnight but allowed to free access to water before experiment. The rats were given VIN-NPs (5 mg/kg, mixed into normal saline) or an equivalent dose of VIN injection, respectively. Blood samples (0.5 mL) of each animal were sampled via the suborbital vein at 0, 0.083, 0.25, 0.5, 1, 2, 4, 6, 8, 12 and 24 h after administration. All the blood samples were immediately centrifuged at 4000g for 10 min to separate the plasma. The plasma obtained was stored at −20 °C until analysis.
The pharmacokinetic parameters were calculated using non compartmental pharmacokinetic model. Pharmacokinetic variables of interest included area under the concentration-time curve from time 0 to ∞ (AUC0–∞), peak concentration (Cmax), terminal elimination rate constant (Ke), half-life of the terminal phase (T1/2) and time to peak concentration (Tmax). A linear-up/log down method of estimation was used for the calculation of AUC0–∞ and was obtained by adding Clast/Ke to AUC0–t. The terminal elimination rate constant (Ke) was determined from the slope of terminal exponential phase of the logarithmic plasma concentration-time curve. The elimination half-life (T1/2) was calculated using 0.693/Ke.
HPLC analysis
The concentrations of VIN in vitro and in vivo were determined by HPLC (Elbary et al. Citation2002). The HPLC system was composed of a model LC-10AT pump (Shimadzu Corporation, Kyoto, Japan) and a model SPD-10A UV detector (Shimadzu Corporation). The analytical column was Diamonsil C18 (200 × 4.6 mm, 5 μm) (Dikma Technologies Inc., Lake Forest, CA). The injection volume was 20 μL; the mobile phase was methanol: 0.1 mol/L ammonium carbonate aqueous solution (90/10, v/v); the flow rate was 1.0 mL/min; the UV detector wavelength was 273 nm; the column temperature was 35 °C.
Plasma samples were processed as follows: a 200 μL plasma sample was mixed with 20 μL of an internal standard (diazepam) solution (2 μg/mL) and 100 μl 0.5M NaOH and vortexed for 1 min. Then, 2 mL of ether anhydrous was added and the mixture was vortexed at room temperature for 5 min. After centrifugation at 3000g for 10 min, the organic layer was transferred to a new tube and evaporated under a stream of high purity nitrogen at 40 °C. The residue was reconstituted with 100 μL of mobile phase by vortexing for 1 min and 20 μL was analyzed by HPLC.
Statistical analysis
All values obtained were expressed as mean ± standard error mean (SEM). Statistical comparisons were performed by analysis of variance and Student’s t-test using SAS Version 8.0 software (North Carolina). A value of P < 0.05 was considered statistically significant.
Results and discussion
HPLC assay validation
The analytical performance of the HPLC system depended on the analytes used for the estimate. In this study, the in vivo assay was linear ranging from 200 to 3500 ng/mL. The standard curve of VIN gave Y = 28.1C − 1.76 (n = 5, r = 0.9996). The intra-day and inter-day precision coefficients of variation (RSD%) in vivo ranged from 9.8 to 13.4% at the three different concentrations (300, 1000 and 3000 ng/mL). The average recoveries of the analytes were >93.2% in vivo. And there were no interfering peaks observed in all the chromatogram. It was noteworthy that the HPLC technique, although simple, was an effective method to analyze VIN in NPs formulation.
Preparation and NPs characterization
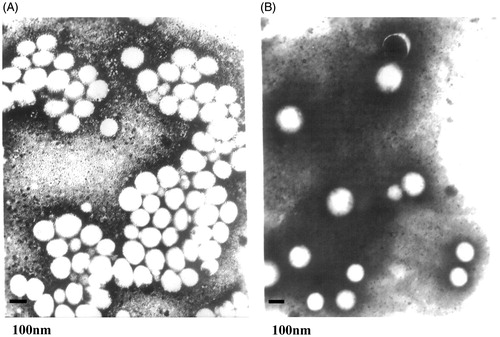
The morphology of VIN-mPEG–PLA NPs was visualized by transmission electron micrograph (TEM). As shown in , the bright entities were surrounded by dark staining, which clearly indicated the presence of spherical particulates. The NPs were discrete; fairly uniform in size. Additionally, the TEM of NPs also presented good spherical shape after 6 months storage ().
Figure 2. 50 000 × magnified TEM of normally prepared VIN-mPEG–PLA NPs.
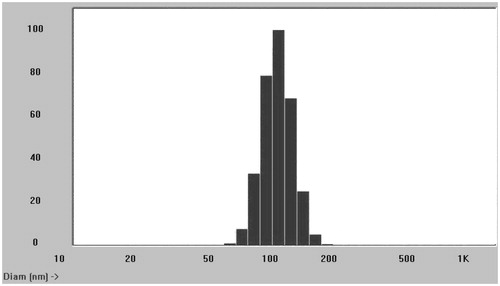
Particle size and zeta potential were main factors for in vivo circulation of nanoparticles (Domínguez et al. Citation1997). The VIN mPEG–PLA NPs were characterized by DLS in terms of mean particle size, PDI and zeta potential. Prepared NPs had a mean size of 110.6 ± 13.2 nm with a PDI 0.26 suggesting good polydispersity (). The zeta potential of NPs was found to be −23.4 ± 1.5 mV. The effect on the size and PDI of mPEG–PLA NPs with respect to ultrasonic power and polymer concentration were also studied. At a lower power of 200 W, the size of the NPs was 223.5 nm, and it was reduced to 110.6 nm at an ultrasonic power of 400 W, but the size maintained with 108.3 nm when the stirring speed was accelerated to 600 rpm. The size also increased slightly with increased mPEG–PLA concentration but no significant difference. The results show that VIN has a relative high entrapment efficiency in mPEG–PLA NPs. The EE and drug loading of prepared NPs were 76.4 ± 6.3 and 9.2 ± 2.2% (n=5), respectively. When the amounts of the emulsifiers increased, the entrapment efficiency decreased due to the solubilization of the emulsifiers.
Figure 3. Mean particle size of prepared VIN mPEG–PLA NPs.
depicts results of stability study of VIN mPEG–PLA NPs. No microscopical physical changes were observed during storage (). Mean particle size, zeta potential and EE values of the NPs carried out at 6 months showed no significant difference. Hence, it was concluded that the VIN mPEG–PLA NPs exhibited good stability.
Table 1. Stability data of VIN mPEG–PLA NPs during 6 months.
In vitro release
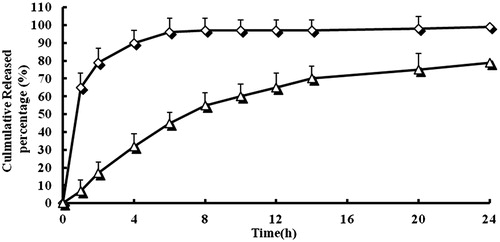
shows the in vitro cumulative release of VIN from the NPs compared to that of the VIN injection. From the release curves of VIN NPs in pH 6.8 PBS, we concluded that NPs showed no burst release at initial stage in medium, which was evidence that there was no unencapsulated drug attaching to the surface of the particles. Moreover, a sustain release profile was demonstrated. The percent of accumulated dissolution of VIN NPs in PBS 6.8 over 24 h was <80%, which was almost 100% of VIN in commercial injections. In fact, 98% of VIN in injections were released in the first 6 h and a very fast release behavior of free VIN was observed. The results suggested that the form of VIN in mPEG–PLA NPs could enter the body circulation to perform sustained release in vivo.
Figure 4. In vitro release profiles of VIN mPEG–PLA NPs from three batches. Release experiments were carried out in PBS (pH 7.4), at 37 ± 0.5 °C. Each point represents the mean value of three different experiments ± SD. ⋄: VIN injection; △: VIN mPEG–PLA NPs.
The in vitro release was kinetically analyzed according to first-order, Baker and Lonsdale, Ritger Peppas and Higuchi diffusion-controlled release mechanism. The correlation coefficient value, R2, is taken into account to decide upon the relevance of the model/curve fit which will best describe the extent of fit. According to the R2 values given by different data fits for VIN NPs, the Ritger Peppas model was to be an ideal fit having R2 = 0.9993. According to Ritger Peppas fit, the release of the drug is decided upon drug diffusion and polymeric matrix erosion.
Pharmacokinetic study
The mean plasma concentration (in value)–time profiles of VIN mPEG–PLA NPs and VIN injections are shown in . The pharmacokinetic parameters were calculated by the pharmacokinetic software 3P97 and summarized in . The Cmax value of VIN mPEG–PLA NPs was 3327.7 ng/ml, which was significantly higher than that obtained with the VIN injection (2543.5 ng/mL) (P<0.05). Plasma half-lives of the NPs groups were also prolonged compared to that of the injection group (5.64 h versus 2.32 h, P<0.05). The AUC0–t after intravenous administration of VIN mPEG–PLA NPs was 13261.5 ± 236.3 ng h/mL, which was ∼2.87-fold higher than that of VIN injection (4625.3 ± 543.5 ng/mL/h). The same phenomenon was found in the value of AUC0-∞. The results indicated that systemic absorption of VIN was significantly enhanced by incorporating into mPEG–PLA NPs compared with VIN injection. There were also significantly different in AUC0–t, AUC0–∞, MRT and CL between the VIN mPEG–PLA NPs and VIN injection (P<0.05). The mPEG–PLA NPs showed a promising potential for enhancing absolute bioavailability of poorly water-soluble drugs. Note: t1/2, Plasma half-lives; Cmax, maximum plasma concentration; AUC0–t, Area under the concentration–time curve from time 0 to the last measurable concentration; AUC0–∞, area under the concentration–time curve extrapolated to infinity; MRT, mean residence time; CL, plasma clearance.
Figure 5. Mean plasma concentration (in value)–time profiles of VIN mPEG–PLA NPs and VIN injections. Each point represents the mean value of six SD rats. ⋄: VIN injection; △: VIN mPEG–PLA NPs.
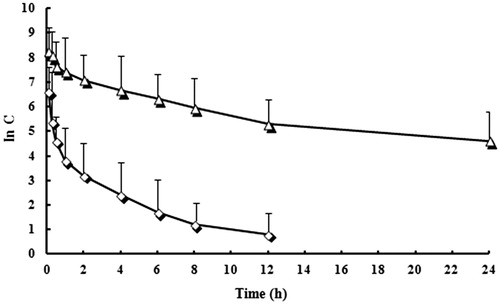
Table 2. Pharmacokinetic parameters of VIN mPEG–PLA NPs and VIN injections (n=6).
Conclusions
In this study, a poorly aqueous-soluble drug VIN was successfully incorporated into mPEG–PLA NPs by an emulsion solvent evaporation method. The physicochemical characterization (morphology, particle size, zeta potential, EE and drug loading) and physical stability were investigated. The in vitro release study showed that the release velocity of mPEG–PLA NPs is always slower compared with the diffusion rate of the VIN injection. In vivo study showed that mPEG–PLA NPs were a promising potential for enhancing absolute bioavailability of poorly water-soluble drugs (VIN).
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Adair JH, Parette MP, Altinoglu EI, Kester M. 2010. Nanoparticulate alternatives for drug delivery. ACS Nano. 4:4967–4970.
- Bönöczk P, Gulyás B, Adam-Vizi V, et al. 2000. Role of sodium channel inhibition in neuroprotection: effect of vinpocetine. Brain Res Bull. 53:245–254.
- Chen C, Yu CH, Cheng YC, et al. 2006. Biodegradable nanoparticles of amphiphilic triblock copolymers based on poly(3-hydroxybutyrate) and poly(ethylene glycol) as drug carriers. Biomaterials. 27:4804–4814.
- Chen GX, Kim HK, Kim ES, Yoon JS. 2006. Synthesis of high-molecular-weight poly (L-lactic acid) through the direct condensation polymeriza tion of L-lactic acid in bulk state. Eur Polym J. 42:468–472.
- Chiu PJ, Tetzloff G, Ahn HS, Sybertz EJ. 1988. Comparative effects of vinpocetine and 8-Br-cyclic GMP on the contraction and 45Ca-fluxes in the rabbit aorta. Am J Hypertens. 1:262–268.
- Discher DE, Ahmed F. 2006. Polymersomes. Annu Rev Biomed Eng. 8:323–341.
- Domínguez A, Fernandez A, González N, et al. 1997. Determination of critical micelle concentration of some surfactants by three techniques. J Chem Educ. 74:1227–1231.
- Dong Y, Feng SS. 2004. Methoxy poly(ethylene glycol)-poly(lactide) (MPEG-PLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials. 25:2843–2849.
- Elbary A, Foda N, El-Gazayerly O, Khatib ME. 2002. Reversed phase liquid chromatographic determination of vinpocetine in human plasma and its pharmacokinetic application. Anal Lett. 35:1041–1054.
- Elsabahy M, Wooley KL. 2012. Design of polymeric nanoparticles for biomedical delivery applications. Chem Soc Rev. 41:2545–2561.
- Fabregat G, Casanovas J, Redondo E, et al. 2014. A rational design for the selective detection of dopamine using conducting polymers. Phys Chem Chem Phys. 16:7850–7861.
- Garlotta DA. 2001. A literature review of poly(lactic acid). J Polym Environ. 9:63–84.
- Hindmarch I, Fuchs HH, Erzigkei H. 1991. Efficacy and tolerance of vinpocetine in ambulant patients suffering from mild to moderate organic psychosyndromes. Int Clin Psychopharmacol. 6:31–43.
- Jain AK, Goyal AK, Gupta PN, et al. 2009. Synthesis, characterization and evaluation of novel triblock copolymer based nanoparticles for vaccine delivery against hepatitis B. J Control Release. 136:161–169.
- Kataoka K, Harada A, Nagasaki Y. 2001. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev. 47:113–131.
- Kievit FM, Zhang M. 2011. Cancer nanotheranostics: improving imaging and therapy by targeted delivery across biological barriers. Adv Mater Weinheim. 23:H217–H247.
- Lavasanifar A, Samuelm J, Kwon GS. 2002. Poly(ethylene oxide)-block-poly(L-amino acid) micelles for drug delivery. Adv Drug Deliv Rev. 54:169–190.
- Layre A, Couvreur P, Chacun H, et al. 2006. Novel composite core-shell nanoparticles as busulfan carriers. J Control Release. 111:271–280.
- Levy MY, Benita S. 1990. Drug release from submicronized o/w emulsion: a new in vitro kinetic evaluation model. Int J Pharm. 66:29–37.
- Miskolczi P, Korma K, Polgár M, Vereczkey L. 1990. Pharmacokinetics of vinpocetine and its main metabolite apovincaminic acid before and after the chronic oral administration of vinpocetine to humans. Eur J Drug Metab Pharmacokinet. 15:1–5.
- Nagarwal RC, Kant S, Singh PN, et al. 2009. Polymeric nanoparticulate system: a potential approach for ocular drug delivery. J Control Release. 136:2–13.
- Nishiyama N, Kataoka K. 2006. Nanostructured devices based on block copolymer assemblies for drug delivery: designing structures for enhanced drug function. Polym Ther II. 193:67–101.
- Polgár M, Vereczkey L, Nyáry I. 1985. Pharmacokinetics of vinpocetine and its metabolite, apovincaminic acid, in plasma and cerebrospinal fluid after intravenous infusion. J Pharm Biomed. Anal 3:131–139.
- Pudleiner P, Vereczkey L. 1993. Study on the absorption of vinpocetine and apovincaminic acid. Eur J Drug Metab Pharmacokinet. 18:317–321.
- Qiu Y, Garren J, Sammara E, et al. 2003. Once-a-day controlled-release dosage form of divalproex sodium II: development of a predictive in vitro drug release method. J Pharm Sci. 92:2317–2325.
- Rancan F, Blume-Peytavi U, Vogt A. 2014. Utilization of biodegradable polymeric materials as delivery agents in dermatology. Clin Cosmet Investig Dermatol. 7:23–34.
- Sadat Tabatabaei Mirakabad F, Nejati-Koshki K, Akbarzadeh A, et al. 2014. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac J Cancer Prev. 15:517–535.
- Shah NB, Vercellotti GM, White JG, et al. 2012. Blood-nanoparticle interactions and in vivo biodistribution: impact of surface PEG and ligand properties. Mol Pharm. 9:2146–2155.
- Subhan Z, Hindmarch L. 1985. Psychopharmacological effects of vinpocetine in normal healthy volunteers. Eur J Clin Pharmacol. 28:567–571.
- Szakács T, Veres Z, Vereczkey L. 2001. In vitro-in vivo correlation of the pharmacokinetics of vinpocetine. Pol J Pharmacol. 53:623–628.
- Veronese FM, Pasut G. 2005. PEGylation, successful approach to drug delivery. Drug Discov Today. 10:1451–1458.
- Walkey CD, Olsen JB, Guo H, et al. 2012. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 134:2139–2147.
- Washington C. 1990. Drug release from microdisperse systems: a critical review. Int J Pharm. 58:1–12.
- Xiong MH, Bao Y, Yang XZ, et al. 2014. Delivery of antibiotics with polymeric particles. Adv Drug Deliv Rev. 78:63–76.
- Zhang X, Li Y, Chen X, et al. 2005. Synthesis and characterization of the paclitaxel/MPEG-PLA block copolymer conjugate. Biomaterials. 26:2121–2128.