
Abstract
Objectives:
Few studies have characterized healthcare resource utilization among patients with idiopathic pulmonary fibrosis. The objective of this study is to assess healthcare resource utilization among patients with idiopathic pulmonary fibrosis as compared to members without this condition.
Methods:
Patients newly diagnosed with idiopathic pulmonary fibrosis were identified from a national administrative claims database (2006–2011) as having ≥2 claims with idiopathic fibrosing alveolitis, or ≥1 claim with idiopathic fibrosing alveolitis and ≥1 claim with post-inflammatory pulmonary fibrosis (earliest claim with idiopathic fibrosing alveolitis denoted the index date), a procedure of lung biopsy or high-resolution computed tomography within ±90 days of the index date, 12-month pre-index continuous enrollment, plus ≥2 confirmatory idiopathic fibrosing alveolitis diagnoses after the procedure. For each idiopathic pulmonary fibrosis patient, three members without the condition were matched by age/gender/region/payer type. Demographic/clinical characteristics were measured during the 1-year pre-index period. Healthcare resource utilization was assessed by quarter during 1-year pre- and post-index periods. Generalized estimating equation models controlling for patient characteristics were constructed to estimate adjusted post-index healthcare resource utilization.
Results:
In total, 1735 patients with idiopathic pulmonary fibrosis and 5205 without (mean age = 71.5 years; 46.1% female) were included. Adjusted results revealed idiopathic pulmonary fibrosis patients were more likely to use healthcare resources than members without the condition 1-year post-index (number of hospitalizations, emergency room visits, and outpatients visits: 0.63 vs 0.31, 0.62 vs 0.48, and 5.7 vs 3.1 per person-year, respectively).
Conclusions:
Healthcare resource utilization is considerably higher among patients with idiopathic pulmonary fibrosis than members without the condition. Effective treatments for patients with idiopathic pulmonary fibrosis are needed to help reduce burden of healthcare resource use.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive and fatal lung disease characterized by symptoms such as breathlessness and severe coughing, with no effective treatment, and a median duration of survival between 2–3 years from diagnosisCitation1–3. Patients with IPF typically have high comorbidity burden including gastrointestinal esophageal reflux disease, coronary artery disease, lung cancer, and obstructive sleep apneaCitation4–9.
The public health burden of IPF has been largely characterized by estimates of incidence, prevalence, and mortalityCitation10,Citation11. In the US, during the period 1997–2005, the prevalence was 27.9 per 100,000 population and the incidence was 8.8 per 100,000 populationCitation10,Citation11. While these measures of occurrence are useful for describing the relative public health burden of IPF, they are not clinically meaningful measures compared to mortality or healthcare resource utilization (HRU) ratesCitation3.
Historically, limited treatments were available for people with IPF. American Thoracic Society guidelines only include strong recommendations toward lung transplantation and long-term oxygen therapy, which represent costly and burdensome therapiesCitation12.
There has been increasing attention and controversy about what outcomes should be used in the design and conduct of clinical trialsCitation3,Citation13. A recent review conducted by a panel of experts concluded that all-cause mortality and non-elective hospitalization were among the most clinically meaningfulCitation3. However, in a recent analysis of mortality data from two clinical trials of patients with mild-to-moderate IPF, researchers found low 1–2-year mortality rates and suggested that mortality may not be a feasible measure for clinical trialsCitation13. Hospitalization rates and other HRU measures are likely more frequent compared to mortality during the period of a clinical trial. However, little is known about the frequency and patterns of HRU among a broad spectrum of patients with IPFCitation14,Citation15, and an assessment of HRU among a large scale of patients with IPF in the real world setting is warranted.
To address this gap we conducted a retrospective database study to describe the clinical characteristics of patients newly diagnosed with IPF, including their comorbidity profiles, and to compare their patterns of HRU to those of individuals without a diagnosis of IPF.
Methods
Data source
This is a retrospective cohort study utilizing commercial administrative claims from PharMetrics Integrated Database (IMS Lifelink), which contains complete medical and pharmacy claims for ∼70 million members from 100 managed care organizations in all four US geographical regions. Enrollees include both commercially insured adults (18–64 years old) and Medicare Advantage population (≥65 years old). This database has been used in several recently published articles to estimate the clinical and economic burden of various medical conditions in the USCitation16,Citation17._ENREF_16 The most recent 5-year (2006–2011) data available at the time of the study execution was used in this analysis.
Sample selection
Newly diagnosed patients with IPF were identified using the following criteria. Patients were required to have at least two claims with associated diagnosis of idiopathic fibrosing alveolitis (IFA) (International Classification of Diseases, Ninth Revision, Clinical Modification [ICD-9-CM] code of 516.3x) on separate dates, or at least one claim with diagnosis of IFA and at least one claim with associated diagnosis of post-inflammatory pulmonary fibrosis (PPF, ICD-9-CM: 515.xx), in any sequence on separate dates; the date of the first claim with IFA was set as the index date. Patients must also have a claim for lung biopsy (ICD-9-CM: 33.28, 34.21; Current Procedural Terminology 4 [CPT-4]: 32095, 32100–32160, 32602) or high-resolution computed tomography (HRCT) of the thorax (ICD-9-CM: 87.41; CPT-4: 71250–71270) within 90 days before or after the index date. Furthermore, patients must have 12-months continuous enrollment before the index date, and at least two claims with IFA after the lung biopsy/HRCT date and not on the same day as the index date to further confirm the IPF diagnosis.
Patients were excluded if at least one of the following criteria was met: (1) < 18 years old as of their first diagnosis of 516.3; (2) had a claim of IFA during the 12-month pre-index period; (3) had a claim for other interstitial lung diseases with known reasons (please see Supplementary Table 1 for a list of ICD-9-CM codes) after the index IPF diagnosis; (4) had a claim for lung cancer (ICD-9-CM code 162.xx, 197.0) before the index IPF diagnosis; and (5) diagnosed with cystic fibrosis (ICD-9-CM code 277.0) at any point in the patient profile.
Patients with cystic fibrosis or pre-index lung cancer were excluded because such patients may have different HRU patterns and their treatment or disease progression will possibly differ from patients with IPF without these conditions. There was no requirement on the enrollment duration for patients with IPF after the index date due to the varied follow-up duration. Newly diagnosed patients with IPF were followed until 12 months after the index date, disenrollment of plan, or end of 2011, whichever was earlier.
Each patient with IPF was matched to three individuals without diagnosis of IPF (non-IPF members) via direct matching to improve statistical efficiencyCitation18. The two cohorts, patients with IPF and non-IPF members, were matched on age (±2 years), gender, region, and payer type. For non-IPF members, the index date was selected to be January 1st of the index year of the matching IPF patients; members had 12 months continuous enrollment before the index date and at least the same length of enrollment after the index date as their matched IPF patients. The follow-up period of non-IPF members was censored based on the length of the follow-up period of their matching IPF patients.
Study measures
Demographic characteristics included age, gender, region, payer type, and plan type as of index date. Prevalences of comorbidities, including components of Charlson Comorbidity Index (CCI) and comorbidities related to IPF as specified in the American Thoracic Society treatment guidelines for IPFCitation12, were derived based on claims during the 12-month pre-index period. Specific comorbidities in the American Thoracic Society guidelines included pulmonary hypertension, hypertension, gastroesophageal reflux, obstructive sleep apnea, obesity, emphysema/chronic obstructive pulmonary disease, depression, asthma, arrhythmia, and coronary artery disease.
HRU was estimated 12 months before and up to 12 months after the index date and summarized by 3-month (quarterly) intervals before and after the index date. Only patients with full enrollment during each quarter and their matching non-IPF members were included in the quarterly assessment. The proportion of patients with at least one hospitalization, emergency room visit, physician office visit, outpatient hospital visit, pulmonologist visit, or oxygen therapy were calculated on a quarterly basis. The mean number of each HRU measure for patients with IPF and non-IPF members was also estimated for each quarter.
Statistical analysis
Descriptive analyses were conducted to assess HRU among patients with IPF and non-IPF members. Means and standard deviations (SD) were reported for continuous variables, and proportions were reported for categorical variables. The Student’s t-test was used to detect the differences for continuous variables (e.g. CCI scores). The Chi-square test was used for categorical variables (e.g. presence of comorbidities); p-values < 0.05 were considered statistically significant.
A generalized linear model (GLM) assuming Poisson distribution and log link function was utilized to estimate the adjusted difference in HRU between cohorts, with generalized estimating equation (GEE) techniques controlling for correlations between repeated measures based on quarter (i.e. flag for first quarter, second quarter, third quarter, and fourth quarter post-index). Other independent variables include age, region, gender, payer type, CCI, comorbidities, and any hospitalization in the 1-year pre-index period. Because it is difficult to interpret the coefficients estimated by the log-transformed regression from the GLM model, the predicted HRU of the two cohorts and the marginal difference between cohorts during the year after the index date were estimated. Specifically, the predicted HRU was estimated by applying the regression coefficients derived from the GLM model to the covariates included in the model with the IPF indicator set to 1 for IPF patients, and estimated again by setting the IPF indicator to 0 for non-IPF members. Marginal difference was the difference between the two estimations from the model equations, reflecting the divergence in HRU attributed by IPF. Bootstrap methods were used to derive 95% confidence intervals (CI) of predicted values.
Results
Of 26,394 patients with at least one claim for IFA during 2006–2011, 1735 patients were identified with IPF (3.6% with lung biopsy, 78.6% with HRCT, and 17.8% with both lung biopsy and HRCT; data not shown) who met the inclusion/exclusion criteria (). Among these 1735 patients, the mean (SD) follow-up duration was 9.7 (3.6) months. Direct matching via a 1:3 matching ratio yielded 5205 non-IPF members. The average age of both cohorts was 71.5 years and 46.1% were female. Approximately one-third lived in the US South (31.5%) or Midwest region (29.3%). Overall, about two-thirds of the study cohorts had commercial health insurance, close to one-fifth had Medicare health insurance, and more than half had preferred provider organization plans. The absolute differences of plan type between patients with IPF and non-IPF members were small, although statistically different due to the large sample size ().
Figure 1. Sample selection of newly diagnosed IPF patients. CF, cystic fibrosis; HRCT, high-resolution computed tomography; IFA, idiopathic fibrosing alveolitis; ILD, interstitial lung diseases; IPF, idiopathic pulmonary fibrosis; PPF, post-inflammatory pulmonary fibrosis.
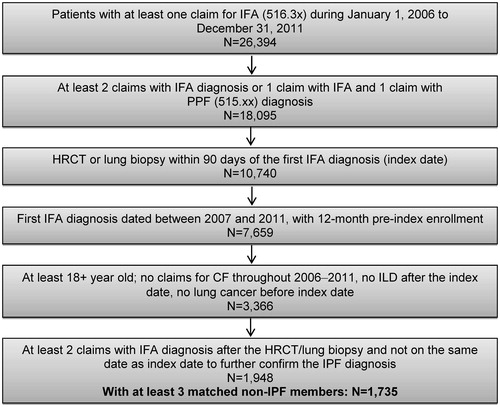
Table 1. Baseline characteristics.
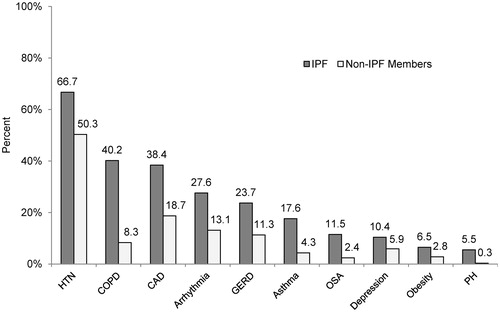
Patients with IPF had a significantly higher mean CCI score compared to non-IPF members (2.0 vs 0.9, p < 0.01) (). The prevalence of all American Thoracic Society-specified comorbidities was higher among patients with IPF compared to non-IPF members. The three most prevalent co-morbid conditions among IPF patients vs non-IPF members were: hypertension: 66.7% vs 50.3%, chronic obstructive pulmonary disease: 40.2% vs 8.3%, and coronary artery disease: 38.4% vs 18.7% (all p < 0.01) (). The greatest relative differences in prevalence of comorbidities between patients with IPF and non-IPF members were noted for pulmonary hypertension (5.5% vs 0.35%, 15.7-times higher), chronic obstructive pulmonary disease (40.2% vs 8.3%, 4.8-times higher), and obstructive sleep apnea (11.5% vs 2.4%, 4.8-times higher).
Figure 2. Baseline prevalence of selected comorbidities. CAD, coronary artery disease; COPD, chronic obstructive pulmonary disease; GERD, gastroesophageal reflux disease; HTN, hypertension; IPF, idiopathic pulmonary fibrosis; OSA, obstructive sleep apnea; PH, pulmonary hypertension. *Prevalence of all IPF-related comorbidities was significantly higher in IPF patients than non-IPF members at p < 0.01. †ICD-9-CM codes associated with these comorbidities are listed in Supplementary Table 2.
Unadjusted resource use was consistently higher during the pre- and post-index periods for all HRU measures among patients with IPF compared to non-IPF members (). In general, differences in HRU were greatest during the first quarter post-index for hospitalizations (28.4% vs 4.6%), emergency room visits (20.5% vs 7.6%), physician office visits (92.7% vs 61.5%), outpatient hospital visits (64.4% vs 25.3%), pulmonologist visits (54.5% vs 1.6%), and oxygen therapy (40.0% vs 3.1%), all p < 0.05. Moreover, there was a consistent pattern of a gradual increase in all HRU measures from the fourth quarter prior to the index date, with peaks during the quarters immediately before and after diagnosis, followed by declines during the second quarter post-index, and a plateau or slow decline in subsequent quarters. In contrast, all HRU measures among non-IPF members were relatively stable during the pre- and post-index periods (). The distribution of unadjusted annual healthcare utilization by healthcare settings, pulmonologist visits, and oxygen therapy is reported in . Overall, patients with IPF had significantly higher resource utilization than non-IPF members in all HRU measures (all p < 0.001). Specifically, the mean differences of use in HRU between patients with IPF and non-IPF members were more pronounced in outpatient hospital visits (7.5 vs 2.7), physician office visits (16 vs 7.8), and oxygen therapies (7.8 vs 0.6).
Figure 3. Unadjusted HRU 1 year before and after index date: proportion of patients with service in different settings. IPF, idiopathic pulmonary fibrosis; QT, quarter.
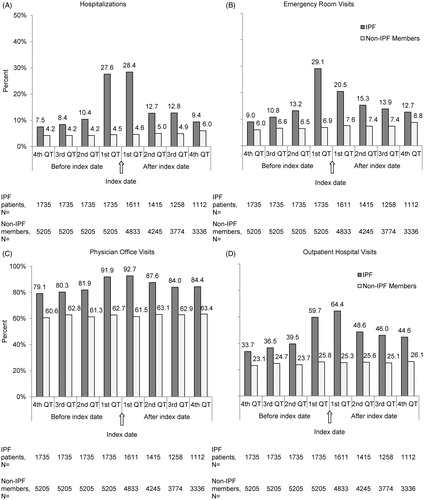

Table 2. Distribution of unadjusted HRU for 1 year post-index: mean number of services per person-year.
On average, during the 1-year post-index period the adjusted rates of HRU for all measures were significantly higher among patients with IPF compared to non-IPF members (). Specifically, the predicted 1-year hospitalization rate for patients with IPF and non-IPF members was 0.63 (95% CI = 0.58–0.66) and 0.31 (0.29–0.33), respectively; the marginal difference (0.32 [0.26–0.35]) was significant between patients with IPF and non-IPF members, as zero was not included in the 95% CI. The magnitude of difference between cohorts before and after the multivariable adjustment is relatively similar for inpatient services, pulmonologist visits, and chest x-rays, while the difference becomes smaller after the adjustment for emergency room, outpatient hospital, and physician office visits. Nonetheless, the HRU remained significantly higher among patients with IPF when compared to non-IPF members, even after multivariable adjustment.
Table 3. Adjusted HRU for 1 year post-index: mean number of services per person-year.
Discussion
In this study it was found that patients with IPF have a high burden of co-morbid conditions and HRU compared to non-IPF members. Co-morbid conditions most frequently associated with IPF were cardiovascular and other respiratory diseases. All HRU measures were consistently higher among patients with IPF than non-IPF members both before and after the diagnosis. After adjustment for different follow-up periods and other covariates, the pattern remained consistent, with HRU measures approximately twice as high among patients with IPF as compared to non-IPF members.
Further, compared to non-IPF members, patients with newly diagnosed IPF had significantly higher HRU in the pre-diagnosis period, and HRU increased and peaked during the quarters immediately before diagnosis, suggesting that patients with suspected symptoms may require diagnostic evaluations for IPF earlier to reduce unnecessary medical encounters.
This study adds to the limited literature on comorbidities and HRU among patients with IPF. The annual, unadjusted, post-index all-cause hospitalization rate in this study (0.7 per person-year) was only slightly higher compared to the previous study by Collard et al.Citation14 (0.5 per person-year). Moreover, we found a higher number of hospitalization days for IPF patients (5.0 vs 3.1 per person-year), a higher rate of emergency room visits (0.8 vs 0.4 per person-year), but a comparable number of pulmonologist visits (2.2 vs 2.1 per person-year). Possible reasons for the observed differences can be attributed to the use of different claims databases (PharMetrics vs Truven Health Analytics MarketScan) and different study periods (2006–2011 vs 2001–2008)Citation14. To our best knowledge, with the application of GLMs with GEE to adjust for correlations between repeated measures and control for differences of patient characteristics between IPF and non-IPF members, this is the first observational study that reports the incremental HRU attributable to IPF in the real-world setting.
Findings from this study support that HRU measures, such as all-cause hospitalization rate, may serve as outcomes of interest among IPF clinical trials. Raghu et al.Citation3 suggested all-cause hospitalization and mortality as clinically relevant end-points for patients with IPF in future clinical trials. However, a recent publication by King et al.Citation13 concluded that all-cause mortality as an outcome in clinical trials would need a substantial sample size or follow-up duration and have dramatic cost implications; all-cause hospitalization as a potential outcome needs to be further investigated. In another newly published article, Brown et al.Citation19 assessed outcomes after hospitalization among patients with IPF treated in a tertiary medical center; the authors reported that the hospitalizations were common in patients with IPF and concluded that all-cause or respiratory hospitalizations could be used as an end-point in IPF clinical trials.
This study provides insights regarding resource utilization burden associated with IPF patients as compared with non-IPF members. It will be informative for future studies to compare HRU outcomes between patients with IPF and patients with another respiratory disease (e.g. COPD). In addition, future studies can assess HRU patterns among a sub-group of patients with IPF who had a surgical lung biopsy, as these patients are more likely to be younger patients with fewer comorbidities.
Several limitations that may affect internal and external validity of this study need to be considered. First, healthcare claims used in this analysis were primarily used for administrative purposes to obtain reimbursement for services provided to health plan members; hence, there is potential for diagnostic and procedural coding inaccuracies, causing diagnostic and procedural misclassifications. Further, no diagnostic test results from HRCT or lung biopsy were available in the database to confirm diagnosis of IPF. Additionally, the ICD-9-CM codes of 516.3x and 515.xx used in this study are not specific to IPF and may include other idiopathic lung diseases resulting in misclassification. To minimize the potential for misclassification, a thorough algorithm was used requiring both relevant diagnosis codes and procedure code for HRCT followed by at least two confirmatory claims with IFA diagnosis and no claim for other interstitial lung diseases after the first claim with IFA. Similar methods to identify patients with IPF have been used by Collard et al.Citation14 and Raghu et al.Citation20. However, this algorithm to identify IPF may lead to an over-estimation of the HRU measures as these selected patients with IPF in the study may be more likely to have increased healthcare utilization. This current study database does not include information to confirm physician specialty; hence, misclassification of physician specialty and an under-estimation of pulmonologist visits are possible if coding errors exist. A previous study has demonstrated that hospitalization is associated with a high mortality rate among patients with IPF, and understanding the impact of mortality on HRU among this patient population is importantCitation19. However, mortality data are not available in the study database and, consequently, its impact on HRU could not be assessed.
The lack of clinical data present in claims data limits the accuracy of proxies used to adjust for disease severity among IPF patients, particularly in the multivariate analyses. Although the study patients were geographically diverse, they may not have been nationally representative. HRU measures may have been under-estimated for the elderly patients who were enrolled in Medicare supplemental insurance plans, because some of their inpatient claims were probably submitted for processing to the Centers for Medicare and Medicaid Services instead of the managed care plans. However, the proportion of hospitalizations and outpatient services was similar across different plan and payer types (results not reported), suggesting this limitation may have a limited impact on study findings.
Conclusions
There is a high burden associated with patients with IPF in terms of comorbidities and HRU. HRU measures, such as all-cause hospitalization, may serve as feasible and clinically meaningful outcomes for future clinical trials of new agents for IPF treatments.
Transparency
Declaration of funding
Boehringer Ingelheim Pharmaceuticals, Inc. (BIPI) provided funding for this study. BIPI is the employer of one of the authors, YY. Beyond those, BIPI played no active role in the study or the study results. BIPI had no influence on the results of the study.
Declaration of financial/other relationships
NW, C-CC, RW, and NB are employed by Evidera, which provides consulting and other research services to pharmaceutical, device, government, and non-government organizations. In these salaried positions, they work with a variety of companies and organizations and are precluded from receiving payment or honoraria directly from these organizations for services rendered. YY is employed by Boehringer Ingelheim Pharmaceuticals, Inc which supported the funding of this analysis. DC served as a consultant for Boehringer Ingelheim Pharmaceuticals, Inc.
Acknowledgments
The authors would like to thank Giovanna Devercelli of BIPI, who contributed to the study design, reviewed and provided comments on the draft manuscript, and oversaw the project management. They would also like to thank Janet Dooley of Evidera for her assistance in the production of this manuscript.
References
- Mapel DW, Hunt WC, Utton R, et al. Idiopathic pulmonary fibrosis: survival in population based and hospital based cohorts. Thorax 1998;53:469-76
- Gulati M. Diagnostic assessment of patients with interstitial lung disease. Prim Care Respir J 2011;20:120-7
- Raghu G, Collard HR, Anstrom KJ, et al. Idiopathic pulmonary fibrosis: clinically meaningful primary endpoints in phase 3 clinical trials. Am J Respir Crit Care Med 2012;185:1044-8
- Bandeira CD, Rubin AS, Cardoso PF, et al. Prevalence of gastroesophageal reflux disease in patients with idiopathic pulmonary fibrosis. J Bras Pneumol 2009;35:1182-9
- Chung MJ, Goo JM, Im JG. Pulmonary tuberculosis in patients with idiopathic pulmonary fibrosis. Eur J Radiol 2004;52:175-9
- Lettieri CJ, Nathan SD, Barnett SD, et al. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest 2006;129:746-52
- Nathan SD, Basavaraj A, Reichner C, et al. Prevalence and impact of coronary artery disease in idiopathic pulmonary fibrosis. Respir Med 2010;104:1035-41
- Ozawa Y, Suda T, Naito T, et al. Cumulative incidence of and predictive factors for lung cancer in IPF. Respirology 2009;14:723-8
- Lancaster LH, Mason WR, Parnell JA, et al. Obstructive sleep apnea is common in idiopathic pulmonary fibrosis. Chest 2009;136:772-8
- Fernandez Perez ER, Daniels CE, Schroeder DR, et al. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest 2010;137:129-37
- Nalysnyk L, Cid-Ruzafa J, Rotella P, et al. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012;21:355-61
- Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788-824
- King TE, Jr. Albera C, Bradford WZ, et al. All-cause mortality rate in patients with idiopathic pulmonary fibrosis. Implications for the design and execution of clinical trials. Am J Respir Crit Care Med 2014;189:825-31
- Collard HR, Ward AJ, Lanes S, et al. Burden of illness in idiopathic pulmonary fibrosis. J Med Econ 2012;15:829-35
- Navaratnam V, Fogarty AW, Glendening R, et al. The increasing secondary care burden of idiopathic pulmonary fibrosis: hospital admission trends in England from 1998 to 2010. Chest 2013;143:1078-84
- Zhu B, Kulkarni PM, Stensland MD, et al. Medication patterns and costs associated with olanzapine and other atypical antipsychotics in the treatment of bipolar disorder. Curr Med Res Opin 2007;23:2805-14
- Baser O, Chalk M, Fiellin DA, et al. Cost and utilization outcomes of opioid-dependence treatments. Am J Manag Care 2011;17(Suppl 8):S235-48
- Janes H, Pepe M. The optimal ratio of cases to controls for estimating the classification accuracy of a biomarker. Biostatistics 2006;7:456-68
- Brown AW, Fischer CP, Shlobin OA, et al. Outcomes after hospitalization in idiopathic pulmonary fibrosis: a cohort study. Chest 2015;147:173-9. doi: 10.1378/chest.13-2424
- Raghu G, Weycker D, Edelsberg J, et al. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2006;174:810-16