ABSTRACT
The National Institute for Occupational Safety and Health (NIOSH) research on direct-reading instruments (DRIs) needed an instantaneous sampling method to provide independent confirmation of the concentrations of chemical warfare agent (CWA) simulants. It was determined that evacuated canisters would be the method of choice. There is no method specifically validated for volatile organic compounds (VOCs) in the NIOSH Manual of Analytical Methods. The purpose of this study was to validate an evacuated canister method for sampling seven specific VOCs that can be used as a simulant for CWA agents (cyclohexane) or influence the DRI measurement of CWA agents (acetone, chloroform, methylene chloride, methyl ethyl ketone, hexane, and carbon tetrachloride [CCl4]). The method used 6-L evacuated stainless-steel fused silica-lined canisters to sample the atmosphere containing VOCs. The contents of the canisters were then introduced into an autosampler/preconcentrator using a microscale purge and trap (MPT) method. The MPT method trapped and concentrated the VOCs in the air sample and removed most of the carbon dioxide and water vapor. After preconcentration, the samples were analyzed using a gas chromatograph with a mass selective detector. The method was tested, evaluated, and validated using the NIOSH recommended guidelines. The evaluation consisted of determining the optimum concentration range for the method; the sample stability over 30 days; and the accuracy, precision, and bias of the method. This method meets the NIOSH guidelines for six of the seven compounds (excluding acetone) tested in the range of 2.3–50 parts per billion (ppb), making it suitable for sampling of these VOCs at the ppb level.
An evacuated canister sampling and analysis method for ppb-level concentrations of VOCs simulating CWAs and possible interferents in air was developed. The analysis was conducted using a gas chromatograph with a mass selective detector. The method was validated using NIOSH guidelines. This method is useful for identifying and quantifying seven specific VOCs in the ppb range.
INTRODUCTION
The National Institute for Occupational Safety and Health (NIOSH) is currently investigating the performance of direct-reading organic vapor instruments when detecting chemical warfare agent (CWA) simulants and possible interferents. The simulant used in this research is cyclohexane and the interferents are acetone, chloroform, methylene chloride, methyl ethyl ketone, hexane, and carbon tetrachloride (CCl4). An independent instantaneous sampling method to determine parts-per-billion (ppb) levels of these compounds was needed for this study. The compounds were selected based on a report by the Chemical Security Analysis CenterCitation1 that indicated exhaust gases, gasoline, kerosene, chlorine bleach, insect repellant, and diesel fuel vapors could be used as interferences in detector evaluation for first responders. All of these, except for chlorine bleach, are complex mixtures of volatile and semi-volatile organic compounds detectable by gas chromatography (GC)/mass spectrometry (MS). The main component of chlorine bleach (sodium hypochlorite) is not detectable by GC-MS. The six interferents (acetone, chloroform, methylene chloride, methyl ethyl ketone, hexane, and CCl4) are common chemicals that are likely to be encountered during emergency response scenarios and represent different classes of organic compounds (straight-chain hydrocarbons, ketones, and chlorinated hydrocarbons) that may elicit different instrument responses. Hexane was selected because it is the straight-chain version of cyclohexane. In addition, hexane and methyl ethyl ketone are found in car and truck exhausts. Methylene chloride was chosen as a substitute for chlorine bleach.
The current NIOSH methods for sampling volatile organic compounds (VOCs) were investigated for use in the CWA simulant research. The first method for screening for VOCs uses thermal desorption tubes containing graphitized carbon and carbon molecular sieve sorbents.Citation2 After sampling, the thermal desorption tubes are desorbed and analyzed by GC-MS. In addition, there are several methods using charcoal tubes for specific classes of VOCs such as ketonesCitation3 and hydrocarbons.Citation4 These methods are for time-weighted sampling and are unsuitable for the CWA simulant research. They have several additional disadvantages when used for field sampling that include noise generated by the pumps, a limited sampling time (20 min to 10 hr on the basis of the minimum and maximum flow rates and volumes), potential for pump or battery failure, the need to charge batteries, etc.
Because evacuated canisters can be used for instantaneous sampling, it was determined that this would be the best method to use in the CWA simulant work. The NIOSH Manual of Analytical Methods (NMAM) does not have a validated evacuated canister method for VOCs. Therefore, it was decided to validate an evacuated canister method for cyclohexane, acetone, chloroform, methylene chloride, methyl ethyl ketone, hexane, and CCl4 following NIOSH guidelinesCitation5 for possible incorporation into the NMAM.
In addition to instantaneous sampling, collecting airborne VOCs directly in evacuated canisters has the following advantages over the current NIOSH methodsCitation6: a wider range of sampling times, whole air sampling, no breakthrough of target compounds, no thermal or solvent desorption, ruggedness, no field calibration necessary, sample stability, less maintenance (e.g., no batteries to charge), and the ability to perform repeat analysis on the same sample. Evacuated canisters can also be equipped with a passive flow regulation system to provide sampling times ranging from 3 min to 1 week, depending on the size of the canister and the characteristics of the flow regulation system.
There are several disadvantages to using canisters for sampling. They are expensive, especially if many are needed. They must be cleaned before each use to prevent cross-contamination, and the cleaning equipment is specialized and expensive. They must be checked for leaks before each use, and the presampling pressure must be recorded because there is a possibility that canisters could leak between being checked and used. Some polar compounds may have an affinity for canister walls at low concentrations. Canisters are not good for polycyclic aromatic hydrocarbons with more than 10 carbon atoms and semi-volatile organic compounds. The advantages of canisters outweigh their disadvantages for sampling VOCs in the low-ppb concentration range. Over a period of years, the cost of canisters and the associated equipment could be comparable to the cost of using thermal desorption tubes. Canister samples can be quantitatively analyzed whereas thermal desorption tubes provide qualitative results.
Several studies and methods use evacuated canisters for air sampling. The U.S. Environmental Protection Agency (EPA) has several methods for sampling and analysis of VOCs in air using stainless-steel pressure vessels with specially prepared interior surfaces such as electropolished (SUMMA) passivated canisters.Citation7,Citation8 The 97 VOCs (a subset of the 189 hazardous air pollutants listed in Title III of the Clean Air Act Amendments of 1990) are separated by GC-MS or by multidetector techniques. EPA defines a VOC as an organic compound having a vapor pressure greater than 10−1 Torr at 25 °C and 760 mm Hg. These methods apply to ambient concentrations of VOCs above 0.5 ppb.
Eklund et al.Citation9 developed and validated a sampling and analytical method to measure ethylene oxide in air using evacuated SUMMA and fused silica-lined (FSL) stainless-steel canisters. The method evaluation involved laboratory and field tests. Ethylene oxide spiked at 2 ppb, and 20 ppb was found to be stable over a period of 15 days in a nitrogen matrix and in laboratory air. The method's limit of detection was 0.25 ppb. Their field studies indicated the method was capable of detecting ethylene oxide (additionally toluene and benzene) in an ambient air matrix.
Batterman et al.Citation10 studied the analysis, stability, and recovery of seven aldehydes (butanal, pentanal, hexanal, octanal, nonanal, and benzaldehyde) and four terpenes (α-pinene, β-pinene, limonene, and 3-carene) in SUMMA canisters at a high and low concentration. Depending upon the compound, the high concentration ranged from 68 (nonanal) to 130 parts per million (ppm) (butanal) in humidified nitrogen. Three sets of low-concentration standards were prepared ranging from 2.9 (nonanal) to 5.4 ppb (butanal) in humidified zero air, dry zero air, and humidified nitrogen. On the basis of the analysis of the low-concentration samples, aldehydes and terpenes stored in electropolished canisters had lower recoveries and shorter half-lives compared with toluene, the non-polar aromatic control. Batterman et al.Citation10 found that losses occurred during short (hours) and long (days to weeks) time scales. The losses occurred during the first hour after filling and continued over the measurement period. The authors suggested the losses could be attributed to dissolution of soluble compounds in the water present in the canister, gas-phase reactions and transformations, heterogeneous reactions on the canister wall, hydrolysis, polymerization, or secondary adsorption. The estimated half-lives of aldehydes and terpenes in the canisters were 18 days in humidified air-filled canisters, 24 days in humidified nitrogen, and 6 days in dry air. Their results indicated the collection and storage of aldehydes and terpenes in canisters require humidification. The absolute accuracy of samples and standards containing these compounds stored in canisters under the aformentinoned conditions may be limited.
Daughtrey et al.Citation11 used two alternative methods to sample CCl4, α-pinene, isoprene, benzene, n-decane, toluene, m-,p-xylene, and n-octanal in outdoor air. The first method was a near-real-time measurement using a two-trap organic vapor concentrator attached to a gas chromatograph/mass spectrometer. This system provided results in a matter of minutes. The second method was to collect the air into SUMMA and Silco canisters. The concentrations ranged from less than 0.80 (n-decane) to 14.9 ppb (isoprene). The canisters were analyzed 1–7 days after collection with the same concentrator/GC-MS system.Citation11 They found only slight differences between the near-real-time and canister samples with clear correlations between the two types of samples. Many of the dependent samples had t-test comparisons that were significant. Scatterplots of these sample comparisons were also displayed, yet no correlation coefficients were reported. They concluded that canisters can be used to sample the studied VOCs in ambient air. They may also be stored up to a week. They did note that collection and analysis of aldehydes cannot be done unless canister cleaning methods are improved.
Evans et al.Citation12 collected headspace samples from high-level radioactive waste tanks known to contain moderately high levels of polar and nonpolar organic compounds using 6-L SUMMA canisters and triple sorbent traps (TSTs). Over a 32-week holding time (under refrigerated [−20 °C] and room temperature conditions), the TSTs and canisters were analyzed for trace organic vapor compounds by GC-MS using cryogenic preconcentration techniques. They detected 31 analytes composed of alkanes (butane to tridecane), alcohols (ethanol to butanol), ketones (acetone to 4-methyl-2pentanone), aromatics (benzene, toluene, etc.), pyridine, tetrahydrofuran, nitriles (acetonitrile to butanenitrole), halogenated hydrocarbons, and alkenes. The concentrations found ranged from less than 1 (1,2,4-trimethylbenzene) to approximately 12,000 (ethanol) ppb. They found the two samplers provided generally equivalent results. Their conclusion was that the canister method was capable of a wide dynamic range, not prone to blank contamination during sampling and transport, and provided results that were representative of the actual tank atmosphere.
Hoshi et al.Citation13 established a method to measure 11 oxygen-containing VOCs (OVOCs) in ambient air. The OVOCs selected based on an inventory of VOCs from stationary sources in Tokyo were isopropyl alcohol, acetone, methyl acetate, methyl tertiary butyl ether (MTBE), n-propanol, methyl ethyl ketone, ethyl acetate, isobutyl alcohol, n-butyl alcohol, methyl isobutyl alcohol, methyl isobutyl ketone, and butyl acetate. They used 6-L FSL canisters to collect (over a 24-hr time period) and store the ambient air samples. The samples were analyzed by GCMS. For the recovery of samples from the canisters, the ambient air samples were used as sampled. The concentrations found ranged from not detected (methyl acetate, MTBE, isobutyl alcohol, and n-butyl alcohol) to 24 μg m−3 (acetone). Their data indicated the method was adequate for the recovery of actual ambient air samples. For the sample storage testing, they added a standard gas so that the samples had a certain concentration of every substance in the matrix of actual ambient air. The concentration range for this testing was 0.05 (MTBE) to 98 μg m−3 (ethyl acetate). The OVOCs appeared to be stable after sampling with no significant losses in the canisters after 14 days. They found that the canister collection-GC-MS method had high accuracy and sensitivity for measuring atmospheric OVOCs.
Kelly et al.Citation14 conducted a study to develop a method for analyzing polar VOCs (PVOCs) and VOCs in SUMMA canisters. For the method development in the laboratory, they used 17 PVOCs and VOCs: 1,3-butadiene, propylene oxide, four alcohols (methanol to 1-butanol) (PVOCs), ethyl acrylate, two nitriles, two ketones (PVOCs), two acetates (PVOCs), two ethers (PVOCs), and two aromatics. The concentration of each compound was 200 ppb in dry high-pressure nitrogen, which was found to be stable in all compounds. They also performed the same analysis on ambient air samples collected over 24-hr intervals on a regular basis over 1 yr at former EPA toxic air monitoring study sites in Boston, MA, and Houston, TX. Of the 17 PVOCs and VOCs used for the method development, 9 were found in the field samples: methanol, ethanol, acetone, 2-propanol, MTBE, methyl ethyl ketone, benzene, 1-butanol, and toluene. The concentrations in these samples ranged from less than 0.2 (MTBE, methyl ethyl ketone, and 1-butanol) to 94 ppb (2-propanol). They concluded that sampling for PVOCs can be implemented into existing field sampling programs that use canisters, although the analysis of the samples needs to be done shortly after collection.
Ochiai et al.Citation15 evaluated a canister-based 1-week sampling method using a mechanical flow controller and 6-L FSL canisters. They took roadside 24-hr and 1-week samples in suburban Tokyo. The method was developed to determine the concentration of 47 VOCs (the 45 compounds found in the EPA TO-14A method gas mixture plus butyl acetate and methyl isobutyl ketone). The method was able to determine parts-per-trillion (ppt) to ppb VOC levels by use of a three-stage preconcentration method followed by GC-MS analysis. In the roadside ambient air samples, they found 34 of the 47 VOCs for which the method was validated. The 24-hr sample concentrations ranged from approximately 6 (1,2-dichloroethane) to 15,000 ppt (toluene). The 1-week sample concentration ranged from 16 (1,2-dichloroethane) to 8900 ppt (toluene). Because 28 of the 34 VOCs found in the field samples have less than ±10% deviation, the authors concluded that the method was applicable for sampling ambient air. The six with greater than ±10% deviation were found in low amounts—6.3 (1,2-dichloroethane) to 43 ppt (acrylonitrile).
Rossner et al.Citation16 compared small (300 mL) evacuated high-purity stainless-steel canisters with a specially designed capillary flow controller (a deactivated capillary of 0.05 mm in diameter and 10 cm in length) with charcoal tubes and diffusive badges. They used these three samplers to sample side-by-side in an 18.1-m3 exposure chamber containing four concentrations of styrene (5, 20, 40, and 50 ppm). Their results indicated that the capillary/ canister sampling device could be an acceptable alternative to sorbent samplers as a personal sampler providing reliable results that are representative of exposures. Ross-ner and WickCitation17 performed a field study to evaluate the feasibility of collecting a 40-hr area sample using the same canisters as Rossner et al.Citation16,Citation17 They compared six canister samplers used as area monitors to evaluate a cleaning operation for an entire workweek to 30 diffusive badges (6 per day) monitoring the same process. The sampling was repeated a second week. The compound of interest was a cleaning solvent (Safety-Kleen premium solvent, which is a light petroleum distillate consisting of a mixture of C9 to C11 compounds). The daily concentration of styrene determined by the badges ranged from 2.6 to 7.4 ppm. The average weeklong badge styrene concentrations for the 2 weeks were 3.6 and 6.1 ppm. The weeklong canister sty-rene concentrations were 3.2 and 5.7 ppm. They found no statistical difference between the time-weighted average for the two sampling methods (P > 0.05).
Ochiai et al.Citation18 evaluated the effect of relative humidity (RH) on the recoveries and stabilities of 58 VOCs, including oxygen-, nitrogen-, and sulfur-containing PVOCs in FSL and SUMMA canisters at lowppb levels. They used the 41 VOCs from the EPA TO-14A method as well as ethanol, isopropyl alcohol, propanol, isobutanol, butanol, acetone, methyl ethyl ke-tone, methyl isobutyl ketone, ethyl acetate, butyl acetate, diethyl ether, tetrahydrofuran, MTBE, ethanethiol, propanethiol, tert-butanethiol, and butanethiol. The canisters contained mixtures of approximately 1.7–2.5 ppb in nitrogen at 1.6, 8, 27, 29, 53, and more than 99% RH. All of the canisters were sampled on the day they were filled. All thiols were not recovered from the SUMMA canisters. The FSL canisters had good recoveries (>86%) for all compounds under all conditions (except for the alcohols at >99% RH because of partitioning into the condensed water). The canisters at RHs greater than 8% were also sampled at days 3, 7, 14, and 28 after filling. In the FSL canisters, all of the nonpolar VOCs were relatively stable under all humidities for the 28 days. Polar VOCs having a relatively low vapor pressure, such as butyl acetate, methyl isobutyl ketone, and the alcohols except ethanol, were unstable under the lower humidities (8 and 27%). A RH greater than 53% was required to guarantee good stabilities (a recovery of >80%) for all compounds except the thiols in both types of canisters. The thiols initially had a good recovery from the FSL canisters but could not be recovered from the FSL canisters by day 14. The authors concluded FSL canisters were more inert than SUMMA canisters. They did note that the use of canisters to sample alcohols requires careful attention to ensure the RH is between 50 and 70%. To use the FSL canisters for thiols, more chemical deactivation of the active sites such as residual silanols must be done.
None of these studies validated a canister method in accordance with the procedures outlined by NIOSH.Citation5,Citation19 The NIOSH method validation procedure consists of determining the limit of detection (LOD) and limit of quantitation (LOQ); the optimum concentration range for the method; the sample stability over 30 days; and the accuracy, precision, and bias of the method. This study evaluated and validated a method using 6-L canisters for sampling. An aliquot was removed from the canisters and preconcentrated, followed by GC-MS analysis for measuring seven specific VOCs (acetone, chloroform, methylene chloride, methyl ethyl ketone [2-butanone], hexane, CCl4, and cyclohexane) with concentrations less than 50 ppb.
MATERIALS AND METHODS
Sample Analysis
To achieve the desired detection limits, ppb-level concentration samples must be concentrated before injection into a gas chromatograph-mass spectrometer for analysis. Because the levels of water vapor and carbon dioxide (CO2) are 4–8 orders of magnitude higher than the target volatile compounds, they must be removed before GC injection to avoid chromatography problems and attenuation of response in the mass spectrometer. The developed method uses a 7100 preconcentrator (Entech Instruments, Inc.) using a microscale purge and trap (MPT) method. This method uses three modules to remove the water and CO2 in the sample as well as to concentrate the VOCs of interest. The air sample was first cryogenically trapped at −150 °C on glass beads (module 1). As the sample is passed through this module, the major constituent gases (nitrogen, oxygen, and argon) pass through the trap while water, CO2, and all of the compounds of interest are condensed onto the glass beads. The glass bead module is then warmed to room temperature before passing a small volume (40 mL) of helium to transfer the CO2 and lower-molecular-weight VOCs of interest to a 35/60 mesh Tenax TA trap (module 2), which was cooled to −30 °C. Most of the water remains condensed on the glass beads in module 1 and is baked off later. The CO2 passes through the Tenax and is pumped away. However, the 40-cm3 transfer volume is not enough to cause even the lightest TO-15 compounds to break through the Tenax trap at the reduced temperature. The third module is an empty focusing trap that is cooled to −160 °C. The second module is then heated to 180 °C and backflushed with helium to transfer the sample from it to module 3. At −160 °C, the VOCs and any remaining water are condensed onto a 1/32-in. OD Silonite-coated stainless-steel transfer line heated to 100 °C to refocus the sample before introduction to the gas chromatograph. Module 3 is then rapidly heated to 150 °C. At this temperature, the VOCs volatilize and are swept onto the GC column. The precolumn and the first two modules are baked out to prepare them for the next sample.Citation20 Laboratory temperature and RH were approximately 22.8 °C and 36% during sampling. Samples were not pressurized before analysis, so the effect of water content on analyte stability should not be significant.
After preconcentration, the sample was then analyzed using an Agilent 6890 GC/5973N mass spectrometer (Santa Clara, CA). The gas chromatograph used a Restek Rtx-1 column (fused silica; nonpolar phase; Crossbond 100% dimethyl polysiloxane; model 10157 0.32 mm × 60 m × 1 μm) and helium flowing at a rate of 1 mL min−1. The GC oven was programmed to have an initial temperature of 35 °C, which was held for 2 min, ramped at 8 °C/min until 170 °C was reached, ramped at 20 °C/min until 220 °C was reached, and held for 3.5 min. More detailed information on the preconcentrator and GC-MS operating parameters can be found in the online supplemental information (published at http://secure.awma.org/onlinelibrary/samples/10.3155-1047-3289.61.8.826_supplmaterial.pdf).
Method Operating Range—LOD/LOQ and Upper Bounds
The first step in determining the operating range of the method was to calculate the LOD following NIOSH guidelines.Citation5 For this calculation, five canisters containing 0.08 (less than the expected LOD of 0.2 ppb), 0.2, 0.5, 1, and 2 ppb (10 times the expected LOD) were prepared using the 4600 Dynamic Dilution system (Entech Instruments, Inc.). The LOD concentrations were prepared in 6-L (Entech Instruments, p/n 29–10622G) FSL evacuated canisters. Before use, the 6-L canisters were heated and cleaned for 30 cycles using a 3100 canister cleaner system (Entech Instruments, Inc.). A cycle consisted of pressuring the canister to 20 psia using humidified ultrahigh-purity nitrogen and then evacuating the canister to 50 mTorr. The 4600 system diluted a 100-ppb in nitrogen TO-15 62 component mixture calibration standard (p/n 34437, Restek Corp.) with ultrahigh-purity nitrogen to deliver the desired concentration. It should be noted that since this study was conducted, Restek has added three additional compounds to their TO-15 component mixture for a total of 65. The canisters used in the LOD determination were analyzed using the autosampler, preconcentrator, and GC-MS system described above.
The 7016CA autosampler (Entech Instruments, Inc.) removed a 500-mL aliquot from the canister and sent it to the preconcentrator. A 500-mL aliquot is the largest aliquot size the 7100 preconcentrator can handle. The use of this size aliquot provides the maximum sensitivity for the all of the analysis. Before subjecting the aliquot to the MPT technique, the preconcentrator added 69 mL of Restek TO-14A internal standard/tuning mixture (p/n 34425). This standard contained 100 ppb each of bromochloromethane, 1,4 difluorobenzene, 1-bromo-4-fluorobenzene, chloro-benzene-d5, and 1,4-difluorobenzene. The 69 mL produced a concentration of 13.8 ppb for each of these four compounds. Each standard was run in duplicate. For this study, only 7 of the 62 compounds in the TO-15 mixture were used: acetone, chloroform, methylene chloride, 2-butanone (methyl ethyl ketone), hexane, CCl4, and cyclohexane.
The LOD was calculated using the guidelines published by NIOSH.Citation5 First, the amount ratio (the concentration in ppb of the analyte divided by the concentration of the internal standards [13.8 ppb]) was plotted against the response ratio (the GC response of the analyte in each sample divided by the GC response of the closest internal standard in the sample) for each LOD concentration (i.e., 0.08, 0.2, 0.5, 1, and 2 ppb). The calibration curve equations (response ratio = slope × amount ratio) were then used to calculate the concentration of the analytes in the samples. The linear regression equation (Y = mX + b) was obtained, where m is the slope, Y is the response ratio, and X is the amount ratio determined for each of the seven compounds of interest. The standard error of the regression, sy , was calculated using Equationeq 1:
Once the LOD was determined, the limit of LOQ was calculated using Equationeq 3.
The upper limit of the method operating range was determined by diluting a 1-ppm TO-15 62 component mixture (Restek, p/n 34436) to 100 ppb in a 6-L canister using the Entech 4600. The stock 1-ppm standard is a mixture produced gravimetrically using National Institute of Science and Technology (NIST)-traceable weights. It should be noted that since this study was conducted, Restek has added three additional compounds to their TO-15 component mixture for a total of 65. A series of aliquots from 100 to 500 mL in 50-mL (equivalent to 10 ppb) increments was analyzed from this 6-L canister.
A curve of aliquot size versus response areas from the mass selective detectors for each compound was plotted. These curves were then used to determine the upper limit of the method operating range. This was determined to be the point at which the response of the mass selective detector became nonlinear (i.e., the mass spectrometer was becoming saturated). The upper limit of the operating range was determined visually as the lowest concentration at which the slope of any of the compounds started to decrease (i.e., the point at which the curve started to flatten out). This procedure was repeated two more times to ensure the reproducibility of the upper limit of the operating range.
Instrument Operating Range Concentration Calibration
The GC-MS system was calibrated over its operating range (zero to the upper limit of the operating range) using six standards. The six standards were prepared by diluting the 1-ppm TO-15 standard to 100 ppb in a 6-L canister using the Entech 4600 Dynamic Dilution system. Different aliquot sizes were used by the preconcentrator to give the various concentrations and were analyzed as described previously. A calibration curve (concentration vs. response) was developed for each analyte. Each concentration was run in triplicate providing a total of 18 points upon which the curve was based. This curve was then used in all subsequent phases of the validation.
Determination of Concentration from GC-MS Response
The response of each analyte was converted to a ppb concentration through the use of relative response factors for each of the seven compounds of interest. For each curve, the amount ratio (the standard concentration in ppb of the analyte divided by the concentration of the internal standards [13.8 ppb]) was plotted against the response ratio (the GC response of the analyte in each sample divided by the GC response of the closest internal standard in the sample). A laboratory blank of ultrahigh-purity nitrogen was used to define the origin. The calibration curve equations (response ratio = slope × amount ratio) were then used to calculate the concentration of the analytes in the samples.
Analyte Recovery
To make this determination, three concentrations (4, 25, and 45 ppb) were prepared in custom-made 200-L Teflon sampling bags (made from American Durafilm fluorinated ethylene propylene [FEP] type A Teflon film [p/n FEP-500-L] sealed with a model W-600T foot-operated bag sealing system [HeatSealers.net]). The 4600 system was used to fill the bags by diluting the 1-ppm TO-15 standard to the desired concentration. Twelve instantaneous 6-L canister samples were taken for each concentration. The analysis of the canister samples was performed within 24 hr of the canister being prepared using a 500-mL aliquot size (i.e., 500 mL were taken from each canister and analyzed). The three diluter concentrations were used as the theoretical concentrations. The results of this experiment are precision and bias values that are used in the accuracy calculation.
Sample Stability
A 20-ppb sample was prepared in a custom-made 200-L Teflon sampling bag by diluting the 1-ppm TO-15 standard using the Entech 4600 Dynamic Dilution system. Thirty instantaneous 6-L evacuated canister samples were collected from the bag and stored under the conditions in the laboratory (∼23 °C and 32% RH). The canisters were randomly analyzed according to the schedule in . The stability of the samples in the canisters was determined by analyzing a 500-mL aliquot from the canisters after 7, 14, 21, and 30 days of postpreparation. The concentration of each sample was determined using the instrument operation range calibration curve for each compound. The NIOSH guidelines state that if the average concentration of the set of canisters analyzed on day 7 differs from the set analyzed on day 0 by more 10%, then the method fails the stability criterion.Citation5 The 7-day time frame was selected to allow samples to be shipped to a laboratory for analysis. In addition, to determine the point at which instability occurs (defined as a change of 10%), the average concentration of the canisters analyzed on days 14, 21, and 30 are compared with the average concentration determined for day 0.Citation5
Table 1. Stability data sampling schedule
Precision, Bias, and Accuracy Evaluation
The precision, bias, and accuracy of the method was determined following the NIOSH guidelines using the concentrations obtained during analyte recovery experiments and sample stability experiments. The method accuracy was calculated using the hyperbolic approximation formula. Precision was tested to ensure homogeneity of pooled precision using Bartlett's test.Citation21 The overall bias estimate was used in the precision calculations. The bias and precision estimates were then used in the accuracy calculations to find the estimated accuracy and its corresponding 90% confidence intervals. Method bias should be less than 10%.Citation5 The bias and precision estimates were then used to compute the best single and 90% confidence interval estimates of the accuracy. The latter is bounded by 5 and 95% confidence limits of the accuracy. If both estimates are less than 25%, there is a 95% confidence that the method accuracy is less than 25% and the method meets the accuracy criterion. If both estimates are greater than 25%, there is a 95% confidence that the method accuracy is greater than 25% and the method does not meet the accuracy criterion. However, if the 90% confidence interval estimate for method accuracy includes the value of 25%, the results are not conclusive.Citation5
RESULTS AND DISCUSSION
Method Operating Range—LOD/LOQ and Upper Bounds
The LOD/LOQ for each of the selected seven TO-15 compounds (methylene chloride, methyl ethyl ketone, hexane, CCl4, and cyclohexane) was calculated (). The LOD value of 0.68 ppb for methyl ethyl ketone is presumably due to the decreased sensitivity of the mass spectrometer for this class of compounds or related to the ability of the preconcentrator to concentrate the same. The LODs for methylene chloride and CCl4 were lower in this study than the method detection limits (MDLs) reported in EPA Method TO-15.Citation8 The LOD for methylene chloride was 0.25 ppb compared with the MDL of 1.38. The LOD for CCl4 was 0.39 compared with the MDL of 0.42 ppb. This difference is not surprising because of varying methods of calculating these values.
Table 2. LOD and LOQ values
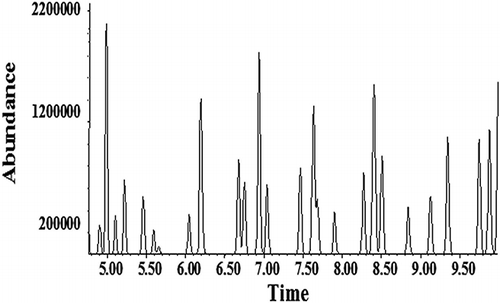
The upper limit of the method was determined by visually inspecting the plots of aliquot size versus peak area (). The visual inspection indicates that the slope becomes nonlinear between the aliquot sizes of 250 and 300 mL, equaling 50 and 60 ppb, respectively. To verify this, the chromatogram of a 300-mL aliquot volume equaling a 60-ppb sample of TO-15 () was examined. It shows poor separation of compounds and peak tailing, indicating that the volume amount of sample is too high. The chromatogram of a 250-mL aliquot volume equaling 50-ppb TO-15 shows much better definition and separation (). Note that the y-axis scales for and are drastically different: The scale of is approximately 6 times higher. On the basis of the chromatogram and the slopes of the lines in , the upper range of the method was determined to be 50 ppb. The GC-MS system was then calibrated from 0 to 50 ppb using the aliquot sizes listed in .
Table 3. Aliquots and concentrations for instrument operating range concentration calibration
Figure 1. Representative graph of aliquot size vs. peak area for ppb concentration method to determine the upper limit of the method's operating range.
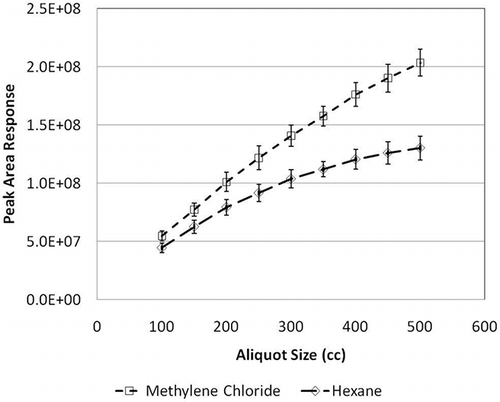
Figure 2. Aliquot volume of 300 mL, equaling a 60-ppb sample of TO-15, shows poor separation of compounds and tailing peaks, indicating the volume amount of sample is too high.
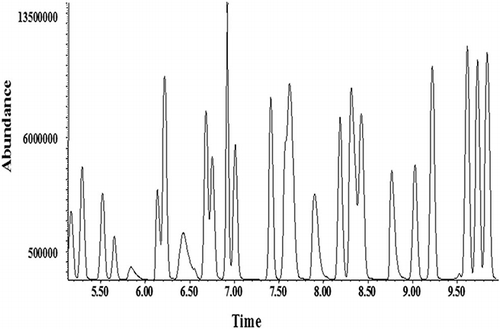
Figure 3. Aliquot volume of 250 mL, equaling a 50-ppb sample of TO-15, showing better separation of compounds.
Analyte Recovery
The three concentrations run for this portion of the study were 4, 25, and 45 ppb. Thirty-six trials were analyzed (3 concentrations × 12 canisters per concentration). The bias and precision results of this evaluation (data not shown) are incorporated into the accuracy calculations. Summary information for data from this section is displayed in the online supplementary material.
Sample Stability Evaluation
The average concentration on day 7 for all compounds except acetone was within 10% of the compound average of day 0 (canisters analyzed immediately after collection; see online supplemental information). The remaining day averages (days 14, 21, and 30) for all compounds except acetone were within 10% of the average of the day 0 samples. The method meets the stability criteria for these six compounds. Acetone stability was variable between days, presumably because of sample preparation or MS response to this analyte. Acetone stability was outside of the 10% limit for days 7 and 30 but came back within acceptable limits for days 14 and 21.
Precision, Bias, and Accuracy
The precision, bias, and accuracy analysis results are given in . The bias for all seven compounds is less than 10%. The bias criterion is met. The best single estimate for the method accuracy for the seven compounds ranged from 9.1% (cyclohexane) to 11.8% (CCl4). The highest upper limit of the 90% confidence interval of the accuracy is less than 25% (13.7% for CCl4). Because both estimates of accuracy are less than 25%, there is a 95% confidence that the method accuracy is not more than 25%. The method meets this criterion. Because accuracy was calculated in EPA Method TO-15 using different nonstatistical methods, the results of this investigation for methylene chloride and CCl4 were different from TO-15 reported values. The best single estimate of accuracy for methylene chloride was 10.6 (95% confidence interval: 9.4 to 12.1), which differs from the 8.6 (n = 30) and 31.4 (n = 3) reported by two different EPA laboratories.Citation8 The best single estimate of accuracy for CCl4 was 11.8 (95% confidence interval: 10.4 to 13.7), which differs from the 12.4 (n = 30) and 9.4 (n = 3) reported by two different EPA laboratories.Citation8
Table 4. Results of precision, bias, and accuracy analysis for the ppb-concentration level method
Because this evacuated canister sampling method met all of the NIOSH recommended validation criteria for sampling six of the seven VOCs, it is suitable for sampling those VOCs. With regards to acetone, the stability needs to be investigated further, but this analyte passed the bias and accuracy part of the method validation. A particular challenge of developing a canister method is replicating the matrix effect because synthetic laboratory standards only poorly simulate ambient or indoor air.Citation10 The use of nitrogen for dilution to produce the various standards is a limitation of the study. The study did not investigate a matrix that would be more representative of those which could be encountered during actual sampling. Further investigation should be done on the method using air for dilution and whether other VOCs may interfere with the sampling and analysis of the compound of interest.
CONCLUSIONS
An evacuated canister sampling and analysis method for ppb-level concentrations of seven VOCs in air using a gas chromatograph with a mass selective detector was developed and validated for six compounds (excluding acetone) using NIOSH guidelines. This evacuated canister method had a bias of less than 10% for chloroform, methylene chloride, methyl ethyl ketone, hexane, CCl4, and cyclohexane. The precision of the method ranged from 2.4 to 2.9 for these compounds. The best single estimate and the 90% confidence interval estimate for the accuracy was less than 25%. The method for six of the seven compounds (excluding acetone), which are potential CWA simulants or interferents, met the NIOSH guidelines for inclusion into the NMAM.
Supplementary Material
Download Zip (56.9 KB)ACKNOWLEDGMENTS
This study was supported by NIOSH. The authors acknowledge Douglas Markle of Entech, Inc., for his assistance in setting up the preconcentration method. The findings and conclusions in this report are those of the authors and do not necessarily represent the views of NIOSH. Mention of commercial product or trade name does not constitute endorsement by the Centers for Disease Control/NIOSH.
REFERENCES
- Morton , D.W. 2008 . Chemical Security Analysis Center, Aberdeen Proving Ground , MD : Personal communication .
- Manual of Analytical Methods (NMAM)—Volatile Organic Compounds (Screening) Method 2549; Department of Health and Human Services National Institute for Occupational Safety and Health (NIOSH) Publication No. 2003-154; NIOSH: Cincinnati, OH, 2003 http://www.cdc.gov/niosh/docs/2003-154/pdfs/2549.pdf (http://www.cdc.gov/niosh/docs/2003-154/pdfs/2549.pdf) (Accessed: 1 September 2010 ).
- Manual of Analytical Methods (NMAM)-Ketones I Method 1300; Department of Health and Human Services National Institute for Occupational Safety and Health (NIOSH) Publication No. 2003-154; NIOSH: Cincinnati, OH, 2003 http://www.cdc.gov/niosh/docs/2003-154/pdfs/1300.pdf (http://www.cdc.gov/niosh/docs/2003-154/pdfs/1300.pdf) (Accessed: 1 September 2010 ).
- Manual of Analytical Methods (NMAM)-Hydrocarbons, BP 36°-216°C Method 1500; Department of Health and Human Services National Institute for Occupational Safety and Health (NIOSH) Publication No. 2003-154; NIOSH: Cincinnati, OH, 2003 http://www.cdc.gov/niosh/docs/2003-154/pdfs/1300.pdf (http://www.cdc.gov/niosh/docs/2003-154/pdfs/1300.pdf) (Accessed: 1 September 2010 ).
- Guidelines for Air Sampling and Analytical Methods and Evaluation; Department of Health and Human Services National Institute for Occupational Safety and Health (NIOSH) Publication No. 95-117; NIOSH: Cincinnati, OH, 1995 http://www.cdc.gov/niosh/nmam/pdfs/chapter-e.pdf (http://www.cdc.gov/niosh/nmam/pdfs/chapter-e.pdf) (Accessed: 1 September 2010 ).
- Wang , D.K.W. and Austin , C.C. 2006 . Determination of Complex Mixtures of Volatile Organic Compounds in Ambient Air: an Overview . Anal. Bioanal. Chem. , 386 : 1089 – 1098 . doi: 10.1007/s00216-006-0575–5
- Compendium Method TO-14A: Determination of Volatile Organic Compounds (VOCs) in Ambient Air Using Specially Prepared Canisters with Subsequent Analysis by Gas Chromatography: Compendium of Methods for the Determination of Toxic Organic Compounds in Ambient Air, 2nd ed.; EPA 625/R-96/010b; U.S. Environmental Protection Agency; Office of Research and Development: Cincinnati, OH, 1999 http://www.epa.gov/ttn/amtic/files/ambient/airtox/to-14ar.pdf (http://www.epa.gov/ttn/amtic/files/ambient/airtox/to-14ar.pdf) (Accessed: 1 September 2010 ).
- Compendium of Methods for the Determination of Toxic Organic Compounds in Ambient Air, Second Edition Compendium Method TO-15: Determination of Volatile Organic Compounds (VOCs) in Air Collected in Specially-Prepared Canisters with Subsequent Analysis by Gas Chromatography, Mass Spectrometry (GC-MS); EPA 625/R-96/010b; U.S. Environmental Protection Agency; Office of Research and Development: Cincinnati, OH, 1999 http://www.epa.gov/ttn/amtic/files/ambient/airtox/to-15r.pdf (http://www.epa.gov/ttn/amtic/files/ambient/airtox/to-15r.pdf) (Accessed: 1 September 2010 ).
- Eklund , B.M. , Williams , C.H. , Bontempo , L.W. , Isbell , M. and Loos , K.R. 2004 . Development and Validation of a Canister Method for Measuring Ethylene Oxide in Ambient Air . Environ. Sci. Technol. , 38 : 4200 – 4205 . doi: 10.1021/es049861o
- Batterman , S.A. , Guo-Zheng , Z. and Baumann , M. 1998 . Analysis and Stability of Aldehydes and Terpenes in Electropolished Canisters . Atmos. Environ. , 32 : 1647 – 1655 . doi: 10.1016/S1352–2310(97)00417–2
- Daughtrey , E.H. Jr. , Oliver , K.D. , Adams , J.R. , Kromiller , K.G. , Lonnenman , W.A. and McClenny , W.A.A. 2001 . Comparison of Sampling and Analysis Methods for Low ppb Levels of Volatile Organic Compounds in Ambient Air . J. Environ. Monitor. , 3 : 166 – 174 . doi: 10.1039/ b007158g
- Evans , J.C. , Huckaby , J.L. , Mitroshkov , A. , Julya , J.L. , Hayes , J.C. , Edwards , J.A. and Sasaki , L.M. 1998 . 32 Week Holding-Time Study of Summa Polished Canisters and Triple Sorbent Traps Used to Sample Organic Constituents in Radioactive Waste Tank Vapor Headspace . Environ. Sci. Technol. , 32 : 3410 – 3417 . doi: 10.1021/es9803153
- Hoshi , J. , Higuchi , M. , Sasaki , Y. and Korenaga , T. 2007 . Determination of Oxygenated Volatile Organic Compounds in Ambient Air Using Canister Collection—Gas Chromatography/Mass Spectrometry . Anal. Sci. , 23 : 987 – 992 . doi: 10.2116/analsci.23.987
- Kelly , T.J. , Callahan , P.J. , Pleil , J. and Evans , G.F. 1993 . Method Development and Field Measurements for Polar Volatile Organic Compounds in Ambient Air . Environ. Sci. Technol. , 27 : 1146 – 1153 . doi: 10.1021/ es00043a014
- Ochiai , N. , Daishima , S. and Cardin , D.B. 2003 . Long-Term Measurement of Volatile Organic Compounds in Ambient Air by Canister-Based One-Week Sampling Method . J. Environ. Monit. , 5 : 997 – 1003 . doi: 10.1039/B307777M
- Rossner , A. , Warner , S.D. , Vyskocil , A. , Tardif , R. and Farant , J.-P. 2004 . Performance of Small Evacuated Canisters Equipped with a Novel Flow Controller for the Collection of Personal Air Samples . J. Occup. Environ. Hyg. , 1 : 173 – 181 . doi: 10.1080/1545962040424483
- Rossner , A. and Wick , D.P. 2005 . A Field Study to Assess the Long-Term Sampling Feasibility of Evacuated Canisters and the Development of a Mathematical Model to Analyze Potential Sampling Bias . J. Occup. Environ. Hyg. , 2 : 474 – 480 . doi: 10.1080/1545962050025779
- Ochiai , N. , Tsuji , A , Nakamura , N. , Daishimaa , S. and Cardin , D.B. 2002 . Stabilities of 58 Volatile Organic Compounds in Fused-Silica-Lined and SUMMA Polished Canisters under Various Humidified Conditions . J. Environ. Monit. , 4 : 879 – 889 . doi: 10.1039/b209210g
- Manual of Analytical Methods (NMAM) Chapter E—Development and Evaluation of Methods; Department of Health and Human Services National Institute for Occupational Safety and Health (NIOSH) Publication No. 2003-154; NIOSH: Cincinnati, OH, 2003 http://www.cdc.gov/niosh/docs/2003-154/pdfs/chapter-e.pdf (http://www.cdc.gov/niosh/docs/2003-154/pdfs/chapter-e.pdf) (Accessed: 1 September 2010 ).
- Casteel, C.; Cardin, D. Enhanced Performance and Dynamic Range for EPA Method TO-15; Application Note 103; Entech Instruments: Simi Valley, CA http://www.entechinst.com/pdf/extended_range_to-15.pdf (http://www.entechinst.com/pdf/extended_range_to-15.pdf) (Accessed: 1 January 2011 ).
- Bartlett , M.S. 1937 . Some Examples of Statistical Methods of Research in Agricultural and Applied Biology . J. Roy. Stat. Soc. Suppl. , 4 : 158 – 159 .