Abstract
Wider use of pharmacological models would facilitate the development of new drugs for Alzheimer's disease (AD), The two main models currently used are based on the cholinergic and glutamatergic hypotheses of AD, Although they lead to some of the attention and memory impairment observed in AD, they do not fully reproduce the AD pattern. The few studies that used a combination modeling approach, ie, the simultaneous administration of several drugs with the aim of impairing several neurotransmitters or different aspects of a single system, have reported no or marginal cumulative effect. On the basis of current understanding of glutamate and acetylcholine involvement in AD pathophysiology, we suggest that models using selective muscarinic-1 (M1) receptor blockers would better mimic the status of the cholinergic system in AD, This kind of model might be suitable for the assessment of drugs that do not act directly on the cholinergic system.
Un empleo más generalizado de modelos farmacológicos facilitaría el desarrollo de nuevos fármacos para la enfermedad de Alzheimer (EA). Los dos modelos principales actualmenie en uso se basan en las hipótesis colinérgica y glutamatérgica de la EA. Aunque ambos modelos se orientan hacia algunos de los deterioros en la atención y en la memoria observados en la EA, ellos no reproducen totalmente el patrón de la EA. Los pocos estudios que utilizan una combination de modelos (ej. la administration simultánea de varios fármacos con el objetivo de afectar algunos neuroiransmisores o diferentes aspectos de un sistema único) no han informado de ningún efecto significativo, a lo más el efecto acumulativo es marginal. De acuerdo con el conocimiento actual del compromiso del glutamato y de la acetílcolina en la fisiopatología de la EA, nosotros sugerimos que los modelos que emplean bloqueadores selectivos de los receptores muscarinicos-1 (M1) podrían imitar mejor la situation del sistema colinérgico en la EA, Esta clase de modelo sería conveniente para la evaluación de fármacos que no actúan directamente en el sistema colinérgico.
Le recours plus fréquent aux modèles pharmacologiques devrait faciliter le développement de nouvelles thérapeutiques dans la maladie d'Alzheimer (MA). Les deux principaux modèles utilisés actuellement sont fondés sur l'hypothèse d'une implication du système cholinergique et du système glutamatergique dans la maladie d'Alzheimer. Mais si l'on peut observer dans ces modèles l'apparition de certains des troubles de l'attention et de la mémoire observés au cours de la MA, ils ne génèrent pas la totalité des manifestations de la pathologie. Dans les quelques études qui ont utilisé un modèle combinant l'administration simultanée de plusieurs molécules dans le but d'altérer différents neurotransmetteurs ou différentes étapes d'un même système, l'effet cumulatif obtenu a été nul ou négligeable. En nous appuyant sur nos connaissances actuelles de l'implication du glutamate et de l'acetylcholine dans la physiopathologie de la MA, nous proposons d'utiliser des inhibiteurs sélectifs des récepteurs muscariniques-1 (M1) pour reproduire au mieux l'état du système cholinergique dans la MA, Ce type de modèle pourrait permettre d'évaluer des molécules qui n'agissent pas directement sur le système cholinergique.
All the professionals involved are convinced that finding effective treatments for Alzheimer's disease (AD) should be a priority for the pharmaceutical industry. AD is a wonderful challenge for industry. However, research and development in this field can also be a risky business. There is currently no consensus on the pathophysiology of AD on which drug development can rely. The clinicopathologic picture that we call AD may actually be a syndrome, with many possible causes. As a consequence, we still have no reliable, positive diagnostic test that can be applied on an individual basis, which leads to the risk of recruiting very heterogeneous patient populations for clinical trials. The low response rate to acetylcholine esterase inhibitors probably illustrates these uncertainties.
Before starting expensive trials, pharmaceutical companies clearly need to assess the validity of the underlying concept in the early phases of development. Part of the answer can come from animal models. However, if a pharmacological effect is observed, it must still be confirmed in humans.
Some advocate early administration to patients, but this is not necessarily the simplest method. The risk of heterogeneous recruitment to clinical trials is an important point. If the goal is to measure clinical improvement, the drug will probably be administered for a long period of time. If the trial intends to assess changes in surrogate markers, these must be defined. Recruiting groups homogeneous for a selected marker can be difficult and time-consuming, and at this phase of development we need to go as fast as possible. Keeping pools of untreated patients at hand for this purpose, and depriving them of currently available drugs, is ethically questionable.
It is easier and faster to work with healthy volunteers, and, better, young healthy volunteers. This requires the use of models, in which the putative drug is evaluated for its ability to reverse either induced cognitive impairment or associated markers (using electroencephalogram [EEG], positron emission tomography [PET] scan, and functional magnetic resonance imaging [fMRI] changes), or both.
The scopolamine model
Scopolamine is a nonselective,Citation1 competitiveCitation2 muscarinic receptor blocker. The scopolamine model has its roots in the cholinergic hypothesis of aging and AD, and has played a major role in its construction, which we will recall briefly here.
From the beginning of the 20th century until the midfifties, scopolamine was used in obstetrics to induce a twilight state and amnesia during childbirth.Citation3 In the sixties and seventies, it became obvious that regions rich in cholinergic afférents, such as the hippocampus, were involved in memory processes (see reference 4 for a review). In 1965, acetylcholine esterase activity was shown to be lowered in AD.Citation5 In 1974, Drachman and LeavittCitation6 administered scopolamine to healthy young volunteers, who then displayed a memory profile very close to that observed in elderly people.
Two to three years later, three independent research teamsCitation7-Citation9 reported a decreased activity of choline acetyltransferase (CAT), the enzyme responsible for acetylcholine (Ach) synthesis, in the cortex of AD patients. This decrease was shown to be correlated with brain lesions and clinical status.Citation10-Citation11 It was soon found that neuronal loss occurs in the forebrain basal nucleus of MeynertCitation12 and medial septal nucleus,Citation13 which are the source of neocortical and hippocampal cholinergic afferent fibers, respectively.Citation14-Citation16 In its early version,Citation4 the cholinergic hypothesis stated nothing about etiological factors, did not address the additional roles that ACh dysfunction may play in other neurobehavioral disturbances of aging and dementia, and did not imply any exclusive or solitary involvement of the cholinergic system in age-related memory loss. It was a kind of “black box” model, in which an unknown pathophysiological process induces deficiency in various neurotransmission pathways thought to be responsible for the cognitive and behavioral aspects of aging and dementia. Despite obvious shortcomings (see references 17-20 for review and discussion), the cholinergic hypothesis legitimized the development of the cholinergic drugs we prescribe today and the administration of scopolamine as a model of investigation for AD research.
The scopolamine model was used in cognitive research to study the clinical correlates of ACh deficiency (see reference 21 for a review). It was applied to elderly subjects and AD patientsCitation22-Citation33 as a marker of cholinergic sensitivity, with the purpose of improving the diagnosis and staging of the disease. It failed, however, to predict cognitive decline on the basis of the subjects' sensitivity.Citation34
Animal studies assessing the reversal of scopolamineinduced memory impairment by various compounds are too numerous to be cited exhaustively. This approach has also been used in humans with the following molecules: physostigmine,Citation35-Citation40 velnacrine,Citation40 choline,Citation41 RO 15-1788,Citation39 moclobemide,Citation42,Citation43 RU 41656,Citation44 L-α-glycerylphosphoryleholine,Citation45 oxiracetam,Citation46 aniracetam and piracetam,Citation47 tenilsetam,Citation48 BMY 21502,Citation49 D-cycloserine,Citation50 SDZ ENS-163,Citation51 and ZK-93426.Citation52 However, the scopolamine model has not become a standard tool in the early assessment of drugs.
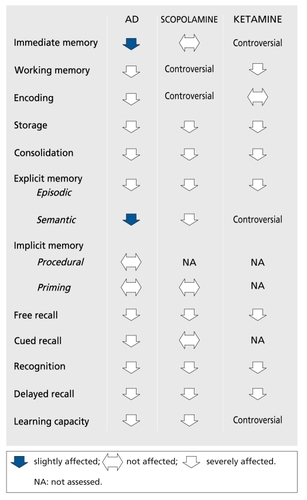
One reason for this is that the cognitive changes induced by scopolamine do not really mimic the AD picture. The details of the differences listed in (based on references 28, 40, and 53-63) are open to discussion, but there is a general agreement on the fact that, as WesnesCitation40 wrote, all the scopolamine-induced deficiencies are also observed in AD, while the reverse is not always true. The same is observed in neurological investigations. The electrophysiological effects of scopolamine (reviewed in reference 64) are close on EEG and similar on visual evoked potentials to those of AD. In PETCitation65-Citation68 and single photon emission computed tomography (SPECT)Citation69 studies, scopolamine induces cerebral blood flow (CBF) and glucose metabolism changes, which are sometimes divergent and region-specific, but in all cases different from the pattern observed in AD.
The ketamine model
Ketamine is a noncompetitive N-methyl-D-aspartate (NMDA) receptor antagonist.Citation70-Citation71 Its administration in order to produce a model is the correlate of the glutamatergic hypothesis of AD (reviewed in reference 72). Two, apparently opposite, glutamatergic hypotheses have been proposed. The excitotoxic hypothesis states that there is a glutamatergic hyperactivity in AD. Domoic acid poisoning in humans was responsible for irreversible memory loss.Citation73 NeuronalCitation74 and astroglialCitation75 glutamate transporter dysfunction in AD could result in excess glutamate in the synaptic cleft and in excitotoxic neuronal damage. This hypothesis is consistent with the beneficial effects of memantineCitation76 and lamotrigineCitation77 in AD patients. Some findings provide a link with the histopathological lesions that are the hallmarks of AD. Kainic acid injection in the rat leads to decreased neuronal amyloid precursor protein (APP) 695 mRNA owing to neuronal death, and increased astrocytic APP 770 mRNA,Citation78 which has been found selectively increased in AD in comparison with other neurodegenerative disordersCitation79 and associated with plaques.Citation80 β- Amyloid sensitizes neurons to glutamate toxicityCitation81 and also enhances glutamate release by macrophages.Citation82 Furthermore, in neuronal culture, glutamate was shown to enhance tau gene expressionCitation83 and induce paired helical filaments similar to those found in AD.Citation84 The hypoglutamatergic hypothesis has been extensively reviewed elsewhere (see Newcomer et al, in this issue). The ketamine model is its application. Ketamine is mainly used in the field of schizophrenia research to provoke psychotomimetic as well as cognitive effects.Citation85-Citation94 These studies did not all assess the same functions or use the same paradigm to assess a particular function. Despite this limitation, when these studies are summarized and the profile of ketamine effects compared with that of AD (Figure1),Citation60-Citation63 the situation is the same as that for the scopolamine model: the functions affected by ketamine are affected in AD, but the reverse is not necessarily the case.
Future directions
The two main models proposed thus lead to some of the attentional and memory impairment observed in AD, but do not fully reproduce the AD pattern. Two options are therefore possible. Since multiple neurotransmitter systems are affected in AD, it has been suggestedCitation95 that combination modeling through simultaneous administration of drugs that impair several neurotransmitters or different aspects of a single system could mimic the AD pattern more closely. The fewpublished studies on this strategy add mecamylamine,Citation96 m-chlorophenylpiperazine (mCPP),Citation97 metergoline, or haloperidolCitation98 to scopolamine, and report noCitation98 or marginalCitation96-Citation97 cumulative effect. Although NMDA antagonists were shown to potentiate the amnesic effect of scopolamine in the rat,Citation58 no study on this combination in humans has been published to date. Beyond the weakness of their effects, combined models are so complex that they become difficult to understand - and particularly difficult to manipulate - in the assessment of cognitive enhancers. Another method is to take advantage of the recent advances in our understanding of AD. Currently available data support the view that neuronal and synaptic loss, rather than secondary neurotransmission disruption, is most likely responsible for cognitive changes in AD.Citation99 They also allow attempts to integrate neurotransmitter changes into a more comprehensive theoretical framework. The cholinergic hypothesis in its current version () focuses on the reciprocal modulatory influences of cholinergic transmission and APP processing (reviewed in references 100 and 101). β-Amyloid (βA) is known to be neurotoxic at high (micromolar) concentrations.Citation102 In vitro, soluble βA at picomolar to nanomolar levels is not toxic but does inhibit synthesis and stimulated release of ACh.Citation103-Citation105 Studies on the possible inhibitory effect of βA on CAT activityCitation104-Citation106 gave divergent results. βA appears to exert its effect on ACh synthesis and release through depletion of ACh precursors. It has been shown to disrupt the activity of pyruvate dehydrogenase,Citation106 which generates acetyl coenzyme A (CoA) from pyruvate and was found to be decreased in the cortex of AD patients,Citation107 and to inhibit highaffinity choline uptake.Citation104 This could have an indirect neurotoxic effect, since cholinergic neurons deprived of choline have been shown to break down phosphatidylcholine from intracellular organelle membranes to provide additional choline.Citation108
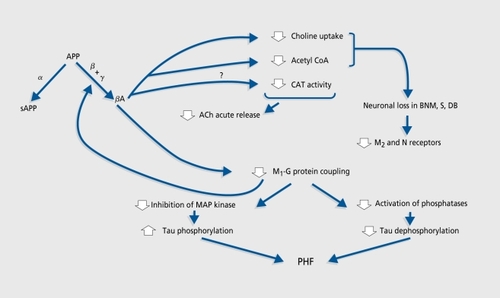
Although there is no general consensus (see reference 109 for review), it is thought that postsynaptic muscarinic Mi acetylcholine receptor (AChR) density is unchanged in AD, while those of presynaptic M2 and nicotinic AChRs are reduced.Citation110,Citation111 It has been shown that activation of protein kinase C through Mj (and M3) AChRs lowers βA production by favoring the nonamyloidogenic processing of APPCitation112,Citation113 Despite their unchanged density, M1 receptors could be dysfunctionalCitation114,Citation115 because of defective coupling to Gq/11 proteins.Citation116 This could lead to increased βA production, which would further impair M1 AChR signal transduction.Citation117 M1 AchR-G protein uncoupling could also favor protein tau phosphorylation and thus paired helical filament formation through disinhibition of mitogen-activated protein (MAP) kinasesCitation118 and decreased efficiency of taudephosphorylation.Citation119
Despite the uncertainties that remain, it is clear that the cholinergic deficiency can no longer be seen as a late consequence of neuropathological changes, but at least as a contributor to the cascade of events leading to fullblown dementia.
The glutamatergic hypothesis has also been revisited. Its current version (see Newcomer et al, in this issue) reconciles the former hyper- and hypoglutamatergic hypotheses by proposing a two-stage process.
In the first stage, βA increases the sensitivity of NMDA receptors to normal concentrations of glutamate, leading to destruction of NMDA-bearing GABAergic (GABA: gamma-aminobutyric acid), noradrenergic (NE), and serotonergic (5-HT) neurons, which have an inhibitory action on basal forebrain cholinergic, anterior thalamic glutamatergic, and cortical neuropeptide Y (NPY) neurons either directly (GABA, NE) or through activation of GABA neurons (5-HT).
This loss of inhibition leads to hyperstimulation of cortical and corticolimbic neurons, which then degenerate, as do the hyperactive cholinergic, glutamatergic, and NPY neurons.
This suggests that for both cholinergic and glutamatergic neurons, the hypoactive, symptomatic stage is preceded by a hyperactive one. These new versions of the cholinergic and glutamatergic hypotheses make it necessary for us to reappraise our models. The goal of an acute pharmacological model is to transiently reproduce the hypoactive, symptomatic stage. According to the scheme proposed by Newcomer et al elsewhere in this issue, NMDA blockers induce transient hyperactivity of basal forebrain cholinergic, anterior thalamic glutamatergic, and cortical NPY neurons. It is likely that the mechanism by which acute administration of NMDA blockers produces memory impairment is different and does not involve the two-stage sequence proposed as a chronic model. The finding that pretreatment with haloperidol reduces ketamineinduced impairment in executive cognitive functionsCitation91 nonetheless suggests that the cognitive effect of NMDA blockade is indirect and nonselective. Higher selectivity, which would also avoid psychotomimetic symptomatology, might be achieved by acting downstream of the NMDA receptor. For the particular posterior cingulate and retrosplenial region, the best choice would be to give m3 and/or kainate receptor blockers. Another target of choice is the hippocampus, in which the most common muscarinic receptor is the mi subtype; the m2 subtype represents 15% and the m3 subtype globally less than 12%.Citation120 Moreover, specific blockade of the m1 receptors would best reproduce their status in AD, where they are hypostimulatcd (because of presynaptic neuronal loss) and dysfunctional. The only molecule which is more or less selective for the mi receptorCitation121 and available for human use is pirenzepine. It is said to cross the blood-brain barrier poorly,Citation122 but very few studies have assessed its central effects in manCitation123-Citation125 and we think it deserves further study.
Do neurotransmitter-based pharmacological models have a future?
The way the cognitive symptoms are produced in AD is complex and many therapeutic strategies already in development address βA metabolism and toxicity,Citation126-Citation128 rather than cholinergic deficiency. However, D-cycloserine, which does not act on the cholinergic system but modulates the NMDA receptor, has been shown to attenuate the effect of scopolamine on memory.Citation50 Moclobemide, a selective monoamine oxidase A (MAOA) inhibitor,Citation42,Citation43 and thyrotropin-releasing hormonc (TRH)Citation129 were also able to partly reverse the scopolamine-induced deficits. In the animal, the same has been observed with estrogensCitation130 and GM1 gangliosides.Citation131 Given these data and the current view that we have on the involvement of the cholinergic deficiency, it is very possible that new compounds, which do not act directly on the cholinergic system, could be effective on cholinergic models. Neurotransmitter-based models still have a place in our armamentarium, although efforts should be made to develop other approaches.
Conclusion
Whatever the model chosen, we must admit that it is e impossible to reproduce the full AD cognitive pattern. Instead, we can try to produce some aspects of memory it impairment, which is considered as the core symptom of:- the disease. The method used should be as simple and selective as possible in order to allow its manipulation. Selective ligands are currently in developmentCitation132; accelerating the toxicological studies of these compounds could allow us to work this way in the near future.
Selected abbreviations and ancronyms
| Ach | = | acetylcholine |
| AChR | = | acetylcholine receptor |
| AD | = | Alzheimer's disease |
| APP | = | amyloid precursor protein |
| βA | = | β-amyloid |
| CAT | = | choline acetyltransferase |
| NMDA | = | N-methyl-D-aspartate |
| NPY | = | neuropeptide Y |
REFERENCES
- BoldenC.CusackB.RichelsonE.Antagonism by antimuscarinic and neuroleptic compounds at the five cloned human muscarinic cholinergic receptors expressed in Chinese hamster ovary cells. J Pharmacol Exp Ther.19925765801346637
- YamamuraHI.SnyderSH.Muscarinic cholinergic receptor binding in the longitudinal muscle of the guinea pig ileum with [3H]quinuclidinyl benzilate. Mol Pharmacol.197410861867
- GaussCJ.Geburten in kunstlichem Dâmmerschlaf. Arch Gynaekol.190678579631
- BartusRT.DeanRL.PontecorvoMJ.FlickerC.The cholinergic hypothesis: a historical overview, current perspective, and future directions.Ann N Y Acad Sci.19854443323582990293
- PopeA.HessHH.LewinE.Microchemical pathology of the cerebral cortex in pre-senile dementias. Trans Am Neurol Assoc.19658915165828492
- DrachmanDA.LeavittJ.Human memory and the cholinergic system: a relationship to aging? Arch Neurol.1974301131214359364
- BowenDM.SmithCB.WhiteP.DavisonAN.Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain.19769945949611871
- DaviesP.MaloneyAJF.Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet.19762140363862
- PerryEK.PerryRH.BlessedG.TomlinsonBE.Necropsy evidence of central cholinergic deficits in senile dementia. Lancet.1977118964712
- PerryEK.TomlinsonBE.BlessedG.BergmannK.GibsonPH.PerryRH.Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. BMJ.1978214571459719462
- MountjoyCQ.RossorMN.IversenLL.RothM.Correlation of cortical cholinergic and GABA deficits with quantitative neuropathological findings in senile dementia. Brain.19841075075186722514
- WhitehousePJ.PriceDL.ClarkAW.CoyleJT.DeLongMR.Alzheimer's disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol.1981101221267283399
- NakanoI.HiranoA.Loss of large neurons in the medial septal nucleus in an autopsy case of Alzheimer's disease. J Neuropathol Exp Neurol.198241341
- MesulamMM.MufsonEJ.LeveyAI.WainerBH.Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol.19832141701976841683
- EverittBJ.SirkiàTE.RobertsAC.JonesGH.RobbinsTW.Distribution and some projections of cholinergic neurons in the brain of the common marmoset, Callithrix jacchus. J Comp Neurol.1988271533558
- AlonsoJR.UHS.AmaralDG.Cholinergic innervation of the primate hippocampal formation. II. Effects of fimbria/fornix transection. J Comp Neurol.19963755275518930785
- BartusRT.DeanRL.FisherSK.Cholinergic treatment for age-related memory disturbances: dead or barely coming of age? In: Crook, T, Bartus R, Ferris S, Gershon S, eds. Treatment Development Strategies for Alzheimer's Disease. Madison, Wis: Mark Powley Associates1986421450
- HaganJJ.The status of the cholinergic hypothesis of dementia. In: Nicholson D, ed. Anti-dementia Agents. Research and Prospects for Therapy. London, UK: Academic Press199485138
- SalterM.BrunoJP.HimmelheberAM.Cortical acetylcholine and attention: neuropharmacological and cognitive principles directing treatment strategies for cognitive disorders. In: Brioni JD, Decker MW, eds. Pharmacological Treatments of Alzheimer's Disease: Molecular and Neurobiological Foundations. New York, NY: Wiley-Liss1997105128
- FrancisPT.PalmerAM.SnapeM.WilcockGK.The cholinergic hypothesis of Alzheimer's disease: a review of progress. J Neurol Neurosurg Psychiatry.19996613714710071091
- LydonRG.Cholinergic neurons and memory: an historical perspective and overview of current research. In: Stone TW, ed. CMS Neurotransmitters and Neuromodulators: Acetylcholine. London, UK: CRC Press1995197232
- RichardsonJS.MillerPS.LemayJS.et al.Mental dysfunction and the blockade of muscarinic receptors in the brain of the normal elderly.Prog Neuropsychopharmacol Biol Psychiatry.198596516544089189
- SunderlandT.TariotP.MurphyDL.WeingartnerH.MuellerEA.CohenRM.Scopolamine challenges in Alzheimer's disease. Psychopharmacology.1985872472493931154
- SunderlandT.TariotPN.MuellerEA.MurphyDL.WeingartnerH.CohenRM.Cognitive and behavioral sensitivity to scopolamine in Alzheimer patients and controls. Psychopharmacol Bull.1985216766793898182
- SunderlandT.TariotPN.CohenRM.WeingartnerH.MuellerEA.MurphyDL.Anticholinergic sensitivity in patients with dementia of the Alzheimer type and age-matched controls. A dose-response study. Arch Gen Psychiatry1987444184263579494
- SunderlandT.TariotPN.NewhousePA.Differential responsivity of mood, behavior, and cognition to cholinergic agents in elderly neuropsychiatrie populations. Brain Res.19884723713893066441
- ZemishlanyZ.ThomeAE.Anticholinergic challenge and cognitive functions: a comparison between young and elderly normal subjects. IsrJ Psychiatry Relat Sci.19912832411800456
- FlickerC.FerrisSH.SerbyM.Hypersensitivity to scopolamine in the elderly. Psychopharmacology.19921074374411615141
- GitelmanDR.ProhovnikI.Muscarinic and nicotinic contributions to cognitive function and cortical blood flow. Neurobiol Aging.1992133133181522945
- RayPG.MeadorKJ.loringDW.ZamriniEW.YangXH.BuccafuscoJJ.Central cholinergic hypersensitivity in aging. J Geriatr Psychiatry Neurol.1992572771317178
- MolchanSE.MartinezRA.HillJL.et al.Increased cognitive sensitivity to scopolamine with age and a perspective on the scopolamine model. Brain Res Brain Res Rev.1992172152261467811
- RabeyJM.NeufeldMY.TrevesTA.SifrisP.KorczynAD.Cognitive effects of scopolamine in dementia. J Neural Transm Gen Sect1996103873881
- TariotPN.PatelSV.HendersonRE.Age-related decline in central cholinergic function demonstrated with scopolamine. Psychopharmacology199612550568724448
- BarkerA.JonesR.PriorJ.WesnesK.Scopolamine-induced cognitive impairment as a predictor of cognitive decline in healthy elderly volunteers: a 6-year follow-up. Int J Geriat Psychiatry.199813244247
- GhoneimMM.MewaldtSP.Studies on human memory: the interactions of diazepam, scopolamine and physostigmine. Psychopharmacology.19775216403551
- AnismanH.Time-dependent changes in activity, reactivity, and responsivity during shock: effects of cholinergic and catecholaminergic manipulations. Neurology.197727783790560649
- LiljequistR.MattilaMJ.Effect of physostigmine and scopolamine on the memory functions of chess players. Med Biol.19795742405
- MewaldtSP.GhoneimMM.The effects and interactions of scopolamine and methamphetamine on human memory. Pharmacol Biochem Behav.197910205210450929
- PrestonGC.WardC.LinesCR.PoppletonP.HaighJR.TraubM.Scopolamine and benzodiazepine models of dementia: cross reversals by RO 151788 and physostigmine. Psychopharmacology.1989984874942570433
- WesnesKA.SimpsonPM.WhiteL.et al.Cholinesterase inhibition in the scopolamine model of dementia. Ann N Y Acad Sci.1991 6402682711776749
- MohsRC.DavisKL.Interaction of choline and scopolamine in human memory. Life Sci.1985371931974010475
- WesnesKA.SimpsonPM.ChristmasL.AnandR.McClellandGR.The effects of moclobemide on cognition. J Neural Transm.198928(suppl)91102
- WesnesK.AnandR.LorscheidT.Potential of moclobemide to improve cerebral insufficiency identified using a scopolamine model of aging and dementia. Acta Psychiatr Scand.1990360(suppl)7172
- PatatA.KleinMJ.SurjusA.HucherM.GranierJ.RU 41656 does not reverse the scopolamine-induced cognitive deficit in healthy volunteers. Eur J Clin Pharmacol.1991412252311684155
- CanalN.FranceschiM.AlberoniM.CastiglioniC.De MolinerP.LongoniA.Effect of L-alpha-glycerylphosphorylcholine on amnesia caused by scopolamine. IntJ Clin Pharmacol Ther Toxicol.1991291031072071257
- PredaL.AlberoniM.BressiS.et al.Effects of acute doses of oxiracetam in the scopolamine model of human amnesia. Psychopharmacology.19931104214267870912
- WesnesK.AnandR.SimpsonP.ChristmasL.The use of a scopolamine model to study the potential nootropic effects of aniracetam and piracetam in healthy volunteers. Int J Geriatr Psychiatry.1990695102
- WesnesK.SimpsonPM.KiddAG.The use of a scopolamine model to study the nootropic effects of tenilsetam (CAS 997) in man. Med Sci Res.19871510631064
- ShrotriyaR.StalloneF.RobinsonD.BradyM.TempleD.Clinical pharmacology of BMY 21 502 - a potential cognitive enhancer. Abstracts of the 17th CINP, Kyoto, Japan 1990 9 10-14 Vol II 230
- JonesRW.WesnesKA.KirbyJ.Effects of NMDA modulation in scopolamine dementia. Ann N Y Acad Sci.19916402412441837979
- BrassEP.PolinskyR.SramekJJ.et al.Effects of the cholinomimetic SDZ ENS-163 on scopolamine-induced cognitive impairment in humans. J Clin Psychopharmacol.19951558627714229
- DukaT.OttH.RohloffA.VoetB.The effects of a benzodiazepine receptor antagonist beta-carboline ZK-93426 on scopolamine-induced impairment on attention, memory and psychomotor skills. Psychopharmacology.19961233613738867876
- GroberE.GitlinHL.BangS.BuschkeH.Implicit and explicit memory in young, old, and demented adults. J Clin Exp Neuropsychol.1992142983161572951
- DanionJM.ZimmermannMA.Willard-SchroederD.et al.Effects of scopolamine, trimipramine and diazepam on explicit memory and repetition priming in healthy volunteers. Psychopharmacology.19901024224242251340
- BishopKl.CurranHV.An investigation of the effects of benzodiazepine receptor ligands and of scopolamine on conceptual priming. Psychopharmacology.19981403453539877014
- RustedJM.Eaton-WilliamsP.WarburtonDM.A comparison of the effects of scopolamine and diazepam on working memory. Psychopharmacology.19911054424451798839
- LydonRG.NakajimaS.Differential effects of scopolamine on working and reference memory depend upon level of training. Pharmacol Biochem Behav.1992436456601438505
- LiHB.MatsumotoK.TohdaM.YamamotoM.WatanabeH.NMDA antagonists potentiate scopolamine-induced amnestic effect. Behav Brain Res.1997832252289062690
- DunneMP.Scopolamine and sustained retrieval from semantic memory. J Psychopharmacol.199041318
- EustacheF.AgnielA.Neuropsychologie clinique des démences: évaluation et prise en charge. Marseille, France: Solal1995
- FleischmanDA.GabrieliJDE.RemingerSL.VaidyaCJ.BennettDA.Object decision priming in Alzheimer's disease. J int Neuropsychol Soc.199844354469745233
- ChristensenH.GriffithsK.MackinnonA.JacombP.A quantitative review of cognitive deficits in depression and Alzheimer-type dementia. J Int Neuropsychol Soc.199736316519448376
- ErgisAM.Van der LindenM.DeweerB.Explicit memory, procedural learning and lexical priming in Alzheimer's disease. Cortex.1994301131268004981
- EbertU.KirchW.Scopolamine model of dementia: electroencephalogram findings and cognitive performance. Eur J Clin invest.1998289449499824440
- GrasbyPM.FrithCD.PaulesuE.FristonKJ.FrackowiakRS.DolanRJ.The effect of the muscarinic antagonist scopolamine on regional cerebral blood flow during the performance of a memory task. Exp Brain Res.19951043373487672026
- CohenRM.GrossM.SempleWE.NordahlTE.SunderlandT.The metabolic brain pattern of young subjects given scopolamine. Exp Brain Res.19941001331437813641
- BlinJ.PierceyMF.GiuffraME.MouradianMM.Metabolic effects of scopolamine and physostigmine in human brain measured by positron emission tomography J Neurol Sci.1994 12344518064320
- MolchanSE.MatochikJA.ZametkinAJ.et al.A double FDG/PET study of the effects of scopolamine in older adults. Neuropsychopharmacology.1994101911987916916
- SunderlandT.EspositoG.MolchanSE.CoppolaR.Differential cholinergic regulation in Alzheimer's patients compared to controls following chronic blockade with scopolamine: a SPECT study. Psychopharmacology.19951212312418545529
- AnisNA.BerrySC.BurtonNR.LodgeD.The dissociative anesthetics, ketamine and phencyclidine, selectively decrease excitation of central neurons by N-methyl-D-aspartate. Br J Pharmacol.198383179185
- YamakuraT.MoriH.MasakiH.ShimojiK.MoshinaM.Differential sensitivities of NMDA receptor channel subtypes to non-competitive antagonists. Neuroreport.199346876908347808
- KornbuberJ.WiltfangJ.The role of glutamate in dementia. J Neural Transm.199853(suppl)277287
- PerlTM.BedardL.KosatskyT.HockinJC.ToddECD.RemisRS.An outbreak of toxic encephalopathy caused by eating mussels contaminated with domoic acid. N Engl J Med.1990322177517801971709
- MasliahE.AlfordM.DeTeresaR.MalloryM.HansenL.Deficient glutamate transport is associated with neurodegeneration in Alzheimer's disease. Ann Neurol.1996407597668957017
- LiS.MalloryM.AlfordM.TanakaS.MasliahE.Glutamate transporteralterations in Alzheimer's disease are possibly associated with abnormal APP expression. J Neuropathol Exp Neurol.1997569019119258260
- TekinS.Aykut-BingôlC.TanridagT.AktanS.Antiglutamatergic therapy in Alzheimer's disease-effects of lamotrigine. J Neural Transm.19981052953039660108
- PanegyresPK.The effects of excitotoxicity on the expression of the amyloid precursor protein gene in the brain and its modulation by neuroprotective agents. J Neural Transm.19981054634789720974
- GôrtelmeyerR.ErblerH.Memantine in the treatment of mild-to-moderate dementia syndrome. A double-blind placebo-controlled study. Drug Res.199242904913
- TanakaS.LiuL.KimuraJ.et al.Age-related changes in the proportion of amyloid precursor protein mRNAs in Alzheimer's disease and other neurological disorders. Brain Res Mol Brain Res.1992153033101331685
- JohnsonSA.McNeillT.CordellB.FinchCE.Relation of neuronal APP751/APP-695 mRNA ratio and neuritic plaque density in Alzheimer's disease. Science.199028548572111579
- MattsonMP.ChengB.DavisD.BryantK.LieberburgI.RydelRE.β-Amy-loid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci.1992123763891346802
- KlegerisA.McGeerPL.Beta-amyloid protein enhances macrophage production of oxygen free radicals and glutamate. J Neurosci Res.1997492292359272645
- EsclaireF.LesortM.BlanchardC.HugonJ.Glutamate toxicity enhances tau gene expression in neuronal cultures. J Neurosci Res.1997493093189260742
- De BoniU.McLachlanDRC.Controlled induction of paired helical filaments of the Alzheimer type in cultured human neurons, by glutamate and aspartate. J Neurol Sci.1985681051182861254
- HarrisJA.BiersnerRJ.EdwardsD.BaileyLW.Attention, learning, and personality during ketamine emergence: a pilot study. Anesth Analg.1975541691721092205
- PanditSK.KotharySP.KumarS.Low dose intravenous infusion technique with ketamine. Anesthesia.198035669675
- GhoneimMM.HinrichsJV.MewaldtSP.PetersenRC.Ketamine: behavioral effects of subanesthetic doses. J Clin Psychopharmacol.1985570773988972
- OyeI.PaulsenO.MaursetA.Effects of ketamine on sensory perception: evidence for a role of W-methyl-D-aspartate receptors. J Pharmacol Exp Ther.1992260120912131312163
- KrystalJH.KarperLP.SeibylJP.et al.Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Arch Gen Psychiatry.1994511992148122957
- KrystalJH.KarperLP.BennetA.et al.Interactive effects of subanesthetic ketamine and subhypnotic lorazepam in humans. Psychopharmacology.19981352132299498724
- KrystalJH.D'SouzaDC.KarperLP.et al.Interactive effects of subanesthetic ketamine and haloperidol in healthy humans. Psychopharmacology.199914519320410463321
- MalhotraAK.FinalsDA.WeingartnerH.et al.NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology.1996 143013078703299
- AdlerCM.GoldbergTE.MalhotraAK.PickarD.BreierA.Effects of ketamine on thought disorder, working memory and semantic memory in healthy volunteers. Biol Psychiatry.1998438118169611670
- NewcomerJW.FarberNB.Jevtovic-TodorovicV.et al.Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology.1999201061189885791
- SunderlandT.MolchanSE.LittleJT.BahroM.PutnamKT.WeingartnerH.Pharmacologic challenges in Alzheimer disease and normal controls: cognitive modeling in humans. Alzheimer Dis Assoc Disord.199711(suppl 4)S23S269339269
- LittleJT.JohnsonDN.MinichielloM.WeingartnerH.SunderlandT.Combined nicotinic and muscarinic blockade in elderly normal volunteers: cognitive, behavioral and physiologic responses. Neuropsychopharmacology.19981960699608577
- BroocksA.LittleJT.MartinA.et al.The influence of ondansetron and mchlorophenylpiperazine on scopolamine-induced cognitive, behavioral, and physiological responses in young healthy controls. Biol Psychiatry.1998434084169532345
- VitielloB.MartinA.HillJ.et al.Cognitive and behavioral effects of cholinergic, dopaminergic, and serotonergic blockade in humans. Neuropsychopharmacology.19971615248981385
- Gomez-lslaT.HymanBT.Connection and cognitive impairment in Alzheimer's disease. In: Hyman BT, Duyckaerts C, Christen Y, eds. Connections, Cognition and Alzheimer's Disease (Research and Perspectives in Alzheimer's Disease). Berlin, Germany: Springer-Verlag1997149166
- LadnerCJ.LeeJM.Pharmacological drug treatment of Alzheimer disease: the cholinergic hypothesis revisited. J Neuropathol Exp Neurol.1998577197319720487
- AuldDS.KarS.QuirionR.P-Amyloid peptides as direct cholinergic neuromodulators: a missing link? Trends Neurosci.19982143499464686
- YoshikawaK.Neurotoxicity of amyloid p-protein and the amyloid pprotein precursor. In: Goate A, Ashall F, eds. Pathobiology of Alzheimer's Disease. London, UK: Academic Press1995145165
- KarS.SetoD.GaudreauP.QuirionR.p-Amyloid-related peptides inhibit potassium-evoked acetylcholine release from rat hippocampal formation. J Neurosci.199616103410408558231
- KarS.IssaAM.SetoD.AuldDS.CollierB.QuirionR.Amyloid p-peptide inhibits high-affinity choline uptake and acetylcholine release in rat hippocampal slices. J, Neurochem.199870217921879572306
- PedersenWA.KloczewiakMA.BlusztajnJK.Amyloid beta protein reduces acetylcholine synthesis in a cell line derived from cholinergic neurons of the basal forebrain. Proc Natl Acad Sci USA.199693806880718755604
- HoshiM.TakashimaA.MuramayaM.et al.Non-toxic amyloid beta peptide 1-42 suppresses acetylcholine synthesis. Possible role in cholinergic dysfunction in Alzheimer's disease. J Biol Chem.1997272203820418999897
- SheuKFR.KimYT.BlassJP.WekslerME.An immunohistochemical study of the pyruvate dehydrogenase deficit in Alzheimer's disease brain. Ann Neurol.1985174444494004169
- MaireJCE.WurtmanRJ.Choline production from choline-containing phospholipids: a hypothetical role in Alzheimer's disease and aging. Prog Neuropsychopharmacol Biol Psychiatry198486376426543397
- PaviaJ.de CeballosML.de la CuestaFS.Alzheimer's disease: relationship between muscarinic cholinergic receptors, p-amyloid and tau proteins. Fundam Clin Pharmacol.1998124734819794144
- WhitehousePJ.MartinoAM.MarcusKA.et al.Reductions in acetylcholine and nicotinic binding in several degenerative diseases. Ann Neurol.198845722724
- NordbergA.AlafuzoffI.WindbladB.Nicotinic and muscarinic subtypes in the human brain: changes with aging and dementia. J Neurosci Res.1992311031111613816
- BuxbaumJD.OishiM.ChenHI.et al.Cholinergic agonists and interleukin-1 regulate processing and secretion of Alzheimer β4 amyloid protein precursor. Proc Natl Acad Sci U S A.19928910075100781359534
- NitschRM.SlackBE.WurtmanRJ.GordonJH.Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science.19922583043071411529
- GreenwoodAF.PowersRE.JopeRS.Phosphoinositide hydrolysis, G alpha q, phospholipase C, and protein kinase C in post mortem human brain: effects of post mortem interval, subject age, and Alzheimer's disease. Neuroscience.1995691251388637611
- JopeRS.SongL.PowerRE.Cholinergic activation of phosphoinositide signalling is impaired in Alzheimer's disease brain. Neurobiol Aging.1997181111208983039
- FlynnDD.WeinsteinDA.MashDC.Loss of high-affinity agonist binding to M, muscarinic receptors in Alzheimer's disease: implication for the failure of cholinergic replacement therapies. Ann Neurol.1991292562622042942
- KellyJF.FurukawaK.BargerSW.et al.Amyloid beta-peptide disrupts carbachol-induced muscarinic cholinergic signal transduction in cortical neurons. Proc Natl Acad Sci U SA.19969367536758
- QianN.RussellM.JohnsonGL.Acetylcholine muscarinic receptor regulation of the Ras/Raf/MAP kinase pathway. Life Sci.19955694594910188797
- SadotE.GurwitzD.BargJ.BeharL.GinzburgI.FisherA.Activation of m, muscarinic acetylcholine receptor regulates the tau phosphorylation in transfected PC12 cells. J Neurochem.1996668778808592166
- SchliebsR.RessnerS.Distribution of muscarinic acetylcholine receptors in the CNS. In: Stone TW, ed. CMS Neurotransmitters and Neuromodulators: Acetylcholine. London, UK: CRC Press19956783
- MoriyaH.TakagiY.NakanishiT.HayashiM.TaniT.HirotsuI.Affinity profiles of various muscarinic antagonists for cloned human muscarinic acetylcholine receptor (MACHR) subtypes and MACHRS in rat heart and submandibular gland. Life Sci.1999642351235810374898
- JaupBH.BlomstrandC.Cerebrospinal fluid concentrations of pirenzepine after therapeutic dosage. ScandJ Gastroenterol.198015(suppl 66)3537
- StacherG.BauerP.SchmiererG.SteinringerH.The effect of intramuscular pirenzepine on oesophageal contractile activity and lower oesophageal sphincter pressure under fasting conditions and after a standard meal. A double-blind study. IntJ Clin Pharmacol Biopharm.197917442448511419
- FinkM.IrwinP.EEG and behavioral effects of pirenzepine in normal volunteers. Scand J Gastroenterol.198066(suppl)3946
- WaltersL.BartelP.SommersDK.BeckerP.The effects of anticholinergics on the photopalpebral reflex, memory and mood. Methods Find Exp Clin Pharmacol.1988104194253419245
- MooreCL.WolfeM.Inhibition of p-amyloid formation as a therapeutic strategy. Exp Opin Ther Patents.19999135146
- SotoC.Plaque busters: strategies to inhibit amyloid formation in Alzheimer's disease. Mol Med Today.1999534335010431167
- SchenkD.BarbourR.DunnW.et al.Immunization with amyloid-p attenuates Alzheimer-disease-like pathology in the PDAPP mouse.Nature.199940017317710408445
- MolchanSE.MellowAM.LawlorBA.et al.TRH attenuates scopolamine-induced memory impairment in humans. Psychopharmacology.199010084892104988
- GibbsRB.BurkeAM.JohnsonDA.Estrogen replacement attenuates effects of scopolamine and lorazepam on memory acquisition and retention. Horm Behav.1998341121259799622
- SilvaRH.FelicioLF.Frussa-FilhoR.Ganglioside GM1 attenuates scopolamine-induced amnesia in rats and mice. Psychopharmacology.19991411111179952034
- EglenRM.ChoppinA.DillonMP.HegdeS.Muscarinic receptor ligands and their therapeutic potential. Curr Opin Chem Biol.1999342643210419852