Abstract
An increasing level of N-methyl-D-aspartate (NMDA) receptor hypofunction within the brain is associated with memory and learning impairments, with psychosis, and ultimately with excitotoxic brain injury. As the brain ages, the NMDA receptor system becomes progressively hypofunctional, contributing to decreases in memory and learning performance. In those individuals destined to develop Alzheimer's disease, other abnormalities (eg, amyloidopathy and oxidative stress) interact to increase the NMDA receptor hypofunction (NRHypo) burden. In these vulnerable individuals, the brain then enters into a severe and persistent NRHypo state, which can lead to widespread neurodegeneration with accompanying mental symptoms and further cognitive deterioration. If the hypotheses described herein prove correct, treatment implications may be considerable. Pharmacological methods for preventing the overstimulation of vulnerable corticolimbic pyramidal neurons developed in an animal model may be applicable to the prevention and treatment of Alzheimer's disease.
La acentuación de la hipofunción del receptor N-metil-D-aspartato (NMDA) en el cerebro se ha asociado con deterioro en la memoria y el aprendizaje, con psicosis y, últimamente con daño exitotóxico. Así como el cerebro envejece, el sistema de receptores NMDA progresivamente se torna hipofuncional, lo que contribuye a una disminución de los rendimientos de memoria y aprendizaje. En aquellos individuos vulnerables para desarrollar la enfermedad de Alzheimer hay otras anormalidades (como amiloidopatías y el estrés oxidativo) que interactúan para aumentar la carga de la hipofunción del receptor NMDA. En estos individuos vulnerables, el cerebro entra en un severo y persistente estado de hipofunción del receptor NMDA, el cual puede conducir a una neurodegeneracíon generalizada, con síntomas mentales asociados y a un posterior deterioro cognitivo. Si se demuestra que la hipótesis descrita previamente es correcta, las implicancias terapéuticas de ella pueden ser considerables. Los métodos farmacológicos para prevenir la sobreestimulación de neuronas piramidales córtico-límbicas vulnerables desarrolladas en un modelo animal pueden ser aplicables a la prevención y al tratamiento de la enfermedad de Alzheimer.
L'augmentation de l'hypofonctionnement des récepteurs du N-méthyl-D-aspartate (NMDA) dans le cerveau est associée à des troubles de la mémoire et de l'apprentissage, qui s'accompagnent de psychose et évoluent vers des lésions cérébrales excitotoxiques. Lors du vieillissement cérébral, le système des récepteurs du NMDA devient progressivement hypofonctionnel, contribuant à la diminution des performances mnésiques et d'apprentissage. Chez les individus prédisposés à développer une maladie d'Alzheimer (MA), d'autres anomalies (telles que les amyloïdopathies et le stress oxydatif) interagissent pour augmenter le degré d'hypofonctionnement (NRHypo) des récepteurs du NMDA. Chez ces individus fragiles, le cerveau s'installe alors dans un état d'hypofonctionnement permanent et sévère qui peut conduire à une neurodégénération étendue associée à des symptômes mentaux et d'autres détériorations cognitives. Si les hypothèses décrites ici s'avèrent exactes, les implications thérapeutiques pourraient être considérables. Les méthodes pharmacologiques de prévention de l'hyperstimulation des neurones corticolimbiques vulnérables, développées dans des modèles animaux, pourraient s'appliquer à la prévention et au traitement de la maladie d'Alzheimer.
The amino acid glutamate (Glu) plays a central role in both the normal and abnormal functioning of the central nervous system (CNS). Glu is recognized to be the main excitatory neurotransmitter in the CNS, estimated to be released at. up to half of the synapses in the brain. In addition, Glu is also an excitotoxin that can destroy CNS neurons by excessive activation of excitatory receptors on dendritic and somal surfaces. Two major classes of Glu receptors, ionotropic and metabotropic, have been identified. Glu exerts excitotoxic activity through three receptor subtypes, which belong to the ionotropic family. These three receptors are named after agonists to which they are differentially sensitive, Ar-methyl-D-aspartate (NMDA), amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), and kainic acid (KA). Of these three, the NMDA receptor has been the most extensively studied and the most frequently implicated in CNS diseases.Citation1
Excessive activation of NMDA receptors (NMDA receptor hyperfunction [NRHyper]) plays an important role in the pathophysiology of acute CNS injury syndromes such as hypoxia-ischemia, trauma, and status epilepticus.Citation1,Citation2 Recently, hyperstimulation of AMPA/KA receptors and consequent excitotoxicity has been proposed to underlie neurodegeneration in amyotrophic lateral sclerosis (ALS, Lou Gerhig's DiseaseCitation3,Citation4)- The role of Glu excitotoxicity in the pathology of several other neuropsychiatrie disorders has been extensively reviewed elsewhereCitation1,Citation5 and will not be the focus of this paper. Instead, we will focus on the consequences of underexcitation of NMDA receptors (NMDA receptor hypofunction [NRHypo]).
Progressive increases in the severity of NRHypo within the brain, which can be induced experimentally in vivo using NMDA receptor antagonist drugs, can produce a range of clinically relevant effects on brain function, which are discussed below. In brief, underexcitation of NMDA receptors, induced by even relatively low doses of NMDA antagonist drugs, can produce specific forms of memory dysfunction. More severe NRHypo can produce a clinical syndrome that includes core features of psychosis. Finally, sustained and severe underexcitation of NMDA receptors in the adult brain is associated with a recently discovered and unconventional form of neurotoxicity, with well-characterized neuropathological features. Neuropathological changes that can be associated with sustained NRHypo include the disruption of neuronal cytoskeletons resulting in structures resembling neurofibrillary tangles (NFTs). These NRHypo-induced structures can occur in multiple brain regions, resembling the distribution of NFTs in Alzheimer's disease (AD). Differences in when NRHypo or an equivalent state is instilled in the brain (eg, early in brain development versus during older adulthood), and differences in the cause of the NRHypo state, can lead to differences in clinical and neuropathological presentations, as discussed in detail elsewhere.Citation6,Citation7
In the following sections, we will describe the role of NMDA receptor function in memory, the effect of NMDA receptor blockade on the expression of psy chosis, and the type of neuronal damage produced by severe and sustained hypoactivation of the NMDA receptor. We will then discuss the complex neural circuitry that, is postulated to be perturbed as a consequence of NRHypo and to underlie the expression of some of the neuropathological and clinical features associated with NRHypo. Next, we will discuss the evidence for decreasing NMDA receptor function in aging, and the role that this may play in the expression of agerelated memory decline. Finally, we describe how agerelated decreases in NMDA receptor activity may also interact with disease-related mechanisms to contribute to the expression of psychosis and to certain neuropathological features in patients with AD.
NMDA glutamate receptors and memory
Hippocampal long-term potentiation
NMDA receptors are now understood to critically regulate a physiologic substrate for memory function in the brain. In brief, the activation of postsynaptic NMDA receptors in most hippocampal pathways controls the induction of an activity-dependent synaptic modification called long-term potentiation (FTP). Citation8,Citation9 The NMDA receptor has been conceptualized as a synaptic coincidence detector that can provide graded control of memory formation.Citation10-Citation12 LTP and other forms of activitydependent synaptic modification share important properties with memory function and have been postulated to underlie the brain's ability to store information.Citation13,Citation14 NMDA antagonist drugs can block both in vivo hippocampal LTP induction and spatial learning at intracerebral concentrations comparable to those that block LTP in vitro.Citation15,Citation16
NMDA receptors are heteromeric complexes consisting of an NR1 subunit in combination with one of several NR2 subunits,Citation17,Citation18 with the NR2 subunit regulating channel gating.Citation19 Gene knockout of the NMDA receptor NR2A subunit in mice reduces both hippocampal LTP and spatial learning.Citation20 NR1-NR2B complexes in vitro have longer excitatory postsynaptic potentials than NR1-NR2A complexes.Citation21 This result suggests that increased expression of NR2B subunits and their incorporation into functional receptor complexes in vivo could increase the time period for NMDA receptors to detect synaptic coincidence, increasing synaptic efficacy and also potentially increasing memory function. Consistent, with this hypothesis, investigators have recently reported that overexpression of NR2B receptor subunits in transgenic mice enhances the activation of NM.DA receptors, facilitating synaptic potentiation as well as learning and memory.Citation22
Animal data
Some studies of NMDA antagonist drug effects on in vivo hippocampal LTP induction have related synaptic changes to measures of memory and learning. However, many studies have been devoted to characterizing the effect of NMDA receptor antagonist drugs on memory and learning. The cognitive effects of NMDA receptor antagonist drugs in animals provide strong support, for the proposal that decreases in NMDA receptor function can decrease memory and learning performance. Both competitive and noncompetitive NMDA antagonists transiently impair spatial learning in ratsCitation15,Citation23-Citation28 and cats,Citation24 including performance on object recognition tasks with a major working memory component.Citation29 Many studies demonstrate NMDA antagonist-induced impairments on spatialCitation30-Citation33 and nonspatialCitation34-Citation38 tasks that can be affected by hippocampal lesions. In rats, the defect in memory function induced by NMDA antagonists involves an impairment in the acquisition or encoding of new information, rather than its retrieval from storage,Citation33,Citation39,Citation40 or alternatively an impairment in the consolidation of “short-term” memory into “long-term” memory.Citation41,Citation42 Similar NMDA antagonist-induced impairments in learning and memory (eg, delayed matching-to-sample impairments) have been reported in nonhuman primates using ketamine, phencyclidine (PCP), and MK-801.Citation43-Citation46 These studies similarly suggest an impairment in the acquisition rather than retention of new information.Citation45
Human data
Subanesthetic doses of PCP selectively and noncompetitively act as an antagonist at NMDA receptors.Citation47 Earlystudies of acute PCP effects on cognitive function in humans reported transient, treatment-related reductions in memory performance, psychomotor processing speed, selective attention, reaction time, and weight discrimination.Citation48-Citation51 Similarly, ketamine anesthesia was reported early on to be associated with transient anterograde amnesia.Citation52 While decreased memory performance has also been reported in chronic PCP as well as ketamine abusers,Citation53,Citation54 these naturalistic reports confound acute NMDA receptor effects and other drug and nondrug effects associated with chronic use. Clinical adverse events associated with PCP quickly ended the use of this agent, in humans, so that, more recent studies of NMDA antagonist, effects in humans have been conducted using a variety of other agents including the Food and Drug Administration (FDA)-approved anesthetic agent, ketamine. Ketamine, like PCP, is a noncompetitive NMDA antagonist, but. it. is at. least. 10 times less potent than PCP in binding to the NMDA receptorCitation55 and in blocking NMDA-mcdiatcd excitotoxicity.Citation56
Acute subanesthetic doses of ketamine produce reliable transient decreases in long-term, explicit, or declarative learning and memory performance that are not accounted for by deficits in attentional performance.Citation57-Citation62 Some, but not all, investigators have also detected decreases in sustained attentional performanceCitation59 and verbal fluency,Citation58,Citation59,Citation63 perhaps as a function of higher plasma ketamine levels at the time of task performance. Decreases in performance on long-term, explicit, or declarative memory tasks are fully consistent with the role of NMDA receptors in the induction of hippocampal LTP and with the results of numerous animal experiments. In addition, however, ketamine has also been consistently reported to decrease performance on tasks measuring verbal and nonverbal working memory performance, involving the short-term storage and manipulation of information.Citation59,Citation63,Citation64
The neuroanatomical and neurophysiological substrates of working memory performance have been intensively studied over the past decade, and hippocampal LTP may not play a major role. Instead, neuronal activity in brain regions that include dorsolateral prefrontal cortex and cingulate cortex may play an important role in supporting this type of memory function.Citation65,Citation66 Relevant clinical neuroscience studies have provided new insights into the role of NMDA receptors in regulating memory function, suggesting that NMDA receptors in different brain regions may be regulating short- versus long-term memoryprocesses. Results from the ongoing multidisciplinary research effort in this area, discussed below, suggest that NRHypo in certain brain regions can perturb a complex neural circuit resulting in increased excitatory inputs to a variety of neocortical targets. Some of these targets include important cortical substrates for working memory such as cingulate cortex. These results suggest that NRHypo-induced increases in unmodulated excitatory input, to cingulate cortex and resulting increases in output, could contribute to disturbances in neural networks underlying working memory function. Future studies should address strategies to decrease excitatory inputs to these cortical areas as a test, of the hypothesis that such increased input, underlies NRHypo-induced impairments in function, and to further understand the regulation of working memory in healthy and disease states.
Psychotomimetic effects of NMDA glutamate receptor antagonists
In the 1950s, the dissociative anesthetic, PCP, was observed to induce a psychotic state in human subjects. 67-68The syndrome produced by PCP includes hallucinations, delusions, idiosyncratic and illogical thinking, poverty of speech and thought, agitation, disturbances of emotion, affect, withdrawal, decreased motivation, decreases in cognitive function, and dissociation.Citation48-Citation50,Citation69-Citation71 However, the link to NMDA glutamate receptors was not suspected until 30 years later when blockade of NMDA glutamate receptors was implicated as the primary mechanism by which PCP disrupts brain function.Citation72,Citation73 Hypofunction of NMDA receptors induced byvarious NMDA antagonist drugs is now known to precipitate a transient psychotic state in normal subjects.Citation58-Citation60,Citation62,Citation71,Citation74-Citation77 Ketamine, a well-studied PCP analog still used in human anesthesia, is known to cause emergence reactions similar to, but not as severe as, those caused by PCP and a clinical syndrome at subanesthetic doses that includes mild positive, negative, and cognitive symptoms resembling schizophrenia.Citation47,Citation59,Citation60,Citation62 Notably, these effects are dose-dependent and memory impairments emerge prior to the expression of psychotic symptoms.Citation62
PCP and related ligands act at a “PCP” receptorCitation78,Citation79 located in the ion channel of the NMDA subtype of glutamate receptor to effect a noncompetitive blockade of NMDA receptor function.Citation72-Citation73 In addition, CPPene (3-[2carboxypiperazine-4-yl]propenyl-1-phosphonate), CPP (3-[2-carboxypiperazin-4-yl]propyl-1-phosphonicacid), and COS 19755 (cis-4-[phosphonomethyl]-2-piperidine-carboxylic acid), agents that block NMDA receptors competitively by acting at the NMDA recognition site outside the NMDA ion channel, have all been shown to cause a similar PCP-like psychosis in normal human volunteers.Citation58,Citation74-Citation76 When PCP and ketamine, the most extensively studied of these agents, are administered to healthy subjects, they better mimic a broad range of psychotic symptoms than amphetamine, lysergic acid diamine (LSD), barbiturates, or N,N-dimethyltryptamine.Citation48,Citation51,Citation80-Citation87 Indeed, PCP-induced psychosis can be clinically indistinguishable from an acute presentation of schizophrenia, complicating appropriate clinical care.Citation88,Citation89 Additional observations have strengthened interest in the effects of NMDA receptor function in relation to adult-onset psychoses. Patients with schizophrenia are unusually sensitive to pharmacological blockade of NMDA receptors, in that administration of PCP to stabilized chronic schizophrenia patients can trigger a recrudescence of acute psychotic symptoms lasting for up to several months.Citation68,Citation90 In contrast, LSD causes only a brief hallucinogenic state that does not appear to last longer in schizophrenia patients than in normal healthy subjects.Citation71 Another important observation is that many adults have displayed agitation and psychotic symptoms upon awakening from PCP- or ketamine-induced anesthesia, whereas pediatric patients at any age prior to adolescence show little or no susceptibility to this NRHypoassociated phenomenon.Citation91-Citation95 It would appear that humans become susceptible to NRHypo-induced psychotic reactions around the same age that various adult-onset psychotic syndromes (eg, schizophrenia) can begin to present. These parallels between the drug-induced NRHypo state and adult-onset psychoses have fueled the hypothesis that an NRHypo-related mechanism may contribute to the pathophysiology of psychosis. On the basis of this evidence, as well as postmortem evidence of changes in glutamate metabolismCitation96 and receptor expressionCitation97,Citation98 in schizophrenia, several authors have recently developed theoretical positions regarding the relative roles of NRHypo and dopamine (DA) receptor hyperfunction as causal factors in schizophrenia.Citation6,Citation47,Citation99-Citation101
The NRHypo hypothesis
Recent novel approaches to the treatment and prevention of drug-induced and idiopathic psychoses have emerged from the NMDA glutamate receptor hypofunction hypothesis.Citation102-Citation106 Simply stated, the hypothesis proposes that NRHypo, the condition induced in the human or animal brain by an NMDA antagonist drug, might also be viewed as a model for a disease mechanism which could explain the expression of psychosis, cognitive impairments, and certain neuropathological findings in patients with neuropsychiatrie disorders like schizophrenia and AD. The disease mechanism itself might involve dysfunction of the NMDA receptor or downstream effects that can be modeled by blocking NMDA receptors. An important consequence of blocking NMDA receptors is excessive release of GluCitation107,Citation108 and acetylcholineCitation109-Citation111 (ACh) in multiple brain regions. It, has been proposed that, this excessive release of excitatory transmitters and consequent, overstimulation of postsynaptic neurons might, explain the cognitive and behavioral disturbances associated with the NRHypo state.Citation100,Citation107,Citation108 It. is assumed that both genetic and nongenetic factors can contribute to the NRHypo state, and that NRHypo can interact with a variety of other disease mechanisms.
Neurotoxic effects of NMDA glutamate receptor antagonists
In order to better understand the mechanisms underlying the clinical effects of NMDA antagonist drugs and the clinical consequences of an NRHypo state, several research groups have begun examining the consequences of drug-induced NRHypo and have shown that one typical consequence is excessive release of GluCitation107,Citation108 and AchCitation109,Citation111 in the cerebral cortex. Therefore, a concerted effort is being made to understand the mechanism by which NRHypo triggers excessive release of excitatory neurotransmitters in the hope that this may provide new insights into the pathophysiology of psychosis and certain cognitive impairments. While moderately increased neurotransmitter release and associated overstimulation of postsynaptic neurons can produce certain cognitive and psychotic symptoms, unremittingly severe and chronic NRHypo and associated excitatory transmitter release can lead to neurodegenerative changes in the brain.
For research purposes, creating a drug-induced NRHypo state in the rodent brain provides an excellent means of identifying neuronal populations that are at risk of being hyperstimulaled and potentially injured as a consequence of the NRHypo state. Described below, our findings indicate that a protracted NRHypo state can trigger neuronal injury throughout many corticolimbic brain regions.Citation112,Citation113 Presumably any of these hyperstimulated neurons can be instrumental in producing NMDA antagonist-induced psychotic symptoms or cognitive impairments. In addition, this animal model provides an opportunity to test pharmacological approaches for pre venting the NRHypo state from hyperstimulating and injuring neurons. Finally, a careful analysis of the pharmacological interventions that are protective can provide insights into the circuitry and receptor mechanisms that, mediate this pathological process.
In experimental animal studies, we have found that, if the increased release of Glu and ACh is pronounced, certain postsynaptic neurons can develop either reversible or irreversible morphological changes, depending on the duration and severity of the NRHypo state.Citation100 Low doses of NM'DA receptor antagonist drugs, such as ketamine, MK-801, tiletamine, PCP, CPP, and CPPene, reliably injure certain ccrcbrocortical neurons.Citation114,Citation116 At. these doses, the injury is confined to the posterior cingulate and retrosplenial (PC/RS) cortex and consists of the formation of intracytoplasmic vacuoles in layer III-IV pyramidal neurons. These changes are transient and resolve by 24 hours.Citation100 While these neurons will continue to express the 72-kDa form of heat shock protein (HSP-72) for up to 2 weeks,Citation115,Citation117 they do not become argyrophilic (de Olmos cupric silver method) or die. In contrast, administration of an NMDA antagonist in high dosage or by continuous infusion for several days induces a prolonged NRHypo state, which causes irreversible injury involving the death of neurons in many cerebrocortical and limbic brain regions.Citation112,Citation118-Citation120 Large to medium-sized pyramidal and multipolar neurons are preferentially affected, although smaller neurons are also involved. The full pattern of damage includes the PC/RS, frontal, temporal, entorhinal, perirhinal, piriform, and prefrontal cortices, the amygdala, and hippocampus.Citation112 At 4 hours, the reaction in PC/RS cortex consists of intracytoplasmic vacuole formation, but in other brain regions a spongiform reaction featuring edematous swelling of spines on proximal dendrites is the most prominent cytopathological change. At 24 to 48 hours, the affected neurons become argyrophilic and immunopositive for HSP-72 and begin to display cytoskeletal abnormalities, including a conspicuous corkscrew deformity of their apical dendrites. In the 72- to 96-hour interval many of the degenerating neurons display conspicuous fragmentation, but cytoplasmic organelles and cytoskeletal elements within the cell body and mainstem dendrites of some cells continue to show mixed signs of viability and degeneration for at least 10 days. Over this period, the degenerative reaction does not elicit a robust glial or phagocytic response and the overall appearance is one of a subacute protracted neurodegenerative process.
Neural circuitry that is disturbed by NRHypo
In a series of recent studies, our groupCitation115,Citation121-Citation124 and othersCitation125-Citation127 have found that, several different classes of drugs effectively block the PC/RS neurotoxic action of NMDA antagonist drugs. Hi ese findings, in conjunction with other work,Citation122,Citation128,Citation129 indicate that, a surprisingly complex neural circuitry is involved in NRHypo neurotoxicity (). In a series of recent, studies,Citation100,Citation128,Citation129 we have found that a key feature of the circuitry that mediates the NRHypo neurotoxic process is that Glu, acting at N.M.DA receptors, functions in this circuit, as a regulator of inhibitory tone. Glu accomplishes this regulatory function by tonically stimulating NMDA receptors on GABAergic interneurons (GABA: gammaaminobutyric acid), which, in turn, inhibit, excitatory projections that, convergently innervate vulnerable cerebrocortical neurons. NMDA receptor-blocking drugs prevent Glu from driving GABAergic inhibitory neurons, and this results in a loss of inhibitory control over two major excitatory projections to the cerebral cortex, one that, is cholinergic and originates in the basal forebrain, and one that is glutamatergic and originates in the thalamus.
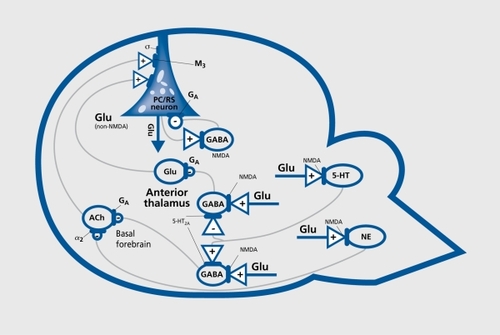
In addition to these basic features, the NRHypo circuitry includes noradrenergicCitation123 and serotonergicCitation130 neurons that, are driven by Glu through NMDA receptors and also perform an inhibitory function so that when NMDA receptors are hypofunctional the inhibitory restraint, contributed by these elements is also lost. One final aspect. that may be quite important for understanding how disinhibition of this circuitry can trigger psychotic reactions is that the vulnerable cerebrocortical neurons are glutamatergic neurons that ordinarily control their own firing by activating an NMDA receptor on a GABAergic neuron in an inhibitory feedback loop. When the NMDA receptor in this feedback loop is hypofunctional (eg, blocked by NMDA antagonist drugs), GABAergic inhibition is lost and the cerebrocortical neurons' control over their own firing is lost at the same time as these neurons are being hyperstimulated by disinhibited glutamatergic and cholinergic excitatory inputs. The expected result, under these conditions would be that the overstimulated cerebrocortical neurons could bombard many other neurons in their projection fields with unmodulated output (ie, noise). This provides a credible hypothesis for the psychotomimetic reactions and working memory impairments induced by NMDA antagonist drugs, and we propose that a similar NRHypo mechanism could contribute to the expression of psychosis and memory impairments in a variety of neuropsychiatrie disorders, including AD.
Treatment implications of NRHypo circuitry model
The ability of certain classes of drugs to prevent the neurotoxic consequences of the NRHypo state in rat brain raises the question whether these drugs might also block the psychotomimetic consequences of the drug-induced NRHypo state in humans, or psychotic symptom formation in certain neuropsychiatrie disorders. Information pertaining to this question, although incomplete, provides some interesting correlations. Agents that promote GABAA neurotransmission prevent the NRHypo state from releasing excessive AchCitation129 (Figure 1) and prevent NRHypo neurotoxicity in the rat cerebral cortex,Citation123 and it is well recognized by anesthesiologists that these agents in sufficient dosage attenuate the psychotomimetic actions of ketamine.Citation91 α2-Adrenergic agonists prevent the NRHypo state from releasing excessive acetylcholineCitation129 (Figure I) and prevent NRHypo neurotoxicity in the rat cerebral cortex,Citation123 and it was recently shown that an α2-adrenergic agonist can prevent ketamine from inducing positive schizophrenia-like symptoms in normal human voluteers.Citation103 Lamotrigine, an agent that may inhibit the excessive release of Glu at non-NMDA receptors (Figure 1), prevents NRHypo neurotoxicity in the rat cerebral cortexCitation131 and was recently shown to prevent, ketamine induced schizophrenia-like symptoms in human voluteers.Citation104 Clozapine and olanzapine, which are effective drugs for treating schizophrenia, are also quite potent in blocking NRHypo neurotoxicity in the rat. cerebral cortex,Citation132 and clozapine has been reported to block ketamine induccd increases in positive symptoms in patients with schizophrenia.Citation133
Age-related decreases in NMDA receptor function may explain age-related decreases in memory
At least, four different laboratories studying three different nonhuman species (mice, rats, and monkeys) have reported that the NMDA receptor transmitter system becomes markedly hypofunctional with advancing age.Citation134-Citation138 Several different age-related changes can contribute to this decrease in function (for a review see reference 138). The most consistently reported decrease in binding parameters has been an age-related decrease in binding to the NMDA site, using agonists and antagonists, in the neocortex and hippocampus of rodents and primates. Depending on the species, this generally appears to reflect a decrease in the number of binding sites, rather than changes in affinity, and generally reflects greater decreases in the cortex than in the hippocampus. Variable age-related changes in binding to the glycine site have been observed. Age-related decreases in binding to the PCP site have also been observed across multiple species, again greater in the neocortex than the hippocampus. This again generally appears to reflect decreases in the number of binding sites, rather than changes in affinity. In humans, a 36% decrease in Bmax for [3H]MK-801, reflecting a decreased number of PCP binding sites, was observed comparing 10 to 20 year olds and individuals in their 90s.Citation139
NMDA receptor subunit expression also changes with aging. NR1 expression has been reported to be decreased in the dentate of aged macaquesCitation140 and in the cortex and hippocampus of rodents.Citation141 NR2B expression also decreases with aging in the hippocampus and cortex of rodents.Citation142,Citation143 It should be recalled that NR1-NR2B complexes in vitro demonstrate longer excitatory postsynaptic potentials than NR1-NR2A complexesCitation21 and that an increased presence of NR1-NR2B complexes in vivo could increase the time period for NMDA receptors to detect, synaptic coincidence, potentially increasing synaptic efficacy and memory function. Thus, an age-related decrease in NR2B expression could account for agerelated shortening of the excitatory postsynaptic potential duration of the NMDA channel.Citation144,Citation145 As mentioned above, overexpression of NR2B receptor subunits in transgenic mice enhances the activation of NMDA receptors, facilitating synaptic potentiation as well as learning and memoir}'.Citation22 This overexpression has also been reported to prevent, age-related decreases in memory and learning performance. These results support, the hypothesis that age-related decreases in NMDA receptor function could account for age-related decreases in memory and learning. This suggests that strategies to prevent, those agerelated changes, or strategies to prevent, downstream or other events related to those changes, could have important therapeutic implications for the prevention or treatment of age-related memory impairments.
NRHypo hypothesis of AD
In addition to age-related increases in NRHypo, it was recently shown in humans that a more severe degree of NRHypo is present in the AD brain than in agematched normal controls.Citation97 Thus, in the aging human brain the stage may already be set for widespread corticolimbic neurodegeneration to occur. All that is required to explain why it occurs to a more severe degree in the AD brain than in the “normal” aging brain is to identify one or more adjunctive conditions peculiar to the AD brain that may serve as catalysts or promoters of the NRHypo state. In our animal model of NRHypo using otherwise healthy brain, no evidence of amyloidosis or amyloid plaque formation is observed. Therefore, we propose that genetic or other predisposing factors peculiar to the AD condition are primarilyresponsible for the amyloidopathy in the AD brain and that when amyloidopathy occurs alongside NRHypo, the pathological process known as AD develops. How then does amyloidosis interact with NRHypo?
Over the past decade, major strides have been made in discovering important genetic abnormalities in AD. Mutations on four different chromosomes, each of which can promote amyloidopathy, have now been identified as etiologic factors in familial AD and the role of apoE genotype as a risk factor in sporadic AD has been established. While recent research has elucidated the basic neurochemistry of beta-amyloid and it is clear that. abnormal deposition of beta-amyloid in the brain occurs early in AD, it. is not. at all clear how beta-amyloid deposition contributes to the neurodegenerative events in AD. Based on evidence that a severe degree of NRHypo is present in the human AD brain, that. NRHypo can induce an AD-like pattern of neurodegeneration in animal brain, and that beta-amyloid deposition may contribute to a worsening of the NRHypo state, we propose NRHypo as a missing link that can help explain major aspects of the pathophysiology of AD that have, until now, remained elusive.
Similarities between the neuronal degeneration seen in NRHypo and in AD
There are important similarities between the overall pattern of NRHypo neurodegeneration and the pattern that has been described in the AD brain by various researchers. The PC/RS cortex, which is the brain region most vulnerable to NRHypo degeneration, was recently reported to be selectively affected early in the course of AD in a PET study of living patients.Citation146 The PC/RS cortex has also been shown to be markedly atrophic late in the disease.Citation147 In contrast, neurodegeneration in the anterior cingulate cortex is less severe in both the AD brainCitation148 and the NRHypo animal model. While it is difficult to make precise anatomical comparisons between the rodent and human brain, the transentorhinal area, considered among the earliest and most severely affected regions in the human AD brain,Citation149 is roughly homologous to the perirhinal cortex in rat brain, which is second only to the PC/RS cortex in its sensitivity to NRHypo neurodegeneration. Other brain regions preferentially affected in both the AD brain and the NRHypo model include portions of the parietal, temporal, enlorhinal, amygdaloid, subicular, hippocampal, and insular cortices. A mild but transient microglial and astrocytic response accompanies the neurodegeneration seen with NRHypo. However, consistent with the known pathology of AD, a robust phagocytic response is conspicuously absent.Citation150
The neurons primarily involved in neurofibrillary tangle (NFT) formation in the AD brain are distributed widely throughout cortical and limbic brain regions, but in each region these neurons tend to be pyramidal or multipolar neurons and in certain cortical regions they are distributed in a bilaminar pattern. This fits the description of the subpopulation of neurons primarily affected in the NRHypo neurodegenerative syndrome.Citation113 Interneurons in the cerebral cortex are also occasionally involved but, the most, prominently affected neurons are mediumsized pyramidal or multipolar neurons in each region.
The tortuousity of dendritic processes in the NRHypo model is accompanied by a parallel pattern of tortuosity of the microtubular cytoskeleton within the distorted dendrite. This suggests that, changes in the external configuration of the dendrite are due to cytoskeletal changes within the dendrite. The cytoskeleton of the injured neurons appears to be undergoing both degenerative and regenerative processes, but the repair effort is not, very successful. As described above the mechanism of injury involves simultaneous hyperactivation of the neuron through several excitatory receptors, including a muscarinic (M3) cholinergic receptor and a glutamatergic (non-NMDA) receptor (Figure 1). Second messenger systems associated with the KA receptor are not known at this time, but it is well known that the M3 muscarinic receptor is coupled to a phosphoinosilide/Ca2+/protein kinase C second messenger system that mediates protein phosphorylation functions. Thus, the erratic pattern of microtubule tortuosity and disarray together with apparent efforts at microtubule regeneration may reflect abnormal hyperactivity of this M3-linked second messenger system and consequent disruption of its protein phosphorylation functions. Since other unknown second messenger systems are also hyperactivated simultaneously in the same neuron, these systems may also contribute to the cytoskeletal disruption pattern.
Neurofibrillary tangles and psychosis could be associated through NRHypo
We are beginning to study the possible relationship between this cytoskeletal disruption process in the NRHypo model and NFTs, its potential counterpart in the AD brain. Thus far, we have not detected ultrastructural evidence of paired helical filaments. Even if such evidence cannot be found, this might signify species specificity of this particular abnormality without disqualifying the NRHypo degenerative process as a valid model of the mechanism giving rise to NFT in the human AD brain. Thus, our findings in the rat are consistent with the conclusion that NRHypo alone can produce many of the neuropathological features of AD. The observations that NRHypo can produce psychotic symptoms, as well as potentially contribute to NFT formation, suggest that psychosis could be associated with NFT burden in AD patients. We recently tested this hypothesis in a postmortem sample of AD patients and found that, after controlling for the severity of dementia, neocortical NFT counts were increased in patients with AD who experienced psychotic symptoms in comparison to patients who did not.Citation151
Amyloidopathy and NRHypo
Most investigators of amyloidosis have tended to focus exclusively on the potential of beta-amyloid to kill neurons by itself without, reference to its potential pathological interaction with NMDA receptors. We think the focus should be redirected. Beta-amyloid alone is not. toxic except at very high concentrations, whereas at substantially lower concentrations beta-amyloid causes cultured neurons to become hypersensitive to Glu or NMDA excitotoxicity.Citation152-Citation154 We propose, therefore, that predisposing factors (eg, apoE4 genotype in sporadic AD, amyloidogenic mutations in familial AD) promote amyloidosis which, in turn, increases the sensitivity of NMDA receptors so that even normal concentrations of Glu can trigger abnormal currents, which on a chronic low-grade excitotoxic basis can destroy NMDA receptor-bearing neurons. Loss of these neurons and their NMDA receptors increases the NRHypo burden in the aging brain.
Other factors such as oxidative stress and disturbances in energy metabolism may also contribute to beta-amyloid's augmentation of neuronal sensitivity to Glu. Disturbances in energy metabolism may cause NMDA receptor hypersensitivity by interfering with a voltage-dependent mechanism by which Mg2+ normally inhibits passage of sodium and calcium currents through the NMDA receptor channel. Because membrane polarization is maintained by energy-dependent mechanisms, impaired energy can trigger partial membrane depolarization, which abolishes the Mg2+ block and allows normal concentrations of transmitter Glu to drive abnormal currents on a chronic basis. Oxidative stressors may act through a similar mechanism, in view of evidence that free radical generation in nitric oxide pathways disrupts glycolytic metabolism, and superoxide radical formation causes hyperactivation of NMDA receptors in cultured neurons. The proposal that oxidative stress may contribute to neurodegeneration in AD is consistent with recent evidenceCitation155 that antioxidant drugs may retard the progression of cognitive deterioration in AD. Whether impaired energy or oxidative stressors are relatively more active in the aging AD brain than the aging normal brain is not. clear at this time. Their presence, even if not, more severe than that in the normal aging brain, would augment, amyloid's ability to sensitize neurons to Glu's excitotoxic potential. Persistent hyperactivation of NMDA receptors would result, in either excitotoxic degeneration of the dendritic spines on which NMDA receptors are located or in degeneration of the entire NMDA receptor-bearing neuron. In either case, NMDA receptors are deleted from the brain and the NMDA receptor system is reduced to a hypofunctional status. NRHypo thus could represent, a residual deficit, condition caused by NRHyper.
AD neurodegeneration: a two-stage process
A major tenet of our proposal is that the NMDA receptor system becomes hypofunctional in either the normal brain or the AD brain after having first gone through an early stage of NRHyper. This hypothesis, consistent with the bulk of available data, assumes that the pattern of massive neurodegeneration in AD tends to follow the pattern of NFT formation, and that the neurons that display NFT at the time of autopsy are injured neurons that would be destined to slowly die and leave behind neurofibrillary debris. However, this hypothesis also assumes that there is a less massive pattern of neuronal degeneration that corresponds to the pattern of amyloid deposition. Our hypothesis suggests that the neurodegenerative events in AD occur in two separate stages, by two separate mechanisms, and according to two separate patterns. These have been difficult to tease apart because the two stages have a significant degree of temporal overlap and the two patterns have significant spatial overlap. We propose that the first neurodegenerative stage entails the deposition of low concentrations of amyloid in the brain and interaction of amyloid with certain NMDA receptors in a manner that increases the sensitivity of these receptors to Glu so that the neurons bearing these receptors will be hyperstimulated and destroyed by endogenous Glu. As these neurons degenerate, amyloid plaques may form and incorporate portions of the degenerating neurons and other neural and glial processes in the immediate environment. The pattern of these early neurodegenerative and reactive events will follow the pattern of distribution of the specific neurons vulnerable to this amyloid/NMDA receptor-mediated neuropathological process. We postulate that it may not be a very conspicuous pattern of neuronal loss because it may be restricted to just the NMDA receptor-bearing neurons in our schematic circuit, that, control the release of transmitters onto the vulnerable pyramidal neuron (Figure 1). In stage I, the neurodegenerative process may produce few if any symptoms, because it. is limited to a. small population of neurons. In addition, we postulate that, the recurrent collateral feedback loop (Figure 1) remains relatively intact, so that, pyramidal neurons, as they begin to receive excessive stimulation, will be prevented from firing erratically onto other neurons and thereby prevented from generating florid symptoms.
The second stage commences when the loss of NMDA receptor-bearing neurons is sufficient, to substantially unleash the disinhibition syndrome in which many primary cerebrocortical and corticolimbic neurons are pathologically hyperstimulated through several signal transduction pathways at the same time. At this point, psychosis and NRHypo-related cognitive disturbances could become evident. We propose that pyramidal neurons in many cortical and limbic brain regions will be affected, and will slowly degenerate and die as the stage II process progresses. Death and deletion of these neurons will disrupt mental functions just as excessive hyperactivation of these neurons will disrupt these functions.
While these neurons are degenerating, we propose that at least some of them develop NFTs on the basis of excessive activation of second messenger pathways associated with muscarinic and/or non-NMDA glutamate receptors. These second messenger systems are coupled to kinases or other possible factors relevant to protein phosphorylation; therefore, hyperactivation of these systems provides a rational explanation for NFT formation, which is believed to result from hyperphosphorylation of microtubule-associated proteins. In stage II, neurodegeneration occurs as a network disturbance. The pattern of degeneration is determined by the pattern of connections within the network, and by the failure of inhibition over certain excitatory pathways within the network, causing specific cortical and limbic neurons innervated by these excitatory pathways to degenerate. This provides a rational explanation for the pattern of degeneration seen in AD.
Selected abbreviations and acronyms
| Ach | = | acetylcholine |
| AD | = | Alzheimer's disease |
| ALS | = | amyotrophic lateral sclerosis |
| AMPA | = | amino-3-hydroxy-5-methyl-4-isoxazole propionic acid |
| GABA | = | gamma-aminobutyric acid |
| Glu | = | glutamate |
| KA | = | kainic acid |
| LTP | = | long-term potentiation |
| NFT | = | neurofibrillary tangle |
| NMDA | = | N-methyl-D-aspartate |
| NRHyper | = | NMDA receptor hyperfunction |
| NRHypo | = | NMDA receptor hypofunction |
| PCP | = | phencyclidine |
| PC/RS | = | posterior cingulate and retrosplenial (cortex) |
Supported in part by NIDA Scientist Development Award for Clinicians DA 00290 (NBF), NIMH Independent Scientist Award MH 01510 (JWN), NIMH Research Scientist Award MH 33894 (JWO), MH 53363 (JWN) from NIMH, DA 05072 (JWO) from NIDA, AG 11355 (JWO) from NIA and NARSAD (JWN, NBF, and JWO).
REFERENCES
- OlneyJW.Excitotoxic amino acids and neuropsychiatrie disorders.Annu Rev Pharmacol Toxicol.19903047712188577
- ChoiDW.Excitotoxic cell death.J Neurobiol.199223126112761361523
- RothsteinJD.Van KammenM.LeveyAIMartinLJ.KunclRW.Selective loss of glial glutamate transporter GLT-1 in amyotropic lateral sclerosis.Ann Neurol.19953873847611729
- IkonomidouC.QinYQ.LabruyereJ.OlneyJW.Motor neuron degeneration induced by excitotoxin agonists has features in common with that seen in the SOD-1 transgenic mouse model of amyotrophic lateral sclerosis.J Neuropathol Exp Neurol.1996552112248786380
- ChoiDW.Glutamate neurotoxicity and diseases of the nervous system.Neuron.198816236342908446
- FarberNB.NewcomerJW.WozniakDF.OlneyJW.The glutamate synapse in neuropsychiatric disorders: focus on schizophrenia and Alzheimer's disease. In: Ottersen OP, Langmoen I, Gjerstad L, eds.Progress in Brain Research. New York, NY: Elsevier.1998116421437
- FarberNB.WozniakDF.PriceMT.et al.Age specific neurotoxicity in the rat associated with NMDA receptor blockade: potential relevance to schizophrenia?Biol Psychiatry.1995387887968750036
- BlissTV.CollingridgeGL.A synaptic model of memory: long-term potentiation in the hippocampus.Nature.199336131398421494
- CollingridgeGL.BlissTVP.Memories of NMDA receptors and LTP.Trends Neurosci.19951854567537406
- StevensCF.SullivanJ.Synaptic plasticity.Curr Biol.19988R151R1539501074
- BearMF.MalenkaRC.Synaptic plasticity: LTP and LTD.Curr Opin Neurobiol.199443893997919934
- BourneHR.NicollR.Molecular machines integrate coincident synaptic signals.Cell.199372(suppl)65758094038
- RisonRA.StantonPK.Long-term potentiation and N-methyl-D-aspartate receptors: foundations of memory and neurologic disease?Neurosci Biobehav Rev.1995195335528684715
- MalenkaRC.Synaptic plasticity in the hippocampus: LTP and LTD.Cell.1994785355388069904
- MorrisRGM.AndersonE.LynchGC.BaudryM.Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5.Nature.19863197747762869411
- DavisS.ButcherSP.MorrisRGM.The NMDA receptor antagonist D-2-amino-5-phosphonopentanoate (D-AP5) impairs spatial learning and LTP in vivo at intracerebral concentrations comparable to those that block LTP in vitro.J Neurosci.19921221341345945
- NakanishiS.Molecular diversity of glutamate receptors and implications for brain function.Science.19922585976031329206
- HollmannM.HeinemannS.Cloned glutamate receptors.Annu Rev Neurosci.199417311088210177
- MonyerH.SprengelR.SchoepferR.et al.Heteromeric NMDA receptors: molecular and functional distinction of subtypes.Science.199225612171221 1350383
- SakimuraK.KutsuwadaT.ItoI.et al.Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit.Nature.19953731511557816096
- MonyerH.BurnashevN.LaurieDJ.SakmannB.SeeburgPH.Developmental and regional expression in the rat brain and functional properties of four NMDA receptors.Neuron.1994125295407512349
- TangTP.ShimizuE.DubeGR.et al.Genetic enhancement of learning and memory in mice.Nature.1999401636910485705
- HandelmanGE.ContrerasPC.O'DonohueTL.Selective memory impairment by phencyclidine in rats.Eur J Pharmacol.198714069733622624
- BalsterRL.ChaitLD.The behavioral pharmacology of phencyclidine.Clin Toxicol.19769513528824088
- McLambRL.WilliamsLR.NanaryKP.WilsonWA.TilsonHA.MK-801 impedes the acquisition of a spatial memory in rats.Pharmacol Biochem Behav.19903741452148213
- MondadoriC.WeiskrantzL.BuerkiH.PetschkeF.FaggGE.NMDA receptor antagonists can enhance or impair learning performance in animals.Exp Brain Res.1989754494562545467
- MorrisRGM.Synaptic plasticity and learning: selective impairment of learning in rats and blockade of long-term potentiation in vivo by the N-methyl-D-aspartate receptor antagonist AP5.J Neurosci.19899304030572552039
- MorrisRGM.DavisS.ButcherSP.Hippocampal synaptic plasticity and NMDA receptors: a role in information storage?Phil Trans R Soc.1990329187204
- PumaC.BaudoinC.BizotJC.Effects of intraseptal infusions of N-methyl-D-aspartate receptor ligands on memory in an object recognition task in rats.Neurosci Lett.1998244971009572594
- DanyszW.WroblewskiJT.CostaE.Learning impairment in rats by N-methyl-D-asparate receptor antagonists.Neuropharmacol.198827653656
- WardL.MasonSE.AbrahamWC.Effects of the NMDA antagonists, CPP and MK-801 on the radial arm maze performance in rats.Pharmacol Biochem Behav.1990357857902189143
- ButelmanER.A novel NMDA antagonist, MK-801, impairs performance in a hippocampal-dependent spatial learning task.Pharmacol Biochem Behav.19893413162696982
- SpanglerEL.BresnahanEL.GarofaloP.MuthNJ.HellerB.IngramDK.NMDA receptor channel antagonism by dizocilpine (MK-801) impairs performance of rats in aversively motivated complex maze tasks.Pharmacol Biochem Behav.1991409499581667826
- TonkissJ.MorrisRGM.RawlinsJNP.Intra-ventricular infusion of the NMDA antagonist AP5 impairs performance on a non-spatial operant DRL task in the rat.Exp Brain Res.1988731811882905273
- CrooksEB.RobinsonGS.HatfieldTJ.GrahamPW.GallagherM.Intraventricular administration of the NMDA antagonist APV disrupts learning of an odor aversion that is potentiated by taste.Soc Neuroscí Abstr.198915464
- JonesKW.BauerleL.DeNobleV.Differential effects of sigma and phencyclidine receptor ligands on learning.Eur J Pharmacol.1990179971022163853
- Parada-TurskaJ.TurskiWA.Excitatory amino acid antagonists and memory: effect of drugs acting at N-methyl-D-aspartate receptors in learning and memory tasks.Neuropharmacol.19902911111116
- PontecorvoMJ.ClissoldDB.WhiteMF.FerkanyJW.N-Methyl-D-aspartate antagonists and working memory performance: comparison with the effects of scopolamine, propranolol, diazepam, and phenylisopropyladenosine.Behav Neurosci.19911055215351657031
- WalkerDL.GoldPE.Effects of the novel NMDA antagonist, NPC 12626, on long-term potentiation, learning and memory.Brain Res.19915492132211832074
- McNamaraRK.SkeltonRW.The neuropharmacological and neurochemical basis of place learning in the Morris water maze.Brain Res Rev.19931833498467349
- KimJ.FanselowM.DeColaJ.Landeira-FernandezJ.Selective impairment of long-term but not short-term conditional fear by the NMDA antagonist APV.Behav Neurosci.19921065915961354443
- KimJ.RisonR.FanselowM.Effects of amygdala, hippocampus, and periaqueductal gray lesions on short- and long-term contextual fear.Behav Neurosci.1993107109310988136063
- ThompsonDM.WinsauerPJ.MastropaoloJEffects of phencyclidine, ketamine and MDMA on complex operant behavior in monkeys.Pharmacol Biochem Behav.1987264014052883665
- ThompsonDM.MoerschbaecherJM.Phencyclidine in combination with d-amphetamine: differential effects on acquisition and performance of response chains in monkeys.Pharmacol Biochem Behav.1984206196276728878
- BuffaloEA.GillamMP.AllenRR.PauleMG.Acute behavioral effects of MK-801 in rhesus monkeys: assessment using an operant test battery.Pharmacol Biochem Behav.1994489359407972299
- FrederickDL.GillamMP.AllenRR.PauleMG.Acute behavioral effects of phencyclidine on rhesus monkey performance in an operant test battery.Pharmacol Biochem Behav.1995527897978587921
- JavittDC.ZukinSR.Recent advances in the phencyclidine model of schizophrenia.Am J Psychiatry.1991148130113081654746
- RosenbaumG.CohenBD.LubyED.GottliebJS.YelenD.Comparison of Sernyl with other drugs: simulation of schizophrenic performance with Sernyl, LSD-25, and amobarbital (Amytal). I. Attention, motor function and proprioception.Arch Gen Psychiatry.1959165165614438905
- DaviesBM.BeechHL.The effect of 1-arylcyclohexylamine (Sernyl) on 12 normal volunteers,J Ment Sci.196010691292413720081
- BakkerCB.AminiFB.Observations on the psychotomimetic effects of Sernyl.Compr Psychiatry.1961226928013864199
- CohenBD.RosenbaumG.LubyED.GottliebJS.Comparison of phencyclidine hydrochloride (Sernyl) with other drugs.Arch Gen Psychiatry.1962639540113880223
- PanditSK.DundeeJW.BovillJG.Clinical studies of induction agents XXXVII: amnesic action of ketamine.Br J Anaesth.1971433623645575186
- JansenKLR.Ketamine - can chronic use impair memory?Int J Addictions.199025133139
- EllisonG.The N-methyl-D-aspartate antagonists phencyclidine, ketamine and dizocilpine as both behavioral and anatomical models of the dementias.Brain Res Rev.1995202502677795658
- HamptonRY.MedzihradskyF.WoodsJH.DahlstromPJ.Stereospecific binding of 3H-phencyclidine in brain membranes.Life Sci.198230214721547109842
- RothmanSM.OlneyJW.Excitotoxicity and the NMDA receptor.Trends Neurosci.198710299302
- HarrisJA.BiersnerRJ.EdwardsD.BaileyLW.Attention, learning and personality during ketamine emergence: a pilot study.Anesth Analg.1975541691721092205
- GhoneimMM.HinrichsJV.MewaldtSP.PetersenRC.Ketamine: behavioral effects of subanesthetic doses.J Clin Psychopharmacol.1985570773988972
- KrystalJH.KarperLP.SeibylJP.et al.Subanesthetic effects of the non-competitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses.Arch Gen Psychiatry.1994511992148122957
- MalhotraAK.PinalsDA.WeingartnerH.et al.NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers.Neuropsychopharmacology.1996143013078703299
- HarborneGC.WatsonFL.HealyDT.GrovesL.The effects of sub-anaesthetic doses of ketamine on memory, cognitive performance and subjective experience in healthy volunteers.J Psychopharmacol.199610134140
- NewcomerJW.FarberNB.Jevtovic-TodorovicV.et al.Ketamine-induced NMDA receptor hypofunction as a model of memory impairment in schizophrenia.Neuropsychopharmacology.1999201061189885791
- AdlerCM.GoldbergTE.MalhotraAK.PickarD.BreierA.Effects of ketamine on thought disorder, working memory and semantic memory in healthy volunteers.Biol Psychiatry.1998438118169611670
- NewcomerJW.SelkeG.MelsonAK.et al.NMDA antagonist-induced decrease in working and declarative memory in healthy humans.Soc Neurosci Abstr.199925633
- SmithEE.JonidesJ.Storage and executive processes in the frontal lobes.Science.19992831657166110073923
- VogtBA.VogtLJ.NimchinskyEA.HofPR.Primate cingulate cortex chemoarchitecture and its disruption in Alzheimer's disease. In: Bloom FE, Bjorklund A, Hokfelt T, eds.Handbook of Chemical Neuroanatomy. Vol 13: The Primate Nervous System. Part I. New York, NY: Elsevier.1997455528
- JohnstoneM.EvansV.BaigelS.Sernyl (CI-395) in clinical anaesthesia.Br J Anaesth.19593143343914407580
- LubyED.CohenBD.RosenbaumG.GottliebJS.KelleyR.Study of a new schizophrenomimetic drug - Sernyl.Arch Neurol Psychiatry19598136336913626287
- LubyED.GottliebJS.CohenBD.RosenbaumG.DominoEF.Model psychoses and schizophrenia.Am J Psychiatry.19621196167
- CorssenG.DominoEF.Dissociative anesthesia: further pharmacological studies and first clinical experience with the phencyclidine derivative CI-581.Anesth Analg.19664529405325977
- DominoEF.LubyED.Abnormal mental states induced by phencyclidine as a model of schizophrenia. In: Domino EF, éd.PCP (Phencyclidine): Historical and Current Perspectives. Ann Arbor, Mich: NPP Books.1981401418
- LodgeD.AnisNA.Effects of phencyclidine on excitatory amino acid activation of spinal interneurons in the cat.Eur J Pharmacol.1982772032047037432
- LodgeD.AramJA.ChurchJ.et al.Excitatory amino acids and phencyclidine-like drugs. In: Hicks TP, Lodge D, McLennan H, eds.Excitatory Amino Acid Transmission. New York: Alan R Liss.19878390
- KristensenJD.SvenssonB.GordhT Jr.The NMDA receptor-antagonist CPP abolishes neurogenic “wind-up pain” after intrathecal administration in humans.Pain.1992512492531484720
- HerrlingP.D-CPPene (SDZ EAA 494), a competitive NMDA antagonist. Results from animal models and first results in humans.Neuropsychopharmacology.199410(3S/Part 1)591S
- GrottaJ.ClarkW.CoullB.et al.Safety and tolerability of the glutamate antagonist CGS 19755 (Selfotel) in patients with acute ischemic stroke. Results of a phase IIa randomized trial.Stroke.1995266026057709405
- MuirKW.GrossetDG.LeesKR.Effects of prolonged infusion of the NMDA antagonist aptiganel hydrochloride (CNS 1102) in normal volunteers.Clin Neuropharmacol.1997203113219260729
- ZukinRS.ZukinSR.Specific [3H]phencyclidine binding in rat central nervous system.Proc Natl Acad Sci U S A.19797653725376291953
- VincentJP.KartalovskiB.GenesteP.KamenkaJM.LazdunskiM.Interaction of phencyclidine (“angel dust”) with a specific receptor in rat brain membranes.Proc Natl Acad Sci U S A.1979764678468241247
- AngristB.GershonS.The phenomenology of experimentally induced amphetamine psychosis - preliminary observations.Biol Psychiatry.19702951075459137
- FreedmanDX.On the use and abuse of LSD.Arch Gen Psychiatry.1968183303474295595
- FreedmanDX.LSD: the bridge from human to animal. In: Jacobs BL, ed.Hallucinogens: Neurochemical, Behavioral, and Clinical Perspectives. New York: Raven Press.1984203226
- AngristB.SathananthanG.WilkS.GershonS.Amphetamine psychosis: behavioral and Biochemical aspects.J Psychiatr Res.19741113234461784
- JanowskyDS.RischSC.Amphetamine psychosis and psychotic symptoms.Psychopharmacol (Berl).1979657377
- LiebermanJA.KaneJM.AlvirJ.Provocative tests with psychostimulant drugs in schizophrenia.Psychopharmacol (Berl).198791415433
- BowersMB.SwigarME.JatlowPI.HoffmanF.GoicoecheaN.Early neuroleptic response: clinical profiles and plasma catecholamine metabolites.J Clin Psychopharmacol.1987783862884237
- GillinJC.KaplanJ.StillmanR.WyattRJ.The psychedelic model of schizophrenia: the case of N,N-dimethyltryptamine.Am J Psychiatry.19761332032081062171
- YesavageJA.FreemanAM III.Acute phencyclidine (PCP) intoxication: psychopathology and prognosis.J Clin Psychiatry.197844664665681304
- ErardR.LuisadaPV.PeeleR.The PCP psychosis: prolonged intoxication or drug-precipitated functional illness?J Psychedelic Drugs.1980122352457431420
- BanTA.LohrenaJJ.LehmannHE.Observations on the action of Sernyl - a new psychotropic drug.Can Psychiatr Assoc J.1961615015713686510
- ReichDL.SilvayG.Ketamine: an update on the first 25 years of clinical experience.Can J Anaesth.1989361861972650898
- MarshallBE.LongneckerDE.General anesthetics. In: Goodman LS, Gilman A, Rail TW, Nies AS, Taylor P, eds.The Pharmacological Basis of Therapeutics. New York: Pergamon Press.1990285310
- KarpHN.KaufmanND.AnandSK.Phencyclidine poisoning in young children.J Pediatr.198097100610097441408
- WelchMJ.CorreaGA.PCP intoxication in young children and infants.Clin Pediatr (Phila).1980195105147389238
- BaldridgeEB.BessenHA.Phencyclidine.Emerg Med Clin North Am.199085415502201519
- TsaiG.PassaniLA.SlusherBS.et al.Abnormal excitatory neurotransmitter metabolism in schizophrenic brains.Arch Gen Psychiatry.1995528298367575102
- UlasJ.CotmanCW.Decreased expression of N-methyl-D-aspartate receptor 1 messenger RNA in select regions of Alzheimer brain.Neuroscience.1997799739829219960
- D'souzaDC.CharneyDS.KrystalJH.Glycine site agonist of the NMDA receptor: a review.CNS Drug Rev.19951227260
- OlneyJW.Endogenous excitotoxins and neuropathological disorders. In: Lodge D, ed.Excitatory Amino Acids in Health and Disease. Chichester, UK: John Wiley & Sons Ltd.1988337351
- OlneyJW.FarberNB.Glutamate receptor dysfunction and schizophrenia.Arch Gen Psychiatry.19955299810077492260
- CoyleJY.The glutamatergic dysfunction hypothesis for schizophrenia.Harvard Rev Psychiatry.19963241253
- FarberNB.NewcomerJW.OlneyJW.Glycine agonists: what can they teach us about schizophrenia?Arch Gen Psychiatry.19995613179892251
- NewcomerJW.FarberNB.SelkeG.MelsonAK.Jevtovic-TodorovicV.OlneyJW.Guanabenz effects on NMDA antagonist-induced mental symptoms in humans.Soc Neurosci Abstr.199824525
- AnandA.CharneyDS.BermanRM.OrenDA.CappielloA.KrystalJH.Reduction in ketamine effects in humans by lamotrigine.Soc Neurosci Abstr.1997231755
- GoffDC.TsaiG.LevitttJ.et al.A placebo-controlled trial of D-cycloserine added to conventional neuroleptics in patients with schizophrenia.Arch Gen Psychiatry.19995621279892252
- Heresco-LevyU.JavittDC.ErmilovM.MordelC.SilipoG.LichtensteinM.Efficacy of high-dose glycine in the treatment of enduring negative symptoms of schizophrenia.Arch Gen Psychiatry.19995629369892253
- MoghaddamB.AdamsB.VermaA.DalyD.Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex.J Neurosci.199717292129279092613
- AdamsB.MoghaddamB.Corticolimbic dopamine neurotransmission is temporally dissociated from the cognitive and locomotor effects of phencyclidine.J Neurosci.199818554555549651235
- GiovanniniMG.MutoloD.BianchiL.MichelassiA.PepeuG.NMDA receptor antagonists decrease GABA outflow from the septum and increase acetylcholine outflow from the hippocampus: a microdialysis study.J Neurosci.199414135813658120631
- HasegawaM.KinoshitaH.AmanoM.HasegawaT.KameyamaT.NabeshimaT.MK 801 increases endogenous acetylcholine release in the rat parietal cortex: a study using brain microdialysis.Neurosci Lett.199315053568469404
- KimSH.PriceMT.OlneyJW.FarberNB.Excessive cerebrocortical release of acetylcholine induced by NMDA antagonists is reduced by GABAergic and α2-adrenergic agonists.Mol Psychiatry.1999434435210483051
- CorsoTD.SesmaMA.TenkovaTl.et al.Multifocal brain damage induced by phencyclidine is augmented by pilocarpine.Brain Res.19977521149106435
- WozniakDF.DikranianK.IshimaruM.et al.Disseminated corticolimbic neuronel degeneration induced in rat brain by MK-801: potential relevance to Alzheimer's disease.Neurobiol Dis.1998530532210069574
- OlneyJW.LabruyereJ.PriceMT.Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs.Science.1989244136013622660263
- OlneyJW.LabruyereJ.WangG.WozniakDF.PriceMT.SesmaMA.NMDA antagonist neurotoxicity: mechanism and prevention.Science.1991254151515181835799
- HargreavesRJ.RigbyM.SmithD.HillRG.IversenLL.Competitive as well as uncompetitive N-methyl-D-aspartate receptor antagonists affect cortical neuronal morphology and cerebral glucose metabolism.Neurochem Res.199318126312697903796
- SharpFR.JasperP.HallJ.NobleL.SagarSM.MK-801 and ketamine induce heat shock protein HSP72 in injured neurons in posterior cingulate and retrosplenial cortex.Ann Neurol.1991308018091838680
- AllenHL.IversenLL.Phencyclidine, dizocilpine, and cerebrocortical neurons.Science.19902472212403696
- EllisonG.Competitive and non-competitive NMDA antagonists induce similar limbic degeneration.Neuroreport.19945268826927696633
- HorvathZC.CzopfJ.BuzsakiG.MK-801-induced neuronal damage in rats.Brain Res.19977531811959125402
- FarberNB.PriceMT.LabruyereJ.et al.Antipsychotic drugs block phencyclidine receptor-mediated neurotoxicity.Biol Psychiatry.1993341191218373932
- PriceMT.FarberNB.LabruyereJ.FosterJ.OlneyJW.Tracing the circuitry that mediates NMDA antagonist neurotoxicity.Soc Neurosci Abstr.1994201532
- FarberNB.OlneyJW.α2-Adrenergic agonists prevent MK-801 neurotoxicity.Neuropsychopharmacology.1995123473497576011
- FarberNB.KimSH.DikranianK.et al.Receptor mechanisms and circuitry underlying NMDA antagonist neurotoxicity.Mol Psychiatry. In press.
- SharpFR.ButmanM.KoistinahoJ.et al.Phencyclidine induction of the hsp70 stress gene in injured pyramidal neurons is mediated via multiple receptors and voltage-gated calcium channels.Neuroscience.199462107910927845588
- SharpFR.ButmanM.WangS.et al.Haloperidol prevents induction of the hsp70 heat shock gene in neurons injured by phencyclidine (PCP), MK801, and ketamine.J Neurosci Res.1992336056161484394
- SharpJW.PetersenDL.LangfordMT.DNQX inhibits phencyclidine (PCP) and ketamine induction of the hsp70 heat shock gene in the rat cingulate and retrosplenial cortex.Brain Res.19956871141247583295
- FarberNB.KimSH.OlneyJW.Costimulation of muscarinic and non-NMDA glutamate receptors reproduces NMDA antagonist neurotoxicity.Soc Neurosci Abstr.1997232308
- KimSH.FarberNB.PriceMT.OlneyJW.Excessive cerebrocortical release of acetylcholine induced by NMDA antagonist is reduced by GABAergic and α2-adrenergic agonists.Mol Psychiatry.1999434435210483051
- FarberNB.HanslickJ.KirbyC.McWilliamsL.OlneyJW.Serotonergic agents that activate 5-HT2A receptors prevent NMDA antagonist neurotoxicity.Neuropsychopharmacology.19981857629408919
- Jevtovic-TodorovicV.OlneyJW.FarberNB.Lamotrigine prevents NMDA antagonist neurotoxicity.Soc Neurosci Abstr.199824745
- FarberNB.FosterJ.DuhanNL.OlneyJW.Olanzapine and fluperlapine mimic clozapine in preventing MK-801 neurotoxicity.Schizophr Res.19962133378998274
- MalhotraAK.AdlerCM.KennisonSD.ElmanI.PickarD.BreierA.Clozapine blunts N-methyl-D-aspartate antagonist-induced psychosis: a study with ketamine.Biol Psychiatry.1997426646689325559
- GonzalesRA.BrownLM.JonesTW.TrentRD.WestbrookSL.LeslieSW.N-methyl-D-aspartate-mediated responses decrease with age in Fischer 344 rat brain.Neurobiol Aging.1991122192251678878
- WenkGL.WalkerLC.PriceDL.CorkLC.Loss of NMDA, but not GABA-A, binding in the brains of aged rats and monkeys.Neurobiol Aging.19911293981646968
- MagnussonKR.CotmanCW.Age-related changes in excitatory amino acid receptors in two mouse strains.Neurobiol Aging.1993141972068391661
- SaransaariP.OjaSS.Dizocilpine binding to cerebral cortical membranes from developing and ageing mice.Mech Ageing Develop.199585171181
- MagnussonKR.The aging of the NMDA receptor complex.Front Biosci.19983e70e809576682
- PiggottMA.PerryEK.PerryRH.CourtJA.[3H]MK-801 binding to the NMDA receptor complex, and its modulation in human frontal cortex during development and aging.Brain Res.19925882772861393579
- GazzaleyAH.SiegelSJ.KordowerJH.MufsonEJ.MorrisonJH.Circuit-specific alterations of N-methyl-D-aspartate receptor subunit 1 in the dentate gyrus of aged monkeys.Proc Natl Acad Sci USA.199693312131258610179
- MagnussonKR.SammondsGE.Age-related changes in the expression of NMDA receptor subunits.FASEB J.1998124365
- ShengM.CummingsJ.RoldanLA.YanYN.YanLY.Changing subunit composition of heteromeric NMDA receptors during development of rat cortex.Nature.19943681441478139656
- OkabeS.CollinC.AuerbachJM.et al.Hippocampal synaptic plasticity in mice overexpressing an embryonic subunit of the NMDA receptor.J Neurosci.199818417741889592097
- CarmignotoG.ViciniS.Activity-dependent decrease in NMDA receptor responses during development of the visual cortex.Science.1992258100710111279803
- HestrinS.Activation and desensitization of glutamate-activated channels mediating fast excitatory synaptic currents in the visual cortex.Neuron.199299919991384578
- MinoshimaS.GiordaniB.BerebtS.FreyKA.FosterNL.KuhlDE.Metabolic reduction in the posterior cingulate cortex in very early Alzheimer's disease.Ann Neurol.19974285949225689
- BraakH.BraakE.BohlJ.Retrosplenial region involvement in Alzheimer's disease.Neurodegeneration.199215357
- BrunA.EnglundE.Regional pattern of degeneration in Alzheimer's disease: neuronal loss and histological grading.Histopathology.198155495647286917
- BraakH.BraakE.Staging of Alzheimer's disease-related neurofibrillary changes.Neurobiol Aging.1995162712847566337
- FixAS.WightmanKA.O'CallaghanJP.Reactive gliosis induced by MK-801 in the rat posterior cingulate/retrosplenial cortex: GFAP evaluation by sandwich ELISA and immunohistochemistry.Neurotoxicology.1995162292397566683
- FarberNB.RubinEH.NewcomerJW.et al.Increased neocortical neurofibrillary tangle density in Alzheimer's disease subjects with psychosis.Arch Gen Psychiatry.2000. In press
- KohJY.YangLL.CotmanCW.Beta-amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage.Brain Res.19905333153202289145
- GrayCW.PatelAJ.Neurodegeneration mediated by glutamate and beta-amyloid peptide: a comparison and possible interaction.Brain Res.19956911691798590049
- PatelAJ.Pretreatment with a sublethal concentration of β-amyloid25-35 potentiates neurodegeneration mediated by glutamate in cultured cortical neurons.Alzheimer's Res.199514144
- SanoM.ErnestoC.ThomasRG.et al.A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's Disease Cooperative Study (see comments).N Engl J Med.1997336121612229110909