Abstract
No animal model to date perfectly replicates Parkinson's disease (PD) etiopathogenesis, and the anatomical organization of the nigrostriatal system differs considerably between species. Human postmortem material therefore remains the gold standard for both formulating hypotheses for subsequent testing in in vitro and in vivo PD models and verifying hypotheses derived from experimental PD models with regard to their validity in the human disease. This article focuses on recent and relevant fields in which human postmortem work has generated significant impact in our understanding of PD. These fields include Lewy body formation, regional vulnerability of dopaminergic neurons, oxidative/nitrative cellular stress, inflammation, apoptosis, infectious and environmental agents, and nondopaminergic lesions.
A la fecha, ningún modelo animal ha podido reproducir perfectamente la etiopatogenia de la enfermedad de Parkinson (EP). Además, la organización anatómica del sistema nigroestriatal difiere considerablemente entre las especies. Es por esto que los estudios postmortem en humanos continúan siendo el gold standard tanto para la formulación de hipótesis que serán probadas posteriormente en modelos de EP in vivo e in vitro, como también para la verificación de hipótesis que deriven de modelos experimentales de EP respecto a su validez para la enfermedad en el hombre. Este artículo se centra en campos recientes y relevantes en los que el trabajo postmortem en humanos ha generado un impacto significativo para nuestra comprensión de la EP. Estos campos incluyen la formación de cuerpos de Lewy, la vulnerabilidad regional de las neuronas dopaminérgicas, el estrés celular de la oxidación y de la nitratación, la inflamación, la apoptosis, los agentes infecciosos y ambientales, y las lesiones no dopaminérgicas.
Aucun modèle animal à ce jour ne reproduit fidèlement l'étiopathogenèse de la maladie de Parkinson (MP). Aussi, l'organisation anatomique du système nigrostrié varie considérablement d'une espèce à l'autre. C'est pourquoi les études post mortem chez l'homme restent la meilleure approche soit pour formuler des hypothèses qui seront ensuite testées dans des modèles in vitro et in vivo de la MP, soit pour vérifier des hypothèses dérivées de modèles expérimentaux de la MP, quant à leur validité en pathologie humaine. Dans cet article, l'accent sera mis sur des domaines de recherche récents et pertinents, tels que la formation de corps de Lewy, la vulnérabilité régionale des neurones dopaminergiques, le stress cellulaire lié à l'oxydation et la nitration, l'inflammation, l'apoptose, les agents infectieux et environnementaux ainsi que les lésions non dopaminergiques, où les études post mortem chez l'homme ont contribué de manière significative à notre compréhension de la MP.
The he history of human postmortem studies in Parkinson's disease (PD) begins at the end of the 1950s with two seminal papers: Carlsson's original suggestion that dopamine (DA) may be a transmitter in the central nervous system (CNS) and be involved in the control of motor function, and thus in the parkinsonian syndromeCitation1 ; and the article by Ehringer and Hornykiewicz,Citation2 which proved the significant, reduction in DA concentration in the neostriatum of patients suffering from sporadic PD. In 1973, these initial observations were followed by demonstration of a correlation between DA cell loss in the substantia nigra pars compacta (SNpc, ) and striatal DA concentrations in PD.Citation3 Interestingly, this study suggested that the classical parkinsonian symptom triad of PD appears when striatal DA concentrations are lowered by approximately 80% ; however, this decline corresponds to only 50% of SNpc DA neuron loss, suggesting that a subset of nigral DA neurons may be morphologically intact, but functionally impaired. This concept is supported by the finding that 15% of melanized neurons in the human SNpc no longer express tyrosine hydroxylase (TH; the rate-limiting enzyme in DA synthesis), but remain morphologically intact:Citation4 This concept of the suffering, ie, metabolically compromised, neuron is important in pathophysiological and therapeutic terms, since it suggests that, a subpopulation of nigral DA neurons are amenable to restorative therapies.
After a general outline on the potential and limitations of human postmortem studies in PD, 1 will explore major questions regarding etiology, pathogenesis, and treatment of PD with reference to human postmortem studies.
The role of human postmortem studies in PD research
There has been considerable debate over the importance of human postmortem studies in PD research. This controversy is based on the many limitations of postmortem research. Human postmortem studies in PD suffer from tissue confounds of aging, end-stage disease, and chronic treatments. Moreover, human postmortem studies cannot answer the question of whether the changes observed are a cause or a consequence of neuronal death in PD. On the other hand, no animal model to date perfectly replicates PD etiopathogenesis, and the anatomical organization of the nigrostriatal system differs considerably between humans and lower species. Thus, human postmortem material remains the gold standard for (i) formulating hypotheses for subsequent, testing in in vitro and in vivo PD models (cell culture, yeast, Drosophila, rodent, and primates models of the disease, based on toxins and/or genetic manipulations); and (ii) verifying hypotheses derived from experimental PD models with regard to their validity in the human disease. This review will emphasize the interaction of findings from postmortem and experimental PD studies.
Genes and PD
The etiology of sporadic PD remains unknown. It is generally believed that, sporadic PD is the result, of complex interactions between genetic susceptibility and environmental factors. In both cases, postmortem studies serve to confirm rather than to generate new hypotheses, with a few notable exceptions.
Genetics is a rapidly growing field in PD research. Three major mutations have been identified to date in affected kindreds: oc-synuclein or Park1Citation5; parkin or Park2Citation6; and DJ-1 or Park7Citation7 A fourth mutated gene product, UCHL1 (Park5), is associated with gene expression; it may not be able to provoke a parkinsonian syndrome alone,Citation8 but may be a susceptibility gene.Citation9 Autopsy specimens from families with these mutations remain rare, but have nevertheless yielded results that have expanded our understanding of sporadic PD, such as the data on α-synuclein.
Three different point mutations in α-synuclein, A.53T, A30P, and E46K, have been associated with PD in separate families with dominantly transmitted PD.Citation5,Citation10,Citation11 These are gain-of-function mutations. There is also evidence that oc-synuclcin promoter variants contribute to the lifetime risk of sporadic PD.Citation12-Citation14 In general, alleles that increase α-synuclein expression are associated with an increased risk for PD. Recent work has shown that, triplication of the α-synuclcin gene is sufficient to cause PD and, in human postmortem brain, is accompanied by doubling of α-synuclein protein expression.Citation15,Citation16 Similarly, postmortem studies in sporadic PD show that α-synuclein mRNA is upregulatcd in the SNpc of affected individuals.Citation17
The link between α-synuclein and sporadic PD is found in Lewy bodies (LBs), the pathological hallmark of PD, since α-synuclein has been shown to be the primary constituent of LBs.Citation18-Citation22 LBs are eosinophilic fibrillar cytoplasmic inclusions in DA neurons that can be detected in both the SNpc and the cortex of PD patients (). LBs are located in the cell body, axons, and dendrites of neurons, and are composed of neurofilaments 7 to 25 nm in diameter; these neurofilaments are believed to be inappropriately phosphorylated, proteolytically truncated, and ubiquitinatcd.Citation23 LBs have been reported to include a wide range of proteins (including ubiquitin, parkin, and tau), heat, shock proteins (HSPs), torsin A, neurofilaments, oxidized/nitrated proteins, proteasomal elements, and others.Citation22,Citation24-Citation31 Interestingly, many proteins that interact with α-synuclein and parkin have also been identified as components of LBs, for instance, parkinassociated endothelin-like receptor (Pael-R; a transmembrane polypeptide),Citation32 synphilin-1,Citation33 and p38 (a structural component of the mammalian am.inoacyl-t.RNA synthetase complex).Citation34 LBs ectopically express the cell cycle protein cyclin B; this may be related to cyclin B's interaction with oc-synuclcin, which predisposes nigral LB-bearing DA neurons to undergo apoptosis.Citation35 Another protein colocalized with α-synuclein in LBs is tissue transglutaminase (tTGase), which induces cross-linking of oc-synuclcin in vitro.Citation36 tTGase inhibition could therefore be a novel therapeutic target in PD, provided that. LB formation is indeed a cytotoxic event.
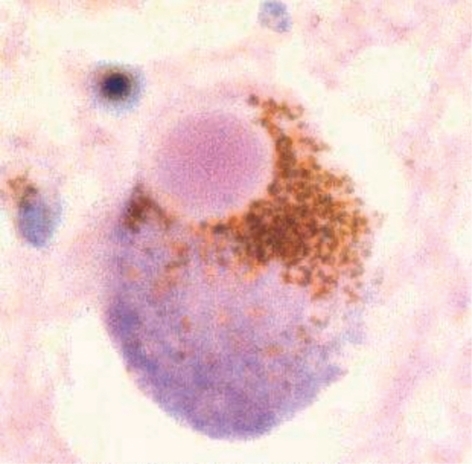
Parkin is widely distributed protein in DA and non-DA neurons in normal human brain and in sporadic PD. It is mostly located in large cytoplasmic vesicles and in the endoplasmic reticulum (RR).Citation37 The initial postmortem studies from five parkin-positive cases initially failed to find LBs - an observation used to argue that parkin is required for LB formation.Citation38,Citation39 However, this conclusion was challenged when Farrer et alCitation40 showed the presence of LBs in one parkin-positive autopsy case. These studies highlight the importance of postmortem study data and their conclusions. They guide our clinical formulations, and thus our experimental and therapeutic approaches.
Finally, studies with DJ-1 are in very early stages. Its distribution has been analyzed in postmortem brain of control and PD subjects in two recent studies. DJ-1 does not colocalize with LBs, but with tau inclusions; it is mainly expressed by astrocytes; and it appears to be sensitive to oxidative stress:Citation11,Citation42 At present, a functional interpretation of these data is lacking.
The role of LBs in DA cell death
There remains much debate over whether LBs are neuroprotective, constitute an age-related epiphenomenon, or are cytotoxic; postmortem end points may supply some answers. Recently, Conway et alCitation43,Citation44 suggested that, accelerated formation of nonfibrillar α-synuclein oligomers is the critical process in PD pathogenesis, ie, LB formation is neuroprotective by sequestering toxic protein species. Once this issue is resolved, drug therapy can be aimed at promoting the healthy process. Two observations from pathological examination of human brain contribute to this dialogue:
SNpc DA neurons containing LB appear to be “healthier” than neighboring neurons,Citation45 whereas the nigral DA neurons undergoing apoptotic-like cell death do not contain somal LBs. Tompkins and HillCitation45 suggested that the majority of SNpc neurons die before or without forming LBs and that SNpc neurons that survive the initial pathological insult suffer damage that leads to LB formation.
It is not uncommon to observe “incidental” LBs at autopsy of aged asymptomatic individuals. An alternative explanation for this finding is that these individuals have not lived long enough to develop a parkinsonian phenotype. Also, if LBs were protective, one might speculate that controls should have more LBs than PD patients, which is clearly not the case.
Alternatively, LBs may occur as an epiphenomenon of the primary pathology and have little or no effect on neuronal viability. In contrast to the observations by Tompkins and Hill,Citation45 Gibb and I .eesCitation46 reported that SNpc neurons with and without somal LBs generally appear to be similarly affected by the disease process. Moreover, cell size and nucleolar size do not differ between LB-positive and LB-negative SNpc neurons.Citation47 Also, dendritic morphological abnormalities found in parkinsonian SNpc arc similar in LB- and non-LB-containing neurons.Citation48 Finally, neurofilament mRNA levels also show a similar level of reduction for both LB- and non-LB-containing neurons.Citation49
We favor the hypothesis that the presence of LBs is an indicator of neuronal distress, although it is impossible to deduce from postmortem work whether I ,Bs are, as such, neurotoxic. The percentage of LB-containing neurons that are positive for caspase-3 and Bax, two proapoptotic proteins, is significantly higher than the percentage of Baxand caspase-3-positive DA neurons not containing LBs,Citation50,Citation51 which indicates that LB-containing DA neurons are more predisposed to undergo apoptosis. Furthermore, aggregated proteins bind HSPs in LBs, and thus prevent their potentially protective chaperone action.Citation52 Entrapment of vital cellular organelles in LBs has been described and may compromise cellular viability.Citation53 Also, LBs may inhibit axonal transport, probably resulting in a “dying back” phenomenon from the synapse to the cell body.Citation52 Clinical neuropathological studies in patients with dementia with LBs (D.LB) report, a correlation between numbers of cortical LBs and the degree of cognitive impairment.Citation54,Citation55 Finally, we have recently conducted a human postmortem study, where the genetic fingerprints of mesencephalic DA neurons containing LBs versus mesencephalic DA neurons not containing LBs were compared in five PD patients. Total RNA from single neurons of both neuronal subpopulations was obtained by immuno-lascr capture microdissection (LCM). Subsequently, RNA arbitrarily primed polymer chain reaction (RAP-PCR) was employed to generate expression profiles from the extracted RNA. Seven expressed sequence tags (HSTs) of interest were selected for further quantitative expression analysis by real-time quantitative reverse transcription PCR (rtq RT-PCR). DA neurons bearing LBs, according to their genetic profile, appeared sicker than their LB-ncgative counterparts, which were preferentially endowed with prosurvival genes. This suggests that inhibition of LB formation indeed represents a therapeutic strategy in PD (Lu and Hartmann, unpublished results).
A differential vulnerability of mesencephalic DA neurons to degeneration
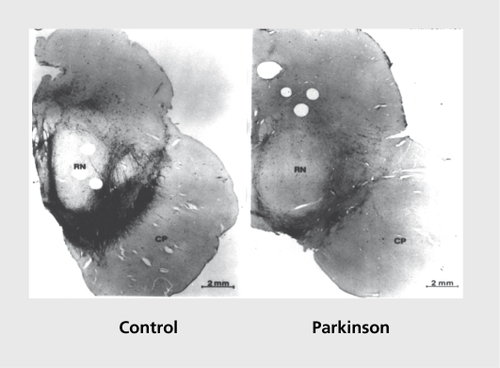
There is evidence that the loss of DA neurons in PD is heterogeneous. DA neurons in the SNpc are affected, as are those in other mesencephalic structures, eg, the ventral tegmental area (VTA) and the central gray substance (COS). These DA neuronal populations display a differential vulnerability to cell death in PD. SNpc DA neurons are most affected, with a cell loss averaging 80% to 90% in PD patients, whereas cell loss in the VTA is intermediate at 40% to 50% . Finally, only 2% to 3% of DA neurons degenerate in the CGS in PD.Citation56 For the PD midbrain, the correlation is simple and direct: the greater the number of pigmented neurons normally present in the DA cell groups, the larger the loss of neurons in the cell groups in the diseased brains. Moreover, within each cell group, nonpigmented neurons are spared relative to the total population of TH-positive neurons and relative to the population of pigmented neurons.Citation4 There is also a regional selective vulnerability of DA neurons within the SNpc to cell death in PD.Citation57-Citation59 On the basis of calbindin D28K (CD28K) immunohistochemistry, the SNpc can be divided into a calbindin -rich region (matrix) and five calbindin-poor pockets (nigrosomes 1-5) (). The depletion of DA neurons begins in nigrosome 1, and then spreads to other nigrosomes and matrix along the rostral, medial, and dorsal axes. Depletion is maximum (98%) in nigrosome 1 , located in the caudal and mediolateral part of the SNpc. Progressively, less cell loss is detectable in more medial and more rostral nigrosomes. A parallel, but lesser, caudorostral gradient, of cell loss is observed for DA neurons included in the matrix. Because the nigro some/matrix analysis refers to compartmental subdivisions within the SNpc, the most obvious conclusion would be that compartmental locality in SNpc itself is a key to differential vulnerability. DA neurons in different, compartments may have different expression patterns of genes implicated in PD pathogenesis. The DA neurons relatively spared from the disease process may be endowed with a range of protective mechanisms, which has sparked research aiming to identify these protective or deleterious mechanisms.
![Figure 3. Summary of midbrain subdivisions illustrated at three representative transverse levels. CGS, central gray substance; M, medial group; Mv, medioventral group (M and Mv constitute the ventral tegmental area [VTA]); A8, dopaminergic group A8; SNpd, substantia nigra pars dorsalis; SNpl, substantia nigra pars lateralis; N, nigrosome; RN, red nucleus; DBC, decussation of the brachium conjunctivum; CP, cerebral peduncle; III, exiting third cranial nerve fibers. Reproduced with permission from reference 58: Damier R Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson's disease. Brain. 1999;122(Pt 8):1437-1448. Copyright©1999. Oxford University Press.](/cms/asset/1e24d72f-f20f-409d-885a-aa08a2b3b15a/tdcn_a_12130563_f0003_oc.jpg)
Whether CD28K determines neuronal vulnerability itself is controversial, and both positive and negative results have been reported. CD28K-positive neurons have been shown to be relatively resistant to degeneration in PDCitation59 and in certain animal models of PD.Citation60-Citation62 There is also a sig
nificant, decrease in CD28K protein and mRNA in the SN, but not in the cerebellum and neocortex of PD patients compared with controls.Citation63 However, on the basis of the viability assessment of midbrain DA neurons in a 1-methyl4-phenyl-1 ,2,3,6-tetrahydropyridine (MPTP) lesion paradigm using CD28K-deficient mice, CD28K-containing neurons are not spared by the pathological process, suggesting that endogenous CD28K is not required for protection of these neurons.Citation64 Thus, CD28K may be a marker of resistance of DA neurons to the degenerative process in PD, but not the causative agent itself.
Perturbation of regulated balance between DAT and VMAT2
It has been proposed that the process underlying PD is the selective degeneration of DA nerve terminals in the striatum expressing dopamine transporter (DAT) and vesicular monoamine transporter 2 (V.M.AT2).Citation65-Citation68 DAT and VMAT2 are essential for normal DA neurotransmission: DAT terminates the actions of DA by rapidly removing it from the synapse; and VMAT2 loads cytoplasmic DA into synaptic vesicles for storage and subsequent release. Cytosolic DA can quickly form reactive oxygen species, and so DA that has been synthesized or transported into the neuron from the extracellular space is rendered harmless by rapid storage in small synaptic vesicles. Hence, DAT activity increases cytoplasmic DA concentrations, whereas VM AT2 activity decreases them. Detailed neuroanatomical analyses of brain from control and PD cases have shown that the regions with the highest DAT/VMAT2 ratio - the caudate and the putamen - are the most sensitive to damage in PD and MPTPinduced parkinsonism.Citation67,Citation69,Citation70 A recent study showed that α-synuclein negatively modulates human DAT activity,Citation71 whereas an earlier study found opposite results.Citation72 The A53T mutation of expression of α-synuclein also reduced levels of VMAT2. Taken together, the defective sequestration of DA mediated by the interplay of DAT, VMAT2, and α-synuclein may be a key event, in the DA cell death in sporadic PD.Citation73
To extend the one gene/one protein approach to the search for the differences in mesencephalic regional vulnerability to cell death, we compared the genetic fingerprints of mesencephalic DA neurons that are particularly prone to degenerate during PD (DA neurons in nigrosome 1 within the SNpc) and mesencephalic DA neurons that are particularly resistant to the disease course (DA neurons in the CGS) in five control subjects. We found that SNpc DA neurons do not per se reveal many distinctive deleterious genes; rather, it appeared as if CGS DA neurons were just embodied with more cellular defenses against, degeneration, suggesting that the transfer of these factors might endow SNpc DA neurons with the same resistance against neuronal death in PD (Lu and Hartmann, unpublished results).
Defects of the ubiquitin-proteasome system in PD
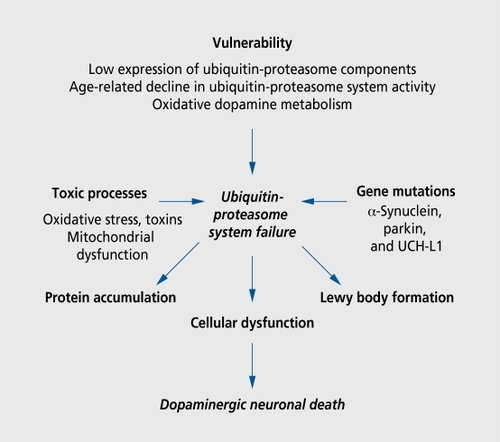
A growing body of evidence suggests that proteolytic stress underlies both familial and sporadic PD. Interest in the role of the proteasomc in the pathophysiology of PD has been triggered by observation of parkin, an E3 ubiquitin liga.se, which tags (potentially neurotoxic) proteins for degradation by the proteasome. Proteasomes are multicatalytic proteases found in the cytoplasm, ER, perinuclear region, and the nucleus of eukaryotic cell.Citation74 The accumulation of oxidized proteins in SNpc suggests that protein clearance is inadequate in this brain region.Citation75 Compared with the brains of age -matched controls, in the brains of subjects with sporadic PD, there is a marked loss of α- but not. β-subunits of 20S proteasome core within nigral DA neurons.Citation76 Levels of PA28 (a multisubunit proteasome activator) are very low in the SNpc of both control and PD subjects.Citation77 These findings point to a primary defect in proteasome -mediated proteolysis of nigral DA neurons in sporadic PD. Dysfunction at any point on the proteasome pathway may result in nigral DA neuronal degeneration, accounting for the particular vulnerability of nigral DA neurons to neurodegeneration. Although low proteasomal activity has been linked to LB formation by some,Citation78 it has been refuted by others.Citation79 The production of abnormal proteins that resist, and inhibit proteolysis (α-synuclein mutations), defects in protein ubiquitination (parkin mutations), reduced deubiquitination (UCH-L1Park,5 mutations), and proteasomal dysfunction (sporadic PD) have all been implicated in the etiopathogenesis of PD ().
Oxidative stress
One of the first, and most relevant hypotheses for PD pathogenesis relates to increased oxidativc/nitrative stress in mesencephalic DA neurons. In PD, DA can auto-oxidize into toxic dopamine-quinone species, superoxide radicals, and hydrogen peroxide. DA auto-oxidizes into neuromclanin, the phenotypic marker of midbrain DA neurons in humans. Accordingly, the neuromelanin content and distribution in the parkinsonian mesencephalon has been linked to the vulnerability of DA neurons to undergo cell death.Citation80 Oxidativc/nitrative stress may result in protein oxidation/nitrationCitation81,Citation82; decreased neuronal glutathione and glutathione peroxidase content, which prevents inactivation of hydrogen peroxide and enhances formation of toxic hydroxyl radicalsCitation83-Citation85; basal lipid peroxidation, which results in membrane damageCitation86; DNA and RNA oxidationCitation75,Citation87; and formation of I LBs.Citation88
A potential signaling pathway between oxidative stress and subsequent cell death has been explored by Hunot ct al.Citation89 They showed that, nuclear translocation of the nuclear factor-KB (NF-κB), which is triggered by oxidative stress and precedes the engagement of an apoptotic program, is increased 70-fold in nigral DA neurons from PD subjects compared with control subjects. Oxidative stress has also been implicated in altered iron, ferritin, and trace metal contents of nigral DA neurons and may increase the susceptibility of these neurons to cell death.Citation90-Citation92 Prior to causing cell death, increased iron in the brain has been suggested to trigger LB formation and initiation of inflammatory responses.Citation93 Interestingly, the detection of redox-activc iron in situ showed a strong labeling of LBs in the SNpc of PD patients, whereas cortical LBs remained unstained; this indicates a fundamental difference between cortical and brain-stem LBs.Citation94 Similarly, Giasson et alCitation95 reported that nitrated α-synuclein is present in the major filamentous building blocks of LBs, underlining the importance of oxidativc/nitrative stress in PD.
Inflammation
The degeneration of DA neurons is associated with a strong glial reaction, which is generally considered to be a nonspecific consequence of neuronal degeneration. However, there is increasing evidence that inflammation is an active phenomenon in PD, continuously triggering DA cell death in this neurodegenerative disorder. The glial reaction in the SN of PD patients is a well-known neuropathological characteristic of the disease. In their seminal study, McGeer and McGeer Citation96 reported a large number of reactive human leukocyte antigen-DR (HLADR)-positive microglial cells in the SN of PD patients. Such a glial reaction has also been described in the affected brain regions in other neurological disorders, such as Alzheimer's disease and brain infarct,Citation97 as well as in animal models of PD.Citation98 These data suggest that, glial activation is not specific to PD and that it most likely represents a common phenomenon in neurodegenerative disorders. However, recent evidence supports the notion that a subpopulation of activated glial cells may be deleterious in PD, particularly for highly dysfunctional neurons that are metabolically compromised. Strong support for this hypothesis came from a study of young drug addicts who developed a parkinsonian syndrome after MPTP intoxication.Citation99 In a recent, study, the same authors reported a postmortem neuropathological study of three subjects with MPTP-induced parkinsonism.Citation100 Interestingly, gliosis and clustering of microglial cells around DA neurons were detected, despite survival times ranging from 3 to 16 years. These findings not only indicate an ongoing nerve cell loss after a time -limited insult, but also suggest, that, activated microglial cells may perpetuate neuronal degeneration. One may thus speculate that after a primary insult of environmental and/or genetic origin, the glial reaction may perpetuate the degeneration of DA neurons.
The mechanism by which microglial cells can amplify injury to nigral DA neurons is not yet known. However, the factors involved in this deleterious effect, are very likely cytokines, including tumor necrosis factor a (TNF-α), interlcukin 1β (II-1β), and interferon γ (IFN-γ). Accordingly, several studies have reported a marked increase of cytokine levels in the brain and cerebrospinal fluid (CSF) of PD patients.Citation101 In addition, a higher density of glial cells expressing TNF-α, II-1γ, and IFN-γ was observed in the SN of PD patients compared with agematched control subjects.Citation102,Citation103 Some of these cells were close to blood vessels and degenerating DA neurons, suggesting their involvement in the pathophysiology of PD. Two mechanisms, which are not mutually exclusive, may explain the deleterious role of cytokines in the parkinsonian SNpc:
Proinflammatory cytokines induce the production of nitric oxide in glial cells.Citation104
TNF-α receptors directly activate DA neurons of the human SN.Citation102
The question of whether inflammation plays a prominent role in PD pathogenesis cannot be resolved by postmortem studies alone, and experimental PD models have much contributed to strengthening this hypothesis, making inflammation a prime candidate for neuroprotective studies in PD patients.Citation98 Importantly, recent primate studies have replicated chronic glial activation in the SNpc following a time-limited MPTP insultCitation105-Citation107 and may thus represent a valuable model to study the long-term consequences of this process.
Apoptosis
There has been much interest, in whether DA neurons in PD die by apoptosis, necrosis, or some other form of cell death. This is because apoptosis is amenable to pharmacological inhibition and may thus be a therapeutic target in PD. The initial human postmortem studies showed conflicting results,Citation51 either because of methodological problems, especially the unreliability of 3' DNA end labeling as a marker of apoptotic nuclear degradation,Citation108 or because the apoptotic changes observed might, in fact represent a perimortem effect rather than the primary disease process.Citation109
Thus, the next step was to look at the cellular transduction pathways mediating apoptosis in PD brains. In this respect, the signaling pathways downstream of the inflammatory release of proinflammatory cytokines, eg, coupled to the TNF type 1 receptor (TNFR1), are of particular interest, given previous work on inflammation triggering cell death in PD. TNF-oc induces trimerization of TNFR1 on binding, which leads to the autoproteolytical activation of caspase-8 via the adaptor molecule TNFRl-associating protein with a death domain and FAS-associated protein with a death domain (FADD). Caspase-8 may in turn either cleave effector caspases, such as caspase-3, directly or amplify the death signal through the mitochondrial release of cytochrome-c into the cytosol.Citation110 Indeed, in a human postmortem study, we showed a significant decrease in the percentage of FADD immunoreactivc DA neurons in the SNpc of PD patients compared with control subjects.Citation111 Furthermore, this decrease correlated with the known selective vulnerability of nigral DA neurons in PD, suggesting that this pathway contributes to the susceptibility of DA neurons to TNF-mediated apoptosis in PD. One step downstream in this proapoptotic signaling cascade, the proportion of melanized neurons displaying caspase-8 activation in PD was also higher in PD than in control subjects.Citation112 Similar results were obtained for caspase-3, where we found (i) a positive correlation between the degree of neuronal loss in DA cell groups affected in the mesencephalon of PD patients and the percentage of caspase-3-positive neurons in these cell groups in control subjects; (ii) a significant decrease in caspase-3-positive pigmented neurons in the SNpc of PD patients compared with control subjects; and (iii) a significantly higher percentage of active caspase-3-positive neurons among DA neurons in PD compared with control subjects.Citation50 Taken together, these studies suggest that the melanized DA neurons expressing the TNFR1 transduction pathway are particularly prone to degeneration in PD if this pathway is activated during the course of the disease.
As regards mitochondrial proteins controlling apoptosis in PD, we have shown a similar distribution of nigral DA neurons immunoreactivc for Bax, a proapoptotic mitochondrial protein, in PD compared with control subjects.Citation113 However, by assessing staining intensity, TattonCitation114 reported increased immunoreactivity for Bax and caspase-3 in nigral DA neurons of PD compared with control subjects. We also studied the mRNA expression of Bcl-xL, a major anti-apoptotic mitochondrial protein in the SNpc of PD patients and controls. We found a significant upregulation of Bcl-xL mRNA expression in nigral DA neurons from PD patients, as assessed by in situ hybridization, which was accompanied by a redistribution of the protein to the mitochondrial outer membrane, as assessed by electron microscopy.Citation115 This process suggests a compensatory upregulation of Bcl-xL in the nigral DA neurons surviving the pathological process in PD. Finally, an experimental link between sublethal activation of apoptotic pathways and LB formation has been suggested by Hashimoto et al,Citation116 who showed that release of cytochrome-c from mitochondria into the cytosol may also function as a stimulator for oc-synuclcin aggregation.
Environmental toxins
The seminal study by Langston et alCitation99 on .MPTP as the causative agent for a PD-like syndrome has triggered numerous studies on the role of environmental toxins in the pathophysiology of PD. Since MPTP inhibits complex I of the mitochondrial respiratory chain, a defect in this protein has been investigated in cases of sporadic PD. In 1990, Schapira et alCitation117 showed that complex I activity is indeed decreased in the SNpc of patients suffering from sporadic PD. Environmental toxins, particularly herbicides and pesticides that inhibit complex I activity, such as rotenone, paraquat, and maneb, have since been studied as potential causative or at least risk factors in PD models and in epidemiological studies.Citation118 However, only limited human postmortem data have been gathered so far. Fleming et alCitation119 screened postmortem brain samples from PD patients and control cases for 16 organochloridc pesticides. They found a positive association of PD and pesticide concentrations for only one pesticide, dieldrin, a lipid-soluble mitochondrial poison. These results were replicated by another group in separate studies with regard to increased dicldrin concentration in PD brain.Citation120-Citation122 However, the mode of action of this pesticide strongly supports current, concepts of oxidative stress and mitochondrial energy impairment, as an important factor in PD pathogenesis. Interestingly, the pesticides dicldrin, paraquat, and rotenone, which are all complex I inhibitors, have been shown to induce an acceleration of α-synuclein fibril formation in vitro, and thus likely Lewy body formation.Citation123
Infection
The idea of a putative role of infectious agents in the etiology of PD can be traced back to 1918, when postencephalitic parkinsonism due to influenza A infection was widespread in Europe. Many decades later, observations of sporadic PD suggest that. LBs harbor viral and bacterial signatures.Citation124 A very recent study has convincingly shown that Nocardia astéroïdes 16S rRNA is present, in LBs from PD patients and points to a role in bacterial infection in protein aggregation.Citation125 These findings, however, need to be confirmed in larger samples. The same authors also showed that one out of two cynomolgus monkeys infected with N. astéroïdes developed intracellular inclusion bodies (immunoreactivc for α-synuclein and ubiquitin); the infected monkey also expressed rRNA for N. astéroïdes. Other viral and bacterial pathogens need to be studied in human postmortem brain tissue of PD patients using more recent virological and bacterial detection methods.Citation126
Non-DA ceil loss in PD
The concept, of a specific neurotransmitter deficiency associated with a specific neurological syndrome potentially amenable to replacement therapy, as exemplified by the initial studies of Carlsson,Citation1 Ehringcr and Homykiewicz,Citation2 and Bemheimer,Citation3 has somehow obscured the fact that PD is not a disease restricted to the nigrostriatal system and involving solely DA as neurotransmitter. In fact, postmortem studies have shown that much of the peripheral and central nervous systems (stellate ganglia, cardiac, and enteric plexus, nucleus basalis of Meynert, amygdala, limbic nuclei of the thalamus, parahippocampal and cingulate gyri, insula, and isocortex) and transmitter systems (serotonin, noradrenaline, and acetylcholine) are affected in PD, albeit, at varying degrees.Citation56 This explains the comorbidity of PD with depression,Citation127 dementia,Citation128 autonomic dysfunction,Citation129 and sleep disorders.Citation130 The common link between degeneration in these structures and/or transmitter systems may be the presence of LBs, which stresses their importance in PD pathogenesis.
The studies of Braak's group are of special interest here. In large series of individuals suffering from PD or in the preliminary stages, the distribution of LBs appears to follow a specific temporal (subdivided into stages 1 to 6) and anatomical distribution: lesions initially occur in the dorsal motor nucleus of the glossopharyngeal and vagal nerves and anterior olfactory nucleus. Thereafter, less vulnerable nuclear grays and cortical areas gradually become affected. The disease process in the brain stem then pursues an ascending course. Cortical involvement, ensues, beginning with the anteromedial temporal mesocortcx. Next, the neocortex is affected, commencing with higher order sensory association and prefrontal areas. First-order sensory association/premotor areas and primary sensory/motor fields are affected last.Citation131,Citation132
Braak et alCitation133 have speculated that PD might originate outside the CNS, caused by an as yet unidentified pathogen capable of passing the mucosal barrier of the gastrointestinal tract and, via postganglionic enteric neurons, entering the CNS along unmyelinated preganglionic fibers generated from the visceromotor projection cells of the vagus nerve. By way of retrograde axonal and transneuronal transport, such a causative pathogen (a toxin and/or infectious agent?) could reach selectively vulnerable subcortical nuclei and, unimpeded, gain access to the cerebral cortex. At present, the experimental arguments supporting this intriguing hypothesis are sparse, especially because the relationship between neuronal degeneration and LB formation is still unclear.Citation134 However, considering current evidence, it is plausible that, the presence of LBs indicates a disease process and reflects neuronal suffering.
Conclusion and perspectives
Human postmortem studies remain the mainstay of our understanding of PD. Despite considerable advances in modeling PD, none of the experimental models available today reflects all the major characteristics of the disease in humans. Human postmortem studies and experimental PD paradigms should be closely associated to study questions related to etiology and/or pathogenesis. Future major research topics will include the role of protein aggregation, LB formation, and protcasomal dysfunction in pathogenesis, and their relationship to DA metabolism, accounting for the selectivity of lesions in PD. The role of environmental toxins and infectious agents in the etiology of PD and in relation to susceptibility genes should also be an area of vigorous research. The microglial reaction and chronic inflammation will also be major therapeutic targets to slow PD progession. Interestingly, an inverse correlation between the intake of nonsteroidal anti-inflammatory drugs (NSAIDs) and the risk for PD has recently been claimed by an extensive epidemiological study.Citation135 In this regard, it would undoubtedly be of great value to study the brains of individuals with a long-standing history of NSAID intake to seek the presence (or absence) of PDlike pathology. With respect, to these questions, we should emphasize the need to collect donor brains in specialized brains banks to supply the field of human postmortem PD research.Citation136 Specifically, brain bank characterization of PD brain samples and other neurodegenerative diseases in the postgenomic era must include the genotype and phenotype of the affected individuals as well as thorough clinical data.
Selected abbreviations and acronyms
| DA | = | dopamine |
| DAT | = | dopamine transporter |
| DLB | = | dementia with Lewy bodies |
| LB | = | Lewy bod |
| MPTP | = | l-methyl-4-phenyl-l,2,3,6-tetrahydropyridine |
| PD | = | Parkinson's disease |
| SNpc | = | substantia nigra pars compacta |
| VMAT2 | = | vesicular monoamine transporter 2 |
REFERENCES
- CarlssonA.The occurrence, distribution and physiological role of catecholamines in the nervous system.Pharmacol Rev.195911(2. Part 2)49049313667431
- EhringerH.HornykiewiczO.Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system [in German].Klin Wochenschr.1960381236123913726012
- BernheimerH.BirkmayerW.HornykiewiczO.JellingerK.SeitelbergerF.Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations.J Neurol Sci.1973204154554272516
- HirschE.GraybielAM.AgidYA.Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease.Nature.19883343453482899295
- PolymeropoulosMH.LavedanC.LeroyE.et al.Mutation in the α-synuclein gene identified in families with Parkinson's disease.Science.1997276204520479197268
- KitadaT.AsakawaS.HattoriN.et al.Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism.Nature.19983926056089560156
- BonifatiV.RizzuP.van BarenMJ.et al.Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonismScience.200329925625912446870
- LeroyE.BoyerR.AuburgerG.et al.The ubiquitin pathway in Parkinson's disease.Nature.19983954514529774100
- MaraganoreDM.LesnickTG.ElbazA.et al. UCHL1 is a Parkinson's disease susceptibility gene.Ann Neurol.20045551252115048890
- KrügerR.KuhnW.MullerT.et al. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's diseaseNat Genet.1998181061089462735
- ZarranzJJ.AlegreJ.Gomez-EstebanJC.et al.The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementiaAnn Neurol.20045516417314755719
- FarrerM.MaraganoreDM.LockhartP.et al.α-Synuclein gene haplotypes are associated with Parkinson's disease.Hum Mol Genet.2001101847185111532993
- TanEK.TanC.ShenH.et al.Asynuclein promoter and risk of Parkinson's disease: microsatellite and allelic size variability.Neurosci Lett.2003336707212493604
- HolzmannC.KrugerR.SaeckerAM.et al.Polymorphisms of the α-synuclein promoter: expression analyses and association studies in Parkinson's disease.J Neural Transm.2003 110677612541013
- FarrerM.KachergusJ.FornoL.et al.Comparison of kindreds with parkinsonism and α-synuclein genomic multiplications.Ann Neurol.20045517417914755720
- SingletonAB.FarrerM.JohnsonJ.et al.α-Synuclein locus triplication causes Parkinson's disease.Science.200330284114593171
- RockensteinE.HansenLA.MalloryM.et al.Altered expression of the synuclein family mRNA in Lewy body and Alzheimer's disease.Brain Res.2000914485611578596
- ArimaK.HiraiS.SunoharaN.et al.Cellular co-localization of phosphorylated tau- and NACP/α-synuclein-epitopes in Lewy bodies in sporadic Parkinson's disease and in dementia with Lewy bodies.Brain Res.1999843536110528110
- BabaM.NakajoS.TuPH.et al.Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies.Am J Pathol.19981528798849546347
- IrizarryMC.GrowdonW.Gomez-lslaT.et al.Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson's disease and cortical Lewy body disease contain α-synuclein immunoreactivity.J Neuropathol Exp Neurol.1998573343379600226
- MezeyE.DehejiaAM.HartaG.et al.A synuclein is present in Lewy bodies in sporadic Parkinson's disease.Mol Psychiatry.199834934999857974
- SpillantiniMG.CrowtherRA.JakesR.et al.α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies.Proc Natl Acad Sci USA.199895646964739600990
- HillWD.LeeVM.HurtigHI.MurrayJM.TrojanowskiJQ.Epitopes located in spatially separate domains of each neurofilament subunit are present in Parkinson's disease Lewybodies.J Comp Neurol19913091501601716646
- IshizawaT.MattilaP.DaviesP.WangD.DicksonDW.Colocalization of tau and α-synuclein epitopes in Lewy bodies.J Neuropathol Exp Neurol.20036238939712722831
- LoweJ.McDermottH.LandonM.MayerRJ.WilkinsonKD.Ubiquitin carboxyl-terminal hydrolase (PGP 9.5) is selectively present in ubiquitinated inclusion bodies characteristic of human neurodegenerative diseases.J Pathol.19901611531602166150
- PollanenMS.DicksonDW.BergeronC.Pathology and biology of the Lewy body.J Neuropathol Exp Neurol.199352183191 7684074
- FornoLS.DeLanneyLE.IrwinI.et al.Electron microscopy of Lewy bodies in the amygdala-parahippocampal region. Comparison with inclusion bodies in the MPTP-treated squirrel monkey.Adv Neurol.1996692172288615131
- GoodPF.HsuA.WernerP.et al.Protein nitration in Parkinson's disease.J Neuropathol Exp Neurol.1998573383429600227
- GiassonBl.DudaJE.MurrayIV.et al.Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions.Science.200029098598911062131
- ShashidharanP.GoodPF.HsuA.et al.TorsinA accumulation in Lewy bodies in sporadic Parkinson's disease.Brain Res.200087737938110986355
- SchlossmacherMG.FroschMP.GaiWP.et al.Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies.Am J Pathol.20021601655166712000718
- MurakamiT.ShojiM.ImaiY.et al.Pael-R is accumulated in Lewy bodies of Parkinson's disease.Ann Neurol.20045543944214991825
- WakabayashiK.EngelenderS.YoshimotoM.TsujiS.RossCA.TakahashiH.Synphilin-1 is present in Lewy bodies in Parkinson's disease.Ann Neurol.20004752152310762166
- CortiO.HampeC.KoutnikovaH.et al.The p38 subunit of the aminoacyl-tRNA synthetase complex is a Parkin substrate: linking protein biosynthesis and neurodegeneration.Hum Mol Genet.2003121427143712783850
- LeeSS.KimYM.JunnE.et al.Cell cycle aberrations by α-synuclein overexpression and cyclin B immunoreactivity in Lewy bodies.Neurobiol Aging .20032468769612885576
- JunnE.RonchettiRD.QuezadoMM.KimSY.MouradianMM.Tissue transglutaminase-induced aggregation of α-synuclein: implications for Lewy body formation in Parkinson's disease and dementia with Lewy bodies.Proc Natl Acad Sci USA.20031002047205212576551
- Zarate-LagunesM.GuWJ.BlanchardV.et al.Parkin immunoreactivity in the brain of human and non-human primates: an immunohistochemical analysis in normal conditions and in parkinsonian syndromes.J Comp Neurol.200143218419611241385
- ShimuraH.HattoriN.KuboS.et al.Immunohistochemical and subcellular localization of Parkin protein: absence of protein in autosomal recessive juvenile parkinsonism patients.Ann Neurol.19994566867210319893
- HayashiS.WakabayashiK.IshikawaA.et al.An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene.Mov Disord.20001588488811009195
- FarrerM.ChanP.ChenR.et al.Lewy bodies and parkinsonism in families with parkin mutations.Ann Neurol.20015029330011558785
- BandopadhyayR.KingsburyAE.CooksonMR.et al.The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson's diseaseBrain.2004127(Pt 2)42043014662519
- RizzuP.HinkleDA.ZhukarevaV.et al.DJ-1 colocalizes with tau inclusions: a link between parkinsonism and dementia.Ann Neurol.20045511311814705119
- ConwayKA.LeeSJ.RochetJC.et al.Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy.Proc Natl Acad Sci U SA 200097571576
- ConwayKA.PchetJC.BieganskiRM.et al.Kinetic stabilization of α-synuclein protofibril by dopamine-α-synuclein adduct.Science.20012941346134911701929
- TompkinsMM.HillWD.Contribution of somal Lewy bodies to neuronal death.Brain Res.199777524299439824
- GibbWR.LeesAJ.The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease.J Neurol Neurosurg Psychiatry.1988517457522841426
- GertzHJ.SiegersA.KuchinkeJ.Stability of cell size and nucleolar size in Lewy body containing neurons of substantia nigra in Parkinson's disease.Brain Res.19946373393418180816
- PattS.GertzHJ.GerhardL.et al.Pathological changes in dendrites of substantia nigra neurons in Parkinson's disease: a Golgi study.Histol Histopathol.199163733801725760
- HillWD.Altered neurofilament expression does not contribute to Lewy body formation.Am J Pathol.19961497287298702011
- HartmannA.HunotS.MichelPP.et al.Caspase-3: a vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson's disease.Proc Natl Acad Sci USA.2000972875288010688892
- HartmannA.HirschEC.Parkinson's disease. The apoptosis hypothesis revisited.Adv Neurol.20018614315311553972
- GalvinJE.LeeVM.SchmidtML.TuPH.IwatsuboT.TrojanowskiJQ.Pathobiology of the Lewy body.Adv Neurol.19998031332410410736
- TuPH.RobinsonKA.de SnooF.et al.Selective degeneration of Purkinje cells with Lewy body-like inclusions in aged NFHLACZ transgenic mice.J Neurosci.199717106471048994061
- HurtigHI.TrojanowskiJQ.GalvinJ.et al.α-Synuclein cortical Lewy body correlate with dementia in Parkinson's disease.Neurology.2000541916192110822429
- MattilaPM.RinneJO.HeleniusH.et al.α-Synuclein-immunoreactive cortical impairment in Parkinson's diseaseActa Neuropathol (Berl).200010028529010965798
- HirschEC.OrieuxG.MurielMP.FrancoisC.FegerJ.Nondopaminergic neurons in Parkinson's disease.Adv Neurol.200391293712449099
- FearnleyJM.LeesAJ.Ageing and Parkinson's disease: substantia nigra regional selectivity.Brain.1991114( Pt 5)228323011933245
- DamierP.HirschEC.AgidY.GraybielAM.The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson's disease.Brain.1999122(Pt 8)1437144810430830
- DamierP.HirschEC.AgidY.GraybielAM.The substantia nigra of the human brain. I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D(28K) immunohistochemistry.Brain.1999122(Pt8)1421143610430829
- GermanDC.ManayeKF.SonsallaPK.et al.Midbrain dopaminergic cell loss in Parkinson's disease and MPTP-induced parkinsonism: sparing of calbindin-D28k-containing cells.Ann N Y Acad Sci.199264842621353337
- GasparP.Ben JellounN.FebvretA.Sparing of the dopaminergic neurons containing calbindin-D28k and of the dopaminergic mesocortical projections in weaver mutant mice.Neuroscience.1994612933057969910
- LiangCL.SintonCM.GermanDC.Midbrain dopaminergic neurons in the mouse: co- localization with calbindin-D28K and calretinin.Neuroscience.1996755235338931015
- lacopinoAM.ChristakosS.Specific reduction of calcium-binding protein (28-kilodalton calbindin-D) gene expression in aging and neurodegenerative diseases.Proc Natl Acad Sci USA.199087407840822140897
- AiraksinenMS.ThoenenH.MeyerM.Vulnerability of midbrain dopaminergic neurons in calbindin-D28k-deficient mice: lack of evidence for a neuroprotective role of endogenous calbindin in MPTP-treated and weaver mice.Eur J Neurosci.199791201279042576
- SangheraMK.ManayeK.McMahonA.SonsallaPK.GermanDC.Dopamine transporter mRNA levels are high in midbrain neurons vulnerable to MPTP.Neuroreport19978332733319351666
- UhlGR.Hypothesis: the role of dopaminergic transporters in selective vulnerability of cells in Parkinson's disease.Ann Neurol.1998435555609585349
- MillerGW.EricksonJD.PerezJT.et al.Immunochemical analysis of vesicular monoamine transporter (VMAT2) protein in Parkinson's disease.Exp Neurol.199915613814810192785
- MillerGW.HeilmanCJ.PerezJT.et al.Immunochemical analysis of dopamine transporter protein in Parkinson's disease.Ann Neurol.1997415305399124811
- ShimadaS.KltayamaS.WaltherD.et al.Dopamine transporter mRNA: dense expression in ventral midbrain neurons.Mol Brain Res.1992133593621352613
- UhlGR.WaltherD.MashD.et al.Dopamine transporter messenger RNA in Parkinson's disease and control substantia nigra neurons.Ann Neurol.1994354944988154880
- WersingerC.SidhuA.Attentuation of dopamine transporter activity by α-synuclein.Neurosci Lett.200334018919212672538
- LeeFJ.LiuF.PristupaZB.NiznikHB.Direct binding and functional coupling of α-synuclein to the dopamine transporters accelerate dopamineinduced apoptosis.FASEBJ.200115916926
- LothariusJ.BrundinP.Pathogenesis of Parkinson's disease: dopamine, vesicles and α-synuclein.Nat Rev Neurosci.2002393294212461550
- VogesD.ZwicklP.BaumeisterW.The 26S proteasome: a molecular machine designed for controlled proteolysis.Annu Rev Biochem.1999681015106810872471
- AlamZl.DanielSE.LeesAJ.et al.A generalised increase in protein carbonyls in the brain in Parkinson's but not incidental Lewy body disease.J Neurochem.199769132613299282961
- McNaughtKS.JennerP.Proteasomal function is impaired in substantia nigra in Parkinson's disease.Neurosci Lett.200129719119411137760
- McNaughtKS.BelezaireR.Isacson0.et al.Altered proteasomal function in Parkinson's disease.Exp Neurol.2003179384612504866
- ArdleyHC.ScottGB.RoseSA.TanNG.MarkhamAF.RobinsonPA.Inhibition of proteasomal activity causes inclusion formation in neuronal and non-neuronal cells overexpressing parkin.Mol Biol Cell.2003144541455612937272
- TofarisGK.RazzaqA.GhettiB.LilleyKS.SpillantiniMG.Ubiquitination of α-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function.J Biol Cnem.20032784440544411
- KastnerA.HirschEC.LejeuneO.Javoy-AgidF.RascolO.AgidY.Is the vulnerability of neurons in the substantia nigra of patients with Parkinson's disease related to their neuromelanin content?J Neurochem.199259108010891494900
- YoritakaA.HattoriN.UchidaK.TanakaM.StadtmanER.MizunoY.Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease.Proc Natl Acad Sci USA.199693269627018610103
- FloorE.WetzelMG.Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay.J Neurochem.1998702682759422371
- KishSJ.MoritoC.HornykiewiczO.Glutathione peroxidase activity in Parkinson's disease brain.Neurosci Lett.1985583433464047494
- JennerP.DexterDT.SianJ.SchapiraAH.MarsdenCD.Oxidative stress as a cause of nigral cell death in Parkinson's disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson's Disease Research Group.Ann Neurol.199232(suppl)S82S871510385
- DamierP.HirschEC.ZhangP.AgidY.Javoy-AgidF.Glutathione peroxidase, glial cells and Parkinson's disease.Neuroscience.199352168433802
- DexterDT.CarterCJ.WellsFR.et al.Basal lipid peroxidation in substantia nigra is increased in Parkinson's disease.J Neurochem.1989523813892911023
- ZhangJ.PerryG.SmithMA.et al.Parkinson's disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons.Am J Pathol.19991541423142910329595
- LeeSJ.α-Synuclein aggregation: a link between mitochondrial defects and Parkinson's disease?Antioxid Redox Signal.2003533734812880487
- HunotS.BruggB.RicardD.et al.Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with Parkinson disease.Proc Natl Acad Sci USA.199794753175369207126
- SoficE.RiedererP.HeinsenH.et al.Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain.J Neural Transm.1988741992053210014
- DexterDT.JennerP.SchapiraAH.MarsdenCD.Alterations in levels of iron, ferritin, and other trace metals in neurodegenerative diseases affecting the basal ganglia. The Royal Kings and Queens Parkinson's Disease Research Group.Ann Neurol.199232(suppl)S94S1001510387
- FaucheuxBA.MartinME.BeaumontC.HauwJJ.AgidY.HirschEC.Neuromelanin associated redox-active iron is increased in the substantia nigra of patients with Parkinson's disease.J Neurochem.2003861142114812911622
- KaurD.AndersenJK.Ironing out Parkinson's disease: is therapeutic treatment with iron chelators a real possibility?Aging Ceil.200211721
- CastellaniRJ.SiedlakSL.PerryG.SmithMA.Sequestration of iron by Lewy bodies in Parkinson's disease.Acta Neuropathol (Bed).2000100111114
- GiassonBl.DudaJE.MurrayIV.et al.Oxidative damage linked to neurodegeneration by selective a-synuclein nitration in synucleinopathy lesions.Science.200029098598911062131
- McGeerPL.McGeerEG.Mechanisms of cell death in Alzheimer disease - immunopathology.J Neural Transm Suppl.1998541 59166
- McGeerPL.SchwabC.ParentA.DoudetD.Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1, 2,3,6tetrahydropyridine administration.Ann Neurol.20035459960414595649
- HunotS.HirschEC.Neuroinflammatory processes in Parkinson's disease.Ann Neurol.200353(suppl 3)S49S5812666098
- LangstonJW.BallardP.TetrudJW.IrwinI.Chronic parkinsonism in humans due to a product of meperidine-analog synthesis.Science.19832199799806823561
- LangstonJW.FornoLS.TetrudJ.ReevesAG.KaplanJA.KarlukD.Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure.Ann Neurol.19994659860510514096
- NagatsuT.MogiM.IchinoseH.TogariA.Cytokines in Parkinson's disease.J Neural Transm Suppl.20005814315111128604
- BokaG.AngladeP.WallachD.Javoy-AgidF.AgidY.HirschEC.Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson's disease.Neurosci Lett.19941721511548084523
- HunotS.DugasN.FaucheuxB.et al.FcepsilonRII/CD23 is expressed in Parkinson's disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-a in glial cells.J Neurosci.1999193440344710212304
- HunotS.BoissiereF.FaucheuxB.et al.Nitric oxide synthase and neuronal vulnerability in Parkinson's disease.Neuroscience.1996723553638737406
- McGeerPL.ItagakiS.AkiyamaH.McGeerEG.Rate of cell death in parkinsonism indicates active neuropathological process.Ann Neurol.1988245745763239957
- HurleySD.O'BanionMK.SongDD.AranaFS.OlschowkaJA.HaberSN.Microglial response is poorly correlated with neurodegeneration following chronic, low-dose MPTP administration in monkeys.Exp Neurol.200318465966814769357
- BarciaC.BahilloAS.Femandez-VillabaE.et al.Evidence of active microglia in substantia nigra pars compacta of parkinsonian monkeys 1 year after MPTP exposure.Glia.20044640240915095370
- TattonWG.Chalmers-RedmanR.BrownD.TattonN.Apoptosis in Parkinson's disease: signals for neuronal degradation.Ann Neurol.200353(suppl 3)S61S70 Discussion. S70-S72.12666099
- KingsburyAE.MardsenCD.FosterOJ.DNA fragmentation in human substantia nigra: apoptosis or perimortem effect?Mov Disord.1998138778849827610
- GreenDR.Apoptotic pathways: the roads to ruin.Ceil.199894695698
- HartmannA.Mouatt-PrigentA.VilaM.et al.Increased expression and redistribution of the antiapoptotic molecule Bcl-xL in Parkinson's disease.Neurobiol Dis.200210283212079401
- HartmannA.TroadecJD.HunotS.et al.Caspase-8 is an effector in apoptotic death of dopaminergic neurons in Parkinson's disease, but pathway inhibition results in neuronal necrosis.J Neurosci.2001212247225511264300
- HartmannA.MichelPP.TroadecJD.et al.Is Bax a mitochondrial mediator in apoptotic death of dopaminergic neurons in Parkinson's disease?J Neurochem.2001761785179311259496
- TattonNA.Increased caspase 3 and Bax immunoreactivity accompany nuclear GAPDH translocation and neuronal apoptosis in Parkinson's disease.Exp Neurol.2000166294311031081
- HartmannA.Mouatt-PrigentA.FaucheuxBA.AgidY.HirschEC.FADD: a link between TNF family receptors and caspases in Parkinson's disease.Neurology.20025830831011805265
- HashimotoM.TakedaA.HsuLJ.TakenouchiT.MasliahE.Role of cytochrome c as a stimulator of α-synuclein aggregation in Lewy body disease.J Biol Cnem.19992742884928852
- SchapiraAH.CooperJM.DexterD.ClarkJB.JennerP.MarsdenCD.Mitochondrial complex I deficiency in Parkinson's disease.J Neurochem.1990548238272154550
- Di MonteDA.The environment and Parkinson's disease: is the nigrostriatal system preferentially targeted by neurotoxins?Lancet Neurol.2003253153812941575
- FlemingL.MannJB.BeanJ.BriggleT.Sanchez-RamosJR.Parkinson's disease and brain levels of organochlorine pesticides.Ann Neurol.1994361001037517654
- CorriganFM.WienburgCL.ShoreRF.DanielSE.MannD.Qrganochlorine insecticides in substantia nigra in Parkinson's disease.J Toxicol Environ Health A.20005922923410706031
- CorriganFM.MurrayL.WyattCL.ShoreRF.Diorthosubstituted polychlorinated biphenyls in caudate nucleus in Parkinson's disease.Exp Neurol.19981503393429527905
- CorriganFM.FrenchM.MurrayL.Qrganochlorine compounds in human brain.Hum Exp Toxicol.1996152622648839217
- UverskyVN.LiJ.FinkAL.Pesticides directly accelerate the rate of asynuclein fibril formation: a possible factor in Parkinson's disease.FEBS Lett.200150010510811445065
- TakahashiM.YarnadaT.A possible role of influenza A virus infection for Parkinson's disease.Adv Neurol.2001869110411554013
- ChapmanG.BeamanBL.LoefflerDA.et al.In situ hybridization for detection of nocardial 16S rRNA: reactivity within intracellular inclusions in experimentally infected cynomolgus monkeys - and in Lewy body-containing human brain specimens.Exp Neurol.200318471572514769363
- KennedyPG.Neurovirological methods and their applications.J Neurol Neurosurg Psychiatry.2003741016102212876227
- RubU.Del TrediciK.SchultzC.et al.Parkinson's disease: the thalamic components of the limbic loop are severely impaired by α-synuclein Immunoposltlve inclusion body pathology.Neurobiol Aging.20022324525411804710
- EmreM.What causes mental dysfunction in Parkinson's disease?Mov Disord.200318(suppl 6)S63S7114502658
- WakabayashiK.TakahashiH.Neuropathology of autonomic nervous system in Parkinson's disease.Eur Neurol.199738(suppl 2)279387796
- LaiYY.SiegelJM.Physiological and anatomical link between Parkinsonlike disease and REM sleep behavior disorder.Mol Neurobiol.20032713715212777684
- BraakH.RubU.GaiWP.Del TrediciK.Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen.J Neural Transm.200311051753612721813
- Del TrediciK.RubU.De VosRA.BohlJR.Braak H. Where does Parkinson disease pathology begin in the brain?J Neuropathol Exp Neurol.20026141342612030260
- BraakH.Del TrediciK.RubU.de VosRA.Jansen SteurEN.BraakE.Staging of brain pathology related to sporadic Parkinson's disease.Neurobiol Aging.20032419721112498954
- HardyJ.CooksonMR.SingletonA.Genes and parkinsonism.Lancet Neurol.2003222122812849210
- ChenH.ZhangSM.HernanMA.et al.Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease.Arch Neurol.2003601059106412925360
- MurphyDD.RavinaB.Brain banking for neurodegenerative diseases.Curr Opin Neurol.20031645946312869803