Abstract
The etiology of most cases of Parkinson's disease (PD) remains unknown. In recent years, however, research has successfully focused on genetic factors contributing to the degeneration of dopaminergic neurons. Causative mutations have been identified in several monogenically inherited forms of the disease. Although these genetic forms of PD are usually rare, the gene discoveries are likely to identify molecular pathways that are also relevant in the sporadic disorder. These studies have led to the identification of (i) the central role of α-synuclein aggregation, secondary to either point mutations or an amplification of the α-synuclein gene; and (ii) the relevance of defects in the proteasomal protein degradation pathway in the molecular pathogenesis of recessive parkin-linked forms of PD. The recent discoveries of two additional recessive forms associated with mutations in the genes DJ-1 and PINK1 have brought the mitochondrial energy metabolism and the cell's defence against toxic free radicals into the focus of research.
Se desconoce la etiología de la mayoría de los casos de enfermedad de Parkinson (EP). En años recientes, sin embargo, la investigación se ha centrado exitosamente en los factores genéticos que contríbuyen a la degeneración de las neuronas dopaminérgicas. Se han identificado mutaciones causales en algunas formas monogénicas heredadas de la enfermedad. Aunque estas formas genéticas de la EP son habitualmente raras, es posible que los descubrimientos genéticos identifiquen vías moleculares que también sean importantes en la enfermedad esporádica. Estos estudios han conducido a la identificación de: (i) el papel central de la agregación de α-sinucleína, la que puede ser secundaria a mutaciones de puntos del gen de α-sinucleina o a una sobre-expresión de éste y (ii) la importancia de los defectos en la vía de degradación de la proteína proteasómica en la patogénesis molecular de las formas recesivas ligadas al gen parkin de la EP. Los descubrimientos recientes de dos formas recesivas adicionales asociadas con mutacíones en los genes DJ-1 y PINK1 han puesto el foco de atención de la investigación en el metabolismo energético mitocondrial y en la defensa de la célula contra los radicales libres tóxicos.
L'étiologie de la plupart des cas de maladie de Parkinson (MP) reste inconnue. Ces dernières années, cependant, la recherche s'est axée avec succès sur les facteurs génétiques impliqués dans la dégénérescence des neurones dopaminergiques. Des mutations causales ont été identifiées dans plusieurs formes héréditaires monogéniques de la maladie. Bien que ces formes génétiques de MP soient habituellement rares, la découverte de ces gènes permettra probablement l'identification de voies moléculaires qui sont aussi mises en jeu dans la forme sporadique de la maladie. Ces études ont conduit à la mise en évidence (1) du rôle central de l'agrégation de l'α-synucléine, secondaire à des mutations ponctuelles ou à la surexpression du gène de l'α-synucléine; et (2) de la pertinence des défauts de la voie de dégradation des protéines protéosomales dans la pathogenèse moléculaire des formes récessives de MP liées au gène parkin. La découverte récente de deux formes récessives supplémentaires liées aux mutations des gènes DJ-1 et PINK1 fait du métabolisme énergétique mitochondrial et de la défense cellulaire contre les radicaux libres toxiques un axe privilégié de la recherche.
Parkinson's disease (PD) is a common neurodegenerative disorder, characterized clinically by the symptoms of akinesia (slowness and poverty of movements), muscular rigidity, rest tremor, and disturbance of postural reflexes. The pathological substrate is a neuronal loss predominantly of dopaminergic neurons of the substantia nigra, in the presence of characteristic eosinophilic inclusions, the Lewy bodies. The cause of most cases of PD is still unknown, but both genetic and environmental factors arc thought to contribute to the development of the disease.
Genetic contributions to the etiology of PD were implicated in early descriptions of the disease.Citation1 Later, the importance of genetic factors was thought to be low due to twin studies, which produced low concordance rates.Citation2,Citation3 However, in more recent years, interest in the genetics of PD has surged, as a consequence of the identification of several monogenicaily inherited forms of the disease. The mapping and cloning of an increasing number of disease genes in these families has provided new insights into the pathogenesis of the disorder (Table I.)Citation4-Citation13
Table I. Genetically defined forms of Parkinson's disease and parkinsonism. LB, Lewy body.
Autosomal-dominant forms of PD
Monogenic forms of PD with autosomal-dominant inheritance appear to be extremely rare. Nevertheless, the identification of disease-causing mutations has had a major impact on our understanding of the pathogenesis of PD.
PARK1
α-Synuclein was the first PD gene to be identified as causing autosomal-dominant parkinsonism in a large ItalianAmerican family (Contursi kindred). The clinical picture was reported to be consistent with typical L-dopa-responsive PD with Lcwy body pathology, but with an unusually early onset (mean 44 years) and rapid disease progression. A point mutation (A53T) in the α-synuclein gene was found in this and several (probably related) Greek families.Citation4 Two additional point mutations, A30P in a German familyCitation14 and E46K,Citation15 were identified later.
Although point, mutations in the α-synuclein gene appear to be a very rare cause of PD,Citation16,Citation17 this finding was of great importance because oc-synuclein was subsequently identified as the principle component of the Lewy body, which is also the pathological hallmark of typical sporadic PD. Consequently, the pathological aggregation of α-synuclein is thought to play a central role in the molecular pathogenesis of PD. This was further substantiated by the recent finding of a triplication of a 2-Mb genomic region containing the α-synuclein gene in a large autosomal-dominant family with PD.Citation7 This genomic aberration leads to an overcxprcssion of the intact α-synuclein gene, indicating the susceptibility of neurons to an overload with this amyloidogenic protein. Interestingly, several affected members of this kindred, as well as later reported individuals with an A53T Citation18 and E46K point mutationCitation15 had prominent dementia and pathological findings consistent with Lewy body dementia, supporting the close relationship of this disease entity with PD. A possible role of α-synuclein gene variants has also been analyzed in sporadic PD. Some, but not all, studies found a polymorphic dinucleotide repeat, polymorphism (NACP-Repl) located about 4 kb upstream of the transcriptional start site of the gene to be associated with sporadic PD. This variant, may influence α-synuclein transcriptional regulation and expression levels, as suggested by CAT (chloramphenicol acetyltransferase) reporter gene assays,Citation19,Citation20 again suggesting that, overexpression of oc-synuclein and subsequent aggregation may be a crucial event in the pathogenesis of PD.
PARK3
Another autosomal-dominant locus has been described (PARK3), located on chromosomal region 2pl3, in a subset, of families with typical PD and I,ewy body pathology.Citation6 Clinical features resemble those of sporadic PD including a similar mean age of onset (59 years in these families).
The disease gene has not yet. been identified. However, in two independent genome-wide linkage analyses in sibpairs, significant association between age at onset of PD and this gene locus was shown with maximum multipoint LOD scores of 2.08 and 3.4, respectively,Citation21,Citation2 suggesting that the PARK3 gene may actually be a disease-modifying locus rather than a true disease gene, similar to the apolipoprotein H locus in Alzheimer's disease.
PARK5: parkinsonism associated with mutations in the gene for UCH-L1.
A missense change in the gene for ubiquitin C-terminal hydrolase 1 (LJCH-L1) has been identified in two affected members of a small family with PD, based on a candidate genc-sequencing approach.Citation8 No other families have been identified with disease-causing mutations, but a common polymorphism in this gene (S18Y) was found to be protective in several association studies, including one meta-analysis.Citation23 The precise role of this gene in the pathogenesis of PD remains to be elucidated.
PARK8
This locus was first, identified in a large Japanese family (Samigahara family) with autosomal-dominant parkinsonism and linked to chromosome 12q..Citation11 The clinical phenotype showed typical PD with good response to L-dopa and mean age at onset of 51 years. Neuropathologically, four affected members showed nonspecific neuronal degeneration in the substantia nigra, but no Lewy body formation. At least 2 out of 21 families of European ancestry also showed significant linkage within this locus.Citation24 Interestingly, in one of these families, various pathologies have been found, including brain-stem Lewy body disease, diffuse Lewy body disease, tau aggregation, and nigral degeneration without distinctive inclusions, indicating that mutations in this gene may be associated with a relatively wide range of pathologies.
Autosomal-recessive forms of parkinsonism
Monogenic forms of recessive parkinsonism caused by mutations in parkin (PARK2), PINK-1 (PARK6), and DJ-1 (PARK7) represent, an important cause of earlyonset parkinsonism (onset before 40 years of age). The clinical phenotype of early-onset parkinsonism is often characterized by dystonia at onset, hyperreflexia, early complications on L-dopa treatment, and slow disease progression.
PARK2: parkinsonism caused by mutations in the parkin gene
Autosomal-recessive juvenile parkinsonism (AR-JP) was first, recognized in Japanese patients with an early-onset form of PD (onset, usually in the second or third decade) and mapped to chromosome 6.Citation25 Mutations have been identified in a large gene in this region called parkin.Citation5
Mutations in the parkin gene account, for 50% of familial and about 15% of sporadic European PD patients with onset, before the age of 45 years.Citation26,Citation27 The proportion of parkin mutations is clearly a function of the age at onset (82% before age 20, but rare over the age of 55 years).Citation26,Citation28 Different parkin mutations are known, including quantitative alterations like exon deletions and duplications and point mutations.
In a study comparing parkin mutation carriers and noncarriers of parkin mutations in a cohort with early-onset parkinsonism, those with a mutation tended to have earlier and more symmetrical onset, slower progression of the disease, and greater response to L-dopa despite lower doses. Lower-limb dystonia at disease onset, occurs in about a third of patients, but this feature docs not appear to be specific to parkin-related disease, and is more correlated with the age at onset, than with genetic status.Citation29 Functional neuroimaging in parkin-linked parkinsonism showed reduced uptake of dopamine tracer bilaterally in the putamen and caudate nucleus, in contrast to the initially unilateral reduction in dopa uptake of sporadic PD patients.Citation30,Citation31 Psychiatric abnormalities have been recognized in PD patients with parkin mutations.Citation32 Phenotypc-genotype studies indicate that the type of mutation may influence the clinical phenotype to a certain degree: patients with at least one missense mutation showed a faster progression of the disease with a higher Unified Parkinson's Disease Rating Scale (UPDRS) motor score than carriers of truncating mutations. Missense mutations in functional domains of the parkin gene resulted in earlier onset.Citation29
It remains unresolved whether parkin mutations also represent a susceptibility factor for late-onset PD. Heterozygous mutations are found in up to 6% in this group,Citation33 but a recent study also detected known sequence variants associated with parkinsonism in more than 3% of healthy elderly individuals.Citation34 On the other hand, clinically asymptomatic individuals with heterozygous parkin mutations showed mildly reduced uptake of fluorodopa in the basal ganglia,Citation35 indicating a possible “first hit” to the nigrostriatal system.
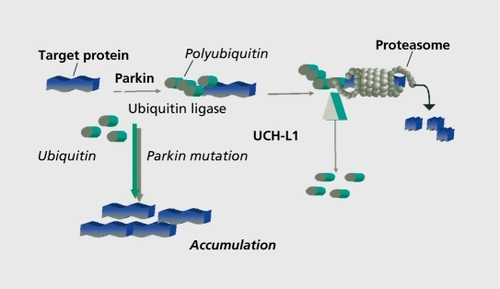
As mutations of the parkin gene cause parkinsonism, in all likelihood, by a loss-of -function mechanism, the study of the normal function of parkin should provide insight into the molecular pathogenesis of the disorder. Several groups have now shown that parkin, a protein that has been found in the cytosole, but also associated with membranes, functions in the cellular ubiquitination/protcin degradation pathway as a ubiquitin ligase ( ).Citation36-Citation38 It is therefore conceivable that the loss of parkin function may lead to the accumulation of a nonubiquitinated substrate, which is deleterious to the dopaminergic cell, but, due to its nonubiquitinated nature, does not form typical Lewy bodies. Several proteins have been shown to interact, with parkin and could possibly be its relevant partner with regard to neurodegeneration: an O-glycosylated form of oc-synuclein.Citation39; a protein associated with synaptic vesicles, CDCrel-1.Citation37; a transmembrane protein, called the pael receptor40; and synphillin-1.Citation41 However, other pathogenetic mechanisms could also be important. Recently, it has been shown that, in parkin knockout mice, a number of genes related to the oxidative metabolism in mitochondria are downregulated. Oxidative damage and dysfunction of components of the mitochondrial respiratory chain could be demonstrated, implicating this pathway in the pathogenesis of PD.Citation42
PARK6: parkinsonism caused by mutations in the gene for PINK1
Very recently, mutations in the gene for PINK1 (PTEN-induced kinase 1) have been found to cause another autosomal-recessive variant of early-onset PD,Citation39 PARK6, which has previously been mapped to chromosomal region lp36.Citation43 Two homozygous mutations affecting the PINK1 kinase domain were identified in three consanguineous PARK6 families: a truncating nonsense mutation and a missense mutation at a highly conserved amino acid. Cell culture studies suggested that PINK1 is a mitochondrially located protein and may exert a protective effect on the cell upon oxidative stress, which is abrogated by the mutations.Citation9 As in families with parkin mutations, PINK1 -associated parkinsonism is of early onset and takes a relatively benign cause.
PARK7: parkinsonism caused by mutations in the gene for DJ-1
Another recessive locus was mapped to chromosome 1 in a consanguineous Italian family.Citation44 These patients had disease onset in their mid-thirties with L-dopa responsiveness, slow progression, and focal dystonic symptoms, such as blepharospasm. Pathogenic mutations were identified in the gene for DJ-1.Citation10 A missense mutation resulting in the substitution of a highly conserved leucine for a proline at position 166 (L166P mutation) and a large homozygous deletion of several exons (exons 1 to 5) were detected in an Italian and a Dutch family, respectively. Both DJ-1 mutations are thought to lead to a loss of function: the deletion leads to a complete loss of the protein, the L166P mutation is thought, to impair the dimerization and the tertiary structure of DJ-1, resulting in a functionally inactive monomeric form of the protein.Citation45,Citation46 The function of DJ-1 is still largely unknown, but there is some evidence that the protein may play a role in the cellular response to oxidative stress, which may render dopaminergic neurons particularly vulnerable. This oxidative stress response may be caused by interactions of DJ-1 with other proteins like protein inhibitor of activated STAT (signal transducer and activator of transcription) (PIASxα), DJ-1 -binding protein (DJBP), and the RNA-binding protein complex; DJ-1 may also regulate the dismutation of peroxides.Citation47,Citation48
The prevalence of pathogenic DJ-1 mutations in youngonset PD patients is certainly much lower than that of parkin, and is estimated to be less than 1 %.Citation49,Citation50 No pathogenic mutation was found in 190 pathologically proven patients with later onset PD.
Identification of susceptibility alleles in nonmendelian PD
Although significant progress has been made in families with mcndelian subtypes of PD, it must be remembered that PD, in the great majority of cases, is a sporadic disorder. The type and the extent of a genetic contribution to nonmendelian PD is still controversial. A populationbased, case-control study indicates that the relative risk for first-degree family members of PD patients is increased only in the order of 2 to 3.Citation51
Most, attempts to identify the susceptibility genes in sporadic PD, have followed a candidate gene approach. On the basis of pathological, pathobiochemical, and epidemiological findings, hypotheses on the etiology of PD can be generated and genetic polymorphisms within - or closely linked to - genes that, are thought to be involved in these pathways have been examined. Unfortunately no consistent, findings have emerged so far.
Major international efforts therefore focus on the examination of large cohorts of affected sibpairs or small nuclear families with the methods of nonparametric linkage analysis, using whole-genome approaches. Several of these studies have been published.Citation12,Citation52-Citation54 Their results indicate that the contribution of any individual locus to PD is likely to be modest, as linkage peaks in these studies generally were rather low and most, of them not reproduced in other studies (with the exception of a locus on chromosome 5 and one on the X chromosome). This is most, likely due to the enormous locus heterogeneity in late-onset PD. Therefore, international collaborations and pooling large patient resources will be necessary to narrow down linkage regions and conduct more advanced studies, such has high-resolution linkage disequilibrium (LD) mapping, which will eventually result, in the identification of the genetic variants responsible.
Conclusion
The genetic findings in rare inherited forms of PD have greatly contributed to our understanding of the clinical, neuropathological, and genetic heterogeneity of PD. The variability of clinical features, such as age at onset, occurrence of dementia, or other associated features, that has been found within single families suggests that a single genetic cause (the pathogenic mutation in a given family) can lead to a spectrum of clinical manifestations. On the other hand, individuals with different genetic defects and different neuropathologies (eg, some of those with mutations in the PARK1 and PARK2 genes) may be clinically indistinguishable from each other and fulfill all presently accepted criteria of idiopathic PD. It is therefore apparent that a new genetic classification of PD is about to emerge, which is only partially congruent with the classic clinical and pathological classification.
There is currently convincing evidence that, genetic factors play an important role in the etiology of at least, a subset of patients with PD. Only a small percentage of cases with dominant or recessive inheritance can probably be explained by mutations in the genes that have been identified so far (the genes for a-synuclcin, ubiquitin carboxy-terminal hydrolase LI, DJ-1, PINK1, and parkin) or by mutations in the as yet unidentified genes on chromosome 1, 2p, and 12. However, the study of wildtype and mutated gene products will provide important, insights into the molecular pathogenesis of nigral degeneration and Lewy body formation. Further intense efforts are still needed to unravel the full spectrum of etiological factors leading to the common sporadic form of this neurodegenerative disorder.
REFERENCES
- GowersWR.Diseases of the Nervous System. Philadelphia, Pa: P. Blakiston, Son & Co;1888
- WardCD.DuvoisinRC.InceSE.NuttJD.EldridgeR.CalneDB.Parkinson's disease in 65 pairs of twins and in a set of quadruplets.Neurology.1983338158246683366
- TannerCM.OttmanR.GoldmanSM.et al.Parkinson disease in twins: an etiologic study.JAMA.19992813413469929087
- PolymeropoulosMH.LavedanC.LeroyE.et al.Mutation in the a-synuclein gene identified in families with Parkinson's disease.Science.1997276204520479197268
- KitadaT.AsakawaS.HattoriN.et al.Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism.Nature.19983926056089560156
- GasserT.Müller-MyhsokB.WszolekZK.et al.A susceptibility locus for Parkinson's disease maps to chromosome 2p13.Nat Genet.1998182622659500549
- SingletonAB.FarrerM.JohnsonJ.et al.α-Synuclein locus triplication causes Parkinson's disease.Science.200330284114593171
- LeroyE.BoyerR.ÀuburgerG.et al.The ubiquitin pathway in Parkinson's disease [letter].Nature.19983954514529774100
- ValenteEM.Abou-SleimanPM.CaputoV.et al.Hereditary early-onset Parkinson's disease caused by mutations in PINK1.Science.20043041158116015087508
- BonifatiV.RizzuP.Van BarenMJ.et al.Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism.Science.200229925625912446870
- FunayamaM.HasegawaK.KowaH.SaitoM.TsujiS.ObataF.A new locus for Parkinson's disease (PARKS) maps to chromosome 12p11.2-q13.1.Ann Neurol.20025129630111891824
- HicksAA.PeturssonH.JonssonT.et al.A susceptibility gene for lateonset idiopathic Parkinson's disease.Ann Neurol.20025254955512402251
- PankratzN.NicholsWC.UniackeSK.et al.Significant linkage of Parkinson disease to chromosome 2q36-37.Am J Hum Genet.2003721053105712638082
- KrügerR.KuhnW.MûllerT.et al.Ala39Pro mutation in the gene encoding (α-synuclein in Parkinson's disease.Nat Genet.1998181061089462735
- ZarranzJJ.AlegreJ.Gomez-EstebanJC.et al.The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia.Ann Neurol.20045516417314755719
- VaughanJR.DurrA.GasserT.et al.The α-synuclein Ala53Thr mutation is not a common cause of familial Parkinson's disease: a study of 230 European cases.Ann Neurol.1998442702739708553
- GasserT.Müller-MyhsokB.WszolekZ.et al.Genetic complexity and Parkinson's disease.Science.19972773883899518367
- SpiraPJ.SharpeDM.HallidayG.CavanaghJ.NicholsonGA.Clinical and pathological features of a Parkinsonian syndrome in a family with an Ala53Thr a-synuclein mutation.Ann Neurol.20014931331911261505
- HolzmannC.KrugerR.SaeckerAM.et al.Polymorphisms of the a-synuclein promoter: expression analyses and association studies in Parkinson's disease.J Neural Transm.2003110677612541013
- Chiba-FalekO.NussbaumRL.Effect of allelic variation at the NACP-Rep1 repeat upstream of the a-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system.Hum Mol Genet.2001103101310911751692
- DeStefanoLewMF.GolbeLI.et al.PARK3 influences age at onset in Parkinson disease: a genome scan in the GenePD study.Am J Hum Genet.2002701089109511920285
- PankratzN.NicholsWC.UniackeSK.et al.PARKS and PARK7 linked to age of onset of Parkinson disease.Neurology.200360P02076
- MaraganoreDM.LesnickTG.ElbazA.et al.UCHL1 is a Parkinson's disease susceptibility gene.Ann Neurol.20045551252115048890
- ZimprichA.Mûller-MyhsokB.FarrerM.et al.The PARKS locus in autosomal dominant parkinsonism: confirmation of linkage and further delineation of the disease-containing interval.Am J Hum Genet.200474111914691730
- MatsumineH.SaitoM.Shimoda-MatsubayashiS.et al.Localization of a gene for an autosomal recessive form of juvenile parkinsonism to chromosome 6q25. 2-27.Am J Hum Genet.1997605885969042918
- LückingCB.DurrA.BonifatiV.et al.Association between early-onset Parkinson's disease and mutations in the parkin gene.N Engl J Med.20003421560156710824074
- KannM.JacobsH.MohrmannK.et al.Role of parkin mutations in 111 community-based patients with early-onset parkinsonism.Ann Neurol.20025162162512112109
- PeriquetM.LatoucheM.LohmannE.et al.Parkin mutations are frequent in patients with isolated early-onset parkinsonism.Brain.2003126(Pt 6)1271127812764050
- LohmannE.PeriquetM.BonifatiV.et al.How much phenotypic variation can be attributed to parkin genotype?.Ann Neurol.20035417618512891670
- PortmanAT.GiladiN.LeendersKL.et al.The nigrostriatal dopaminergic system in familial early onset parkinsonism with parkin mutations.Neurology.2001561759176211425950
- LeendersKL.OertelWH.Parkinson's disease: clinical signs and symptoms, neural mechanisms, positron emission tomography, and therapeutic interventions.Neural Plast.200189911011530892
- YamamuraY.HattoriN.MatsumineH.KuzuharaS.MizunoY.Autosomal recessive early-onset parkinsonism with diurnal fluctuation: clinicopathologic characteristics and molecular genetic identification.Brain Dev.200022{suppl 1)8791
- KleinC.HedrichK.WellenbrockC.et al.Frequency of parkin mutations in late-onset Parkinson's disease.Ann Neurol.200354415416
- LincolnSJ.MaraganoreDM.LesnickTG.et al.Parkin variants in North American Parkinson's disease: cases and controls.Mov Disord.2003181306131114639672
- HilkerR.KleinC.GhaemiM.et al.Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene.Ann Neurol.20014936737611261512
- ShimuraH.HattoriN.KuboS.et al.Familial parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Wat.Genet.200025302305
- ZhangY.GaoJ.ChungKK.HuangH.DawsonVL.DawsonTM.Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1.Proc Natl AcadSci USA.2000971335413359
- GasserT.Parkin and its role in Parkinson's disease. In: Ebadi M, Pfeiffer R, eds.Parkinson's Disease. Boca Raton, Fia: CRC Press LLC;2004
- ShimuraH.SchlossmacherMG.HattoriN.et al.Ubiquitination of a new form of a-synuclein by parkin from human brain: implications for Parkinson's disease.Science.200129326326911431533
- YangY.NishimuraI.ImaiY.TakahashiR.LuB.Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila.Neuron.20033791192412670421
- ChungKK.ZhangY.LimKL.et al.Parkin ubiquitinates the a-synucleininteracting protein, synphilin-1: implications for Lewy body formation in Parkinson disease.Nat Med.200171144115011590439
- PalacinoJJ.SagiD.GoldbergMS.et al.Mitochondrial dysfunction and oxidative damage in parkin-deficient mice.J Biol Chem.2004279186141862214985362
- ValenteEM.BentivoglioAR.DixonPH.et al.Localization of a novel locus for autosomal recessive early-onset parkinsonism, park6, on human chromosome 1p35-p36.Am J Hum Genet.20016889590011254447
- van DuijnCM.DekkerMC.BonifatiV.et al.PARK7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36.Am J Hum Genet.20016962963411462174
- MooreDJ.ZhangL.DawsonTM.DawsonVL.A missense mutation (L166P) in DJ-1, linked to familial Parkinson's disease, confers reduced protein stability and impairs homo-oligomerization.7,.Neurochem.20038715581567
- TaoX.TongL.Crystal structure of human DJ-1, a protein associated with early-onset Parkinson's disease.J Biol Chem.2003278313723137912761214
- TairaT.SaitoY.NikiT.Iguchi-ArigaSM.TakahashiK.ArigaH.DJ-1 has a role in antioxidative stress to prevent cell death.EMBO Rep.20045430
- HodY.PentyalaSN.WhyardTC.El-MaghrabiMR.Identification and characterization of a novel protein that regulates RNA-protein interaction.J Cell Biochem.19997243544410022524
- Abou-SleimanPM.HealyDG.QuinnN.LeesAJ.WoodNW.The role of pathogenic DJ-1 mutations in Parkinson's disease.Ann Neurol.20035428328612953260
- HealyDG.Abou-SleimanPM.ValenteEM.et al.DJ-1 mutations in Parkinson's disease.J Neurol Neurosurg Psychiatry.20047514414514707326
- MarderK.TangMX.MejiaH.et al.Risk of Parkinson's disease among firstdegree relatives: a community-based study.Neurology.1996471551608710070
- ScottWK.NanceMA.WattsRL.et al.Complete genomic screen in Parkinson disease: evidence for multiple.genes. JAMA.200128622392244
- PankratzN.NicholsWC.UniackeSK.et al.Genome screen to identify susceptibility genes for Parkinson disease in a sample without parkin mutations.Am J Hum Genet.20027112413512058349
- DeStefanoGolbeLI.MarkMH.et al.Genome-wide scan for Parkinson's disease: the GenePD study.Neurology.2001571124112611571351