Abstract
Brain neurotransmitter dysfunctions involved in the pathophysiological processes of psychiatric disorders are likely to be reflected by concomitant alterations in sleep continuity and architecture. Since the corrective effects of psychotropic drugs on dysfunctional neurotransmission systems can be evidenced through polysomnographic recordings, one may consider sleep as a kind of “window” on the neurobiology of psychiatric disorders. During the last 10 years, major breakthroughs in our understanding of sleep-wake mechanisms have provided some indications on how psychotropic drugs could influence the sleep-wake cycle. In this review, recent inroads into the understanding of sleep regulatory neural mechanisms are introduced and discussed in terms of the effects of psychotropic drugs. The relationship between the patho-physiological process of a disease, its consequence on sleep, and the corrective effect of a psychotropic drug are exemplified by two psychopathological states: substance withdrawal and major depression. One may conclude that polysomnographic recordings are a unique noninvasive tool to analyze brain functioning, and are particularly well suited to evaluating the objective effects of new psychotropic drugs.
Es probable que las disfunciones de la neurotransmisión cerebral involucradas en los procesos fisiopatológicos de los trastornos psiquiátricos se reflejen en alteraciones concomitantes de la continuidad y arquitectura del sueño. Ya que los efectos correctores de los psicofármacos en los sistemas de neurotransmisión disfuncionales pueden ser evidenciados a través de registros polisomnográficos, es posible considerar al sueño como un tipo de “ventana” hacia la neurobiología de los trastornos psiquiátricos. Durante los últimos diez años los principales progresos en nuestra comprensión de los mecanismos del sueño-vigilia han originado algunas sugerencias acerca de cómo los psicofármacos podrían afectar el ciclo sueño-vigilia. En esta revisión se presentan y discuten avances recientes en la comprensión de los mecanismos neurales reguladores del sueño a partir de los efectos de los psicofáarmacos. La relación entre los procesos fisiopatológicos de una enfermedad, su consecuencia en el sueño y los efectos correctores de un psicofármaco se ejemplifican en dos estados psicopatológicos: la privación de sustancias y la depresión mayor. Se puede concluir que los registros polisomnográficos constituyen una herramienta no invasora original para analizar el funcionamiento cerebral y son especialmente adecuados para evaluar los efectos objetivos de nuevos psicofármacos.
Les dysfonctionnements de la neurotransmission impliqués dans la physiopathologie des troubles psychiatriques se reflètent très probablement dans les altérations concomitantes de la continuité et de l'architecture du sommeil. Comme les médicaments psychotropes corrigent les dysfonctionnements des systèmes de neurotransmission, leurs effets peuvent être mis en évidence par des enregistrements poly-somnographiques. Le sommeil peut donc être considéré, en quelque sorte, comme un reflet de la neurobiologie des troubles psychiatriques. Au cours des 10 dernières années, des avancées majeures dans nos connaissances sur les mécanismes veille-sommeil ont permis d'apporter quelques éléments d'explication sur la manière dont les médicaments psychotropes pouvaient influer sur le cycle veille-sommeil. Dans cette revue de la littérature, les découvertes récentes dans notre compréhension des mécanismes neuronaux impliqués dans la régulation du sommeil sont présentées et discutées en fonction des effets des médicaments psychotropes. Les relations entre le processus physiopathologique d'une maladie, ses conséquences sur le sommeil et les effets correcteurs d'un médicament psychotrope sont illustrés par deux exemples: le sevrage d'une substance et la dépression majeure. En conclusion, retenons que les enregistrements polysomnographiques représentent un outil unique, non invasif, permettant d'analyser le fonctionnement cérébral et particulièrement bien adapté pour l'évaluation des effets objectifs d'un nouveau médicament psychotrope.
As much as one third of the adult population reports difficulty sleepingCitation1-Citation3 and the widespread use of prescribed hypnotic medication, as well as nonprescription remedies, Is an Indirect reflection of this high frequency of sleep complaints.Citation2,Citation4 Sleep disturbance is considered as the second most common symptom of mental distress.Citation5 Individuals reporting disturbed sleep are more likely to report emotional distress and recurrent health problems.Citation1 In fact, disturbed sleep is a common finding in psychiatric illnesses. Some patients will even attribute their daytime psychiatric symptoms to abnormal sleep and believe that Improved sleep will solve their problems. In some cases, the psychological symptoms associated with a primary sleep disorder could. Indeed Improve with adequate therapy, for Instance, the altered states of consciousness or depression encountered. In some patients with sleep apnea could Indeed Improve with nasal continuous positive airway pressure treatment. In primary psychiatric disorders, the sleep complaint usually parallels the state of the disorder, and sleep improves when the psychiatric symptoms improve.
Another point is that alterations of sleep by psychiatric conditions are likely to have underlying brain neurotransmitter dysfunction directly involved in the patho-physiological process of the disease. Indeed, neurotransmission disturbances, such as those encountered in mental disorders, are reflected in spontaneous alteration of sleep continuity and architecture. The corrective effect on dysfunctional neurotransmission systems of psychotropic drugs, such as antidepressants, is also evidenced through polysomnographic recordings. Sleep can thus be considered as a kind of window on the neurobiology of psychiatric disorders. The first section of this review will introduce recent inroads into understanding sleep-regulatory neural mechanisms. The following sections deal with the way psychotropic drugs interact with mechanisms involved in sleep-wake regulation. Finally, the relationship between the pathophysiological process of a disease, its consequence on sleep, and the corrective effect of a psychotropic drug will be exemplified.
Sleep basics
Electrophysiological recordings of human brain reveal three distinct state of existence: wakefulness, rapid eye movement (REM) sleep, and non-REM (NREM) sleep. The distinction between sleep and wakefulness is attributed to the synchronization and desynchronization of thalamocortical circuits.Citation6,Citation7 Wake-like or “desynchronized” (low-amplitude and high-frequency) electroencephalographic (EEG) activity with clusters of REM and very low levels of muscle tone characterize REM sleep. NREM sleep includes all sleep except REM sleep, and is by convention divided into four stages corresponding to increased depth of sleep as indicated by the progressive dominance of “synchronized” EEG activity (also known as low-voltage high-amplitude delta or slow-wave activity); in this respect, sleep stages 3 and 4 are collectively labelled as delta sleep or slow-wave sleep (SWS). Recurrent cycles of NREM and REM sleep of about 90 min characterize normal human sleep. In the successive cycles of the night, the duration of stages 3 and 4 decrease, and the proportion of the cycle occupied by REM sleep tends to increase. The REM episodes occurring late in the night have more eye movement bursts than REM episodes occurring early in the night.Citation8
Sleep-wake alternation is classically viewed as resulting from the interaction of two regulating processes (circadian-C and homeostatic-S).Citation9 The propensity to sleep or be awake at any given time is a consequence of a sleep debt (process S) and its interaction with wake-promoting signals coming from the circadian clock (process C) located in the suprachiasmatic nucleus (SCN). This wake-promoting signal opposes the sleep need, which progressively increases from morning awakening, ensuring an even degree of alertness throughout the day.Citation10 At sleep onset, an imbalance between the two opposing influences favor sleep-promoting signals, and the sleep need and its electrophysiological signature, slow-wave activity, is at its highest level. Throughout sleep and up to final morning awakening, there is a progressive decline in slow-wave activity reflected by an increase in REM sleep proportion across successive REM/NREM cycles. During the last decade, research lent support to the idea that three interacting neuronal systems (a wakepromoting system, an NREM-promoting system, and a REM-promoting system) are involved in this complex regulation construct.
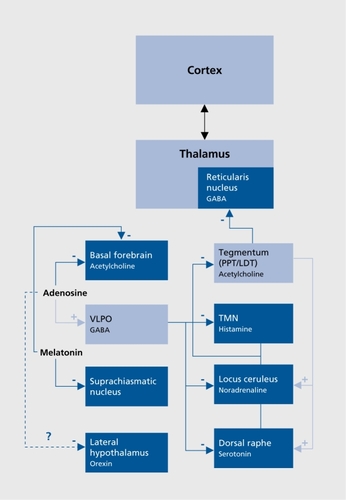
Different structures sending widespread cortical projection and located in the brain stem, the hypothalamus, and the basal forebrain constitute the wake-promoting or arousal system ().Citation11,Citation12 Glutamatergic brain stem reticular neurons, cholinergic neurons of the basal forebrain, and monoaminergic transmission are largely implicated in the arousal system.Citation13 It has been shown that serotonergic (dorsal raphe nuclei [DRN]), noradrenergic (locus ceruleus [LC]) and histaminergic (tuberomammillary nucleus [TMN]) activity is high during wakefulness, decreases during NREM stages, and becomes almost silent during REM sleep.Citation14 The role of the dopaminergic system is less well established; however, recent studies indicated that lesions of wake-active dopaminergic cells in the ventral periaqueductal gray reduce wakingCitation15 and that dopamine D1 D2, and D3 receptor agonists increase waking and reduce REM and NREM sleep.Citation16-Citation18 Orexin (also known as hypocretin) neurons located in the perifornical region of the lateral hypothalamus seem to play a particularly important role in arousal since they project not only over the entire isocortex, but also to additional arousal systems, including the aforementioned monoaminergic and cholinergic systems.Citation19,Citation20 The role of orexin in arousal regulation is further exemplified with narcolepsy, a sleep disorder characterized by excessive daytime sleepiness and deficiency of the orexin system.Citation21-Citation23
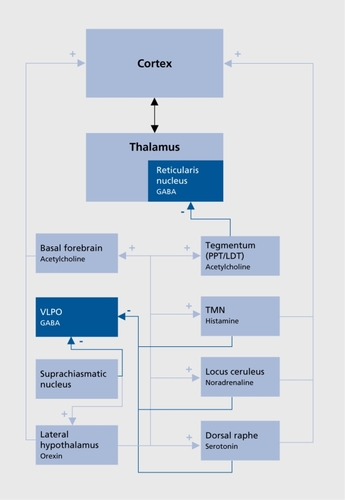
An NREM-promoting system has been evidenced in the hypothalamus (), Electrophysiological recordings have identified GABAergic (GABA, γ-aminobutyricacid) SWS-active neurons in a specific area, the ventrolateral preoptic nucleus (VLPO), where lesions produce insomnia in animals and humans.Citation24 These cells also contain galanin and project to all monoaminergic systems, inhibiting activity during NREM sleep, and receive inputs from multiple brain systems that regulate arousal and autonomic and circadian functions.Citation25 Recent research implicates adenosine in the homeostatic regulation of sleep via actions on the VLPO and other sleep regulatory regions such as the basal forebrain.Citation26 Adenosine functions as a natural sleeppromoting agent, accumulating during period of sustained wakefulness and decreasing during sleep; It has been shown to promote SWS through direct inhibitory effects on cholinergic neurons of the basal forebrainCitation26 and have indirect stimulatory effects on the VLPO.Citation27,Citation28 A further inhibition of wake-promoting mechanism could occur through orexinergic neurons, since a study identified Gi protein-coupled adenosine A1 receptors on this group of neurons.Citation29
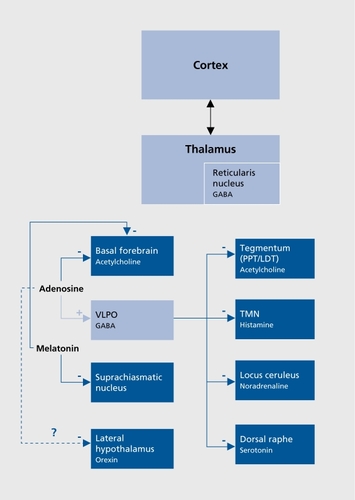
Regarding the circadian influence on the sleep-wake rhythm, recent studies suggested that the SCN regulates sleep-wake mechanisms through the dorsomedial hypothalamus, a key output nucleus of the SCN that inhibits VLPO and stimulates orexin-containing neurons in the lateral hypothalamus.Citation30,Citation31 Melatonin, the hormone of the pineal gland secreted at night and concerned with biological timing, could mediate its sleep-inducing effect through inhibitory influence on SCN neuronsCitation32 and cholinergic neurons of the basal forebrain.Citation33
The REM-promoting system comprises “REM-on” cholinergic neurons located in the laterodorsal tegmental (LDT) and pediculopontine tegmental (PPT) nuclei (). The McCarley and Hobson reciprocal interaction model, first proposed in 1975, and regularly revisited,Citation14 posits a bidirectional inhibitory influence between these REM-on neurons and both the serotonergic DRN and the noradrenergic LC, called “REM-off” neurons. Transition from NREM to REM occurs when activity in the aminergic REM-off neurons ceases. Cholinergic LDT/PPT REM-on neurons are then involved in the initiation of cortical desynchronization through excitatory inputs to the thalamus and in the occurrence of muscle atonia and REMs. During REM sleep, the excitatory input from the REM-on neurons to the DRN and LC leads to a gradual increase in the activity of the REMoff neurons, which in turn inhibit REM-on neurons until the REM episode ends. GABAergic and glutamatergic modulations of this aminergic-cholinergic interplay have been proposed in the revised version of the model.Citation14
The effects of drugs on wake-and sleep-inducing mechanisms
In the following sections, we will review the effects of psychotropic drugs on the three interacting neuronal systems that have been proposed to play a key role in sleep-wake regulation (the wake-promoting system, the NREM-promoting system, and the REM-promoting system). The first four sections deal with drugs acting on wake- or NREM sleep-promoting neurons, while the following section concerns drugs acting on the REMpromoting system with special reference to antidepressant drugs. Whether drugs induce wakefulness (“waking drugs”) or sleep (“hypnosedative drugs”) depend on their liability to stimulate or inhibit wake- or NREM sleep-promoting neurons. Before going further, it should be stressed that the net effects of a hypnosedative drug inhibiting wake-promoting neurons will be very similar to the effects of a drug stimulating NREM-promoting neurons. The converse is true for waking drugs: the effects of a drug inhibiting NREM-promoting neurons will parallel those induced by a drug stimulating wakepromoting neurons. Finally, it should be recognized that a distinction between drugs acting on wake- or NREMpromoting neurons is somewhat arbitrary, due to the close reciprocal negative feedback existing between these two groups of neurons.Citation7
Some drugs directly influence both wake-promoting neurons and sleep-promoting neurons, but in an opposite way; this is the case for compounds influencing adenosine transmission such as caffeine. Caffeine is a psychoactive substance enhancing vigilance performance on psychomotor tasks and significantly affecting sleep at a dose of more than 100 to 150 mg.Citation34,Citation35 It is now widely accepted that the vigilance mechanism of caffeine acts via the antagonism of adenosine receptors. The physiology of the adenosinergic transmission has been recently reviewed,Citation36 as well as its implication in sleepwake mechanisms.Citation26 Adenosine, formed by breakdown of adenosine triphosphate (ATP), is present both intraand extracellularly, and the balance is maintained by membrane transporters, but when energy expenditure exceeds energy production, adenosine levels increase in the extracellular space. In humans, adenosine exerts most of its effects through activation of two high-affinity receptors (the A1 coupled to “inhibitory” G1 proteins and the A2A coupled to “stimulatory” Gs protein). A1 receptors are involved in the inhibitory effect of adenosine on the wake-active cholinergic neurons of the basal forebrain, while there are some indications that A2A receptors could influence the dopaminergic control of wake-promoting mechanisms.Citation37 Adenosine may also disinhibit sleep-active VLPO neurons by removing GABAergic inhibitory inputs, possibly via A1 receptors.Citation27,Citation28 The caffeine-induced increase in vigilance level results from the blockade of A1 and A2A receptors. Accordingly, it is thought that caffeine exerts its effects through two complementary mechanisms: inhibition of wake-promoting cholinergic and dopaminergic influence and disinhibition of sleep-promoting neurons of the VLPO.
It thus emerges that there is a potential role of adenosine A1 and A2A receptor antagonists as arousal stimulators and agonists as sleep promoters. Preclinical studies with such compounds have reported promising results,Citation26 but no clinical trials have been published to date. Since direct adenosine agonists may have marked side effects such as hypotension and bradycardia,Citation36 the use of substances that indirectly modulate the level of endogenous adenosine, such as adenosine uptake inhibitorCitation38 or adenosine kinase inhibitor,Citation39 may be preferable to the use of direct adenosine agonists.
Drugs enhancing the activity of wake-promoting neurons
Amphetamine-like drugs and modafinil are the two most popular wake-promoting medications used for the treatment of narcolepsy, a sleep disorder characterized by excessive daytime sleepiness. Amphetamine, methylphenidate, and cocaine are known to act pharmacologically by blocking the reuptake and enhancing the release of noradrenaline, dopamine, and serotonin within the synaptic cleft of monoamine synapses.Citation40 The exact mechanism by which amphetamine-like stimulants induce their wake-promoting effects remains to be elucidated, but there is growing evidence that the dopaminergic system is mostly implicated.Citation41 For instance, it has recently been demonstrated that dopamine transporter knockout mice were totally insensitive to the wake-promoting properties of classical stimulants suggesting that amphetamine-like compounds require the dopamine transporter for their wake-promoting effects.Citation42
Despite numerous reports of its neuropharmacological action on the central nervous system (CNS), the wake-promoting mechanism of action of modafinil remains uncertain. Using c-Fos immunochemistry in cats, it has been shown that amphetamine-like drugs do not share with modafinil the same pattern of c-Fos activation in the brain. Amphetamine and methylphenidate activate neurons mainly in the cortex and the striatum, whereas modafinilinduced wakefulness was mainly associated with activated neurons in the hypothalamus.Citation43,Citation44 Another study involving c-Fos labelling highlighted Fos activation mainly in the TMN and in orexin-containing neurons of the perif ornical nucleus.Citation45 This suggests that modafinil induces wakefulness by mechanisms distinct from amphetamine-like drugs. It has been suggested that modafinil-induced arousal could be related to noradrenergic transmission, since modafinil affects the firing of the LCCitation46 and its arousal effects are blocked by α1 and β adrenergic receptor antagonists.Citation47 One study shows that modafinil increases noradrenergic release in the hypothalamus, but also both dopaminergic and serotonergic transmission in the cortex, suggesting that the effects of modafinil are not entirely mediated through noradrenergic transmission.Citation48
Besides amphetamine-like drugs and modafinil, the development of drugs acting through the histaminergic or orexinergic system is an area of active research in the field of new therapeutic approach for the treatment of major sleep-wake disorders, such as hypersomnia and narcolepsy H3 receptors are an important target for arousal control and treatment of excessive daytime somnolence, since they are both autoreceptors controlling histamine-containing neuron activity and heteroreceptors, modulating the release of other neurotransmitters including acetylcholine, dopamine, and noradrenaline in brain regions that are crucial for the maintenance of wakefulness.Citation49,Citation50 Administration of H3 receptor antagonists and inverse agonists induced a total suppression of slow- wave activity and spindles and a marked enhancement of fast rhythm, thus eliciting waking and increasing vigilance.Citation51,Citation52 Moreover, recent studies have shown that H3 receptor blockade enhances cognition in rats.Citation53 These studies suggest that the potential benefit of H3 receptor antagonists and inverse agonists are not limited to promoting wakefulness because they could also improve general level of vigilance and cognitive responses in nonsomnolent individuals.Citation50 However, no clinical trials have yet been published showing that H3 receptor blockade promotes wakefulness in humans.
The pharmacology of the orexin system is, up to now, also limited to animal data. Orexins are a pair of neuropeptides, orexin-A and orexin-B, derived from a common precursor peptide, whose actions are mediated by two G protein-coupled receptors termed orexin receptor type 1 (OX1R) and orexin receptor type 2 (OX2R).Citation54 Both receptors are expressed in serotonergic neurons of the DRN, cholinergic neurons of the LDT/PPT, and the hypothalamus, while OXtR is specific to the noradrenergic neurons of the LC and OX2R to histaminergic neurons of the TMN.Citation55,Citation56 Considering that canine narcolepsy results from a mutation of the OX2R geneCitation57 and the phenotypic differences between OX1R and OX knockout mice, there are some indications that the lack of an orexin signal via OX2R contributes to the pathogenesis of narcolepsyCitation58 Since orexin is below detectable limits in the cerebrospinal fluid of human narcolepsy patients,Citation21,Citation22 whose brains exhibit a nearly complete loss of neurons expressing orexinCitation22,Citation23 an orexin agonist should be able to compensate for orexin deficiency, and therefore should be efficient in promoting wakefulness. However, no available clinical data so far support the effectiveness of this approach in treating sleep disorders.
Sleep-inducing drugs that impair the activity of wake-promoting neurons
Many psychotropic drugs, known as sedatives, interfere with wake propensity mechanisms. Indeed, drugs inhibiting cholinergic, noradrenergic, dopaminergic, serotonergic, or histaminergic neurotransmission have shown various sedative effects. Potent sedative drugs used in psychiatry to treat psychomotor agitation, such as phenothiazine derivatives, often antagonize several of these systems. Antagonizing only one of these alerting systems, such as the histaminergic system with first-generation histamine H1 antagonists used for the treatment of allergies, procures merely mild-to-moderate sedation. However, as stated in the previous section, a renewed interest in histaminergic function, and more specifically in histamine H3 receptors, has grown during the last decade. Thus, H3 receptor agonists should lower histamine release in neuronal terminals as well as the release of other wake-promoting neurotransmitters, such as acetylcholine, dopamine, and nor-adrenaline. Preliminary results indicate that H3 agonists increase SWS in animalsCitation59,Citation60 and it can thus be expected that clinically suitable agonists will improve sleep in some types of insomnia.
In the same way, and according to the key involvement of the orexin system in the orchestration of arousal (see above), orexin antagonists could be potential sleepinducing drugs. Moreover, some studies have suggested that the orexin neurotransmission may be associated with the high-arousal stress-like statesCitation61,Citation62 that typically characterize insomniac patients. Indeed, patients having complaints of insomnia show electrophysiological and psychomotor evidence of increased daytime arousalCitation63-Citation66 as well as indications of other stress-related reactions such as increased hypothalamic-pituitary-adrenal axis (HPA) activity,Citation67 and increased sympathetic tone.Citation68 In a first attempt to record in vivo the activity of orexin neurons, Mileykovskiy et alCitation69 showed that, as expected, orexin neurons displayed very low discharge levels in both REM and NREM sleep, but that discharge rates were not significantly elevated during quiet waking, suggesting that orexin neurotransmission is not associated with waking per se. In contrast, high discharge rates were observed in active and/or alert waking. This further supports the potential clinical value of drugs antagonizing the orexin system in the treatment of stress-related sleep disorders, such as insomnia.
Wake-promoting mechanisms and treatment of sleep disturbances in nicotine and alcohol withdrawal
Sleep disturbances following substance withdrawal, such as nicotine or alcohol, reflect complex hyperarousal states involving stress-related disturbances due to the craving phenomenon and peculiar substance-induced neurotransmission imbalance. For instance, polysomnographic recordings performed during the week following nicotine withdrawal in heavy cigarette smokers have shown increased sleep disruption.Citation70,Citation71 It should, however, be stressed that even before withdrawal, current smokers subjectively complain of decreased sleep time and a fragmented sleep, mostly during the second part of the night.Citation71-Citation74 These observations probably relate to the tobacco withdrawal state occurring each night in heavy smokers rather than to nicotine itself. Indeed, the cholinergic system is a major constituent of the wake-promoting system and it contributes to cortical arousal through its ascending components.Citation13 The involvement of nicotine acetylcholine receptors in these cholinergic effects is suggested by studies showing that nicotine injections increase waking,Citation75 and that mice lacking the β2 subunit gene of the nicotine acetylcholine receptor, a major component of high affinity nicotine-binding sites in the brain, exhibited a reduced fragmentation of NREM sleep through microarousals.Citation76 It is also worth noting that 24-h transdermal nicotine delivery system (nicotine patch [NP]), when administered in nonsmoking healthy volunteers has a sleep-disrupting effect.Citation77,Citation78 However, during tobacco withdrawal, 24-h NP induced an improvement of sleep fragmentation and an increase in the proportion of SWS in cigarette smokers, thus reflecting the fact that nighttime nicotine administration decreases rather than increases arousal level in cigarette smokers.Citation71 This was further demonstrated by a study comparing a 16-h NP (applied only when awake) with a 24-h NP (applied continuously); the results show that microarousals were significantly more decreased by the 24-h NP compared with the 16-h NP, and only the former was found to increase SWS, suggesting a more potent protective effect of the 24-h NP on the tobaccowithdrawal-induced sleep fragmentation.Citation79 The sleep disturbances encountered with the 16-h NP were probably related to an insufficiently compensated withdrawal state (nicotine level is too low to balance tobacco withdrawal).
Postdetoxification sleep disturbances in alcohol dependence may reflect the alcohol-induced alterations of a number of neurochemical systems that are believed to regulate sleep.Citation80 Acute alcohol intake decreases neuronal excitability through its potentiation of inhibitory GABAergic mechanisms and its attenuation of excitatory glutamatergic mechanisms.Citation80-Citation82 Over time, with chronic alcohol use, these neurotransmitter systems adapt, in order to maintain homeostasis and optimize brain functioning, and tolerance develops. However, with discontinuation of alcohol, a withdrawal-associated neural hyperexcitability occurs, favoring arousal and thus interfering with sleepregulating mechanisms in addition to other negative symptoms.Citation80-Citation82 Although the most commonly used strategy to renormalize neuronal excitability is to increase GABAergic transmission, influencing glutamatergic transmission could also reduce postwithdrawal neuronal hyperexcitability. Research on alcoholism has recently focused on the glutamatergic system as preclinical studiesCitation83,Citation84 and human laboratory studies,Citation82 provided compelling evidence for a role of the glutamate system in alcohol dependence. Moreover, drugs targeting the glutamatergic systems such as acamprosate are emerging as novel pharmacotherapeutic options for treating alcohol dependence.Citation85-Citation87 Indeed, a magnetic resonance imaging study showed that acamprosate lowers glutamatergic neurotransmission in human subjects.Citation88 In a polysomnographic study, it was found that acamprosate treatment, initiated 1 week before alcohol withdrawal in alcohol-dependent subjects, enhanced sleep continuity during acute and protracted alcohol withdrawal by increasing time spent in sleep stage 3 and decreasing wakefulness after sleep onset (Staner L et al, unpublished data), while it prolonged REM sleep latency. Studies in healthy subjects have shown that acamprosate is devoid of any sedative effects per se.Citation89 Thus, the present results bring support to the idea that lowering the glutamate-related hyperarousal could influence postwithdrawal sleep disturbances. In accordance with this, in the same group of patients, daytime assessments by EEG and magnetoencephalography also indicate that acamprosate attenuates electrophysiological signs of CNS hyperexcitabilityCitation90
Sleep-inducing drugs that enchance the activity of NREM sleep-promoting neurons
The most prescribed hypnotic drugs, benzodiazepines and benzodiazepine-related drugs such as Zolpidem and zaleplon, have been shown to allosterically and positively modulate the action of GABA via direct interaction with their recognition sites, ie, by increasing the affinity of GABA for its own GABAA sites. GABAA receptors are formed by the assembly of five protein subunits among the 18 subunits that have been identified by cloning techniques: α (6 isoforms, α1 to α6), α (3 isoforms, βx to β3), γ (3 isoforms, γ1 to γ3), p (3 isoforms, px to p3), δ (1 isoform), ε (1 isoform), and θ (1 isoform).Citation91 However, most GABAA receptors are believed to be composed of two α, one β, and two γ subunits. Receptors containing the α1, α2, α3, or α5 subunits in combination with any of the β subunit and the γ2 are most prevalent in the brain, the α1β2γ2 being the most prevalent subunit combination (60% of all GABAA receptors). Zolpidem and zaleplon are distinguished from classical benzodiazepine by binding selectively to GABAA receptors containing the α1 subunit, a subtype of GABAA receptors thought to mediate sedative, anticonvulsive, and amnesic effects of benzodiazepine drugs, whereas α2-containing GABAA receptors relate to anxiolytic and myorelaxant effects.Citation91 Different mechanisms could explain the hypnosedative effects of drugs enhancing GABAA neurotransmission. Firstly, GABA is the major inhibitory neurotransmitter system in the mammalian CNS, and GABAA receptors are ubiquitous in the CNS. Secondly, in the thalamus, these drugs could reinforce the inhibitory influence of GABAergic neurons of the reticular nucleus on the relay nuclei, which are the crossing points of all sensorimotor afferents going to the cortex. The reinforcement of inhibitory influence on relay nuclei has been proposed to underlie the decrease of high-amplitude delta slow-wave activity and the concomitant increase in sigma spindling activity during NREM sleep induced by drugs enhancing GABAA neurotransmission.Citation92 Thirdly, since VLPO sleeppromoting neurons are GABAergic, drugs enhancing GABAA neurotransmission will reinforce the VLPO inhibitory effects on all wake-promoting structures.
Recent studies in a point-mutated mouse model have suggested that effects of benzodiazepines on sleep-onset latency and NREM sleep microstructure are mediated through different subtypes of GABAA receptors. Indeed, α2-containing GABAA receptors could relate to the reduction of NREM delta activity, while α1-containing GABAA receptors could be implicated in the shortening of sleep-onset latency induced by benzodiazepines.Citation93-Citation95 Consequently, it may be suggested that sleep could be used a useful tool for the appraisal of α1 GABAA-mediated sedative versus α2, GABAA-mediated anxiolytic properties of a benzodiazepine drug.
Other compounds enhancing GABAergic transmission could be valuable hypnotic drugs, some of which are currently in development. The drugs in question are another α1-containing GABAA-enhancing drug (indiplon), GABA analogues such as gabapentin, a GABA reuptake inhibitor (tiagabine), and a GABAA agonist (gaboxadol).Citation96 These agents, except gaboxadol, nonspecifically enhance GABAergic transmission through GABAA, GABAB, and GABAC receptors. It should be stressed that the hypnotic effects of GABAB and GABAC ligands are not qualitatively similar to those obtained with GABAA ligands.Citation97
Major depression, REM sleep, and antidepressant drugs
More than 90% of depressed patients complain about difficulties in falling asleep, sleep disruption, or earlymorning awakenings.Citation98 Well-established sleep EEG findings are disturbed sleep continuity (lengthening of sleep latency and increased wake after sleep onset resulting in decreased time spent asleep), deficit of SWS, especially during the first sleep cycle, and REM sleep disinhibition. The latter, also known as “increased REM sleep pressure,” is described as a greater amount of REM sleep, mostly in the beginning of the night (also reflected by a shortened REM onset latency) and as an increase in the actual number of REMs during this sleep stage (REM density).Citation99,Citation100 Many studies have suggested that the REM sleep disinhibition profile is not pathognomonic for major depression, but provides evidence of antidepressant-responsive conditions. Thus, beyond depression, shortened REM sleep latencies have been more reliably reported in conditions for which antidepressant drugs are recognized as effective, such as obsessive-compulsive disorder,Citation101 panic disorder,Citation102 generalized anxiety disorder,Citation103 or borderline personality disorder.Citation104 Polysomnographic recordings in some patients with anorexia nervosaCitation105 and alcohol dependenceCitation106 could also demonstrate a shortened REM latency, but a depressive comorbidity was clearly present.
In 1982, McCarley posited that an imbalance between aminergic and cholinergic influences underlie REM sleep disinhibition in depressive disorder.Citation107 Conventional supports for the imbalance theory are based on the fact that the REM sleep suppressant effect of antidepressant drugs might be attributed to facilitation of noradrenergic and/or serotonergic function or cholinergic blockade. In some cases, as with most tricyclic antidepressants, all three mechanisms may be involved. Antidepressant drugs devoid of clear-cut REM-suppressant effects (ie, bupropion, mirtazapine, nefazodone, tianeptine, trazodone, and trimipramine) share one characteristic: their potency to inhibit noradrenergic or serotonergic uptake is absent, doubtful, or moderate.Citation108
There are several other arguments in favor of the aminergic/cholinergic imbalance theory. A recent [18F]deoxyglucose positron emission tomography (FDGPET) study by Nofzinger et alCitation109 of waking to REM sleep changes reported that, compared with healthy subjects, depressed patients showed increased activation of the brain stem reticular formation limbic and anterior paralimbic cortex, and the executive cortex during REM sleep. The authors suggested that their findings could reflect the disinhibition of the REM-on cholinergic neurons either directly (brain stem activation) or indirectly (through cortical projections). Other evidence comes from studies administering different cholinergic-enhancing drugs (physostigmine, arecoline, RS86) in depressed patients. These compounds induced, to various degrees, stronger signs of REM sleep disinhibition than in healthy controls, as well as, for some of them, an increased rate of awakenings and arousals.Citation110 Other convincing arguments come from the monoamine depletion paradigms. αa-Methyl-para-tyrosine, which inhibits catecholamine synthesis, provoked REM sleep abnormalities in humans.Citation111 Rapid tryptophan depletion induced by a tryptophanfree drink also disin_ hibited REM sleep without changing mood in individuals recovered from depression.Citation112-Citation115 Bhatti et alCitation116 extended these observations to healthy volunteers (in these subjects, a tryptophanfree drink decreased REM latency, increased REM expressed as percentage of total sleep time, and increased REM density), findings that were only partially replicated by Voderholzer et al.Citation117
Conclusion
Polysomnographic recordings constitute a unique noninvasive tool to analyze brain function. Neurotransmission disturbances, such as those encountered in mental disorders, are reflected in alterations of sleep continuity and architecture. If we assume a neurobiological link between sleep and these disorders, the recent explosion of basic findings on the functional neuroanatomy of sleep-wake regulation and the cellular basis of the various sleep rhythms should raise new issues about our understanding of psychiatric disorders. Sleep laboratory investigations are a useful aid for the development of new psychotropic drugs, since their influence on a particular neurotransmission system could be reflected in the polysomnographic profile they induce. Moreover, this profile can be compared with the polysomnographic profiles of reference drugs.
Selected abbreviations and acronyms
| DRN | = | dorsal raphe nucleus |
| GABA | = | γ-aminobutyric acid |
| LC | = | locus ceruleus |
| LDT | = | laterodorsal tegmental (nucleus) |
| NP | = | nicotine patch |
| NREM | = | non-rapid eye movement |
| PTT | = | pediculopontine tegmental (nucleus) |
| REM | = | rapid eye movement |
| SCN | = | suprachiasmatic nucleus |
| SWS | = | slow-wave sleep |
| TMN | = | tuberomammillary nucleus |
| VLPO | = | ventrolateral preoptic nucleus |
REFERENCES
- BixlerEO.KalesA.SoldatosCR.KalesJD.HealeyS.Prevalence of sleep disorders in the Los Angeles metropolitan area.Am J Psychiatry.197913612571262314756
- MellingerGD.BaiterMB.UhlenhuthEH.Insomnia and its treatment. Prevalence and correlates.Arch Gen Psychiatry.1985422252322858188
- OhayonM.Epidemiological study on insomnia in the general population.Sleep.199619(suppI)S7S158723370
- BaiterMB.BauerML.Patterns of prescribing and use of hypnotic drugs in the United States. In: Clift AD, ed.Sleep Disturbances and Hypnotic Drug Dependence. New York, NY: Excerpta Medica;1975
- National Center for Health Statistics.Selected Symptoms of Psychological Distress. US Public Health Service Publication 1000, Series 11, Number 37. Washington DC: US Department of Health, Education and Welfare;1970
- SteriadeM.Arousal: revisiting the reticular activating system.Science.19962722252268602506
- Pace-SchottEF.HobsonJA.The neurobiology of sleep: genetics, cellular physiology and subcortical networks.Nat Rev Neurosci.2002359160512154361
- LeschDR.SpireJP.Clinical electroencephalography. In: Thorpy MJ, ed.Handbook of Sleep Disorders. New York, NY: Marcel Dekker;1990131
- DijkDJ.CzeislerCA.Paradoxical timing of the arcadian rhythm of sleep propensity serves to consolidate sleep and wakefulness in humans.Neurosci Lett199416663688190360
- EdgarDM.DementWC.FullerCA.Effect of SCN lesions on sleep in squirrel monkeys: evidence for opponent processes in sleep-wake regulation.J Neurosci.199313106510798441003
- SaperCB.SherinJE.ElmquistJK.Role of the ventrolateral preoptic area in sleep induction. In, Hayaishi O, Inoue S, eds.Sleep and Sleep Disorders: From Molecule to Behaviour. New York, NY: Academic Press;1997
- StanerL.LuthringerR.MâcherJP.Développement de molécules actives dans l'insomnie: actualités et aspects méthodologiques.Rev Neurol (Paris).2003159(11 suppl)6S486S5514646800
- JonesBE.Arousal systems.Front Biosci.20038S438S45112700104
- HobsonJA.Pace-SchottEF.StïckgoldR.Dreaming and the brain: toward a cognitive neuroscience of conscious states. In: Pace Schott EF, Solms M, Blagrove M, Harnad S, eds.Sleep and Dreaming. Cambridge, UK: Cambridge University Press;2003150
- LuJ.ChouTC.SaperCB.Identification of wake active neurons in the ventral periaqueductal gray (PAG). Presented at: Society of Neuroscience Meeting, 2002 November 2-7, Orlando Fla. Abstract 878.1
- MontiJM.FernandezM.JantosH.Sleep during acute dopamine agonist SKF 38393 or D1 antagonist SCH 23390 administration in rats.Neuropsychopharmacology.199031531622141985
- TrampusM.FerriN.AdamiM.OnginiE.The dopamine receptor agonists, A 68930 and SKF 38393, induce arousal and suppress REM sleep in the rat.Eur J Pharmacol.199323583878100197
- IsaacSO.BerridgeCW.Wake promoting actions of dopamine D1 and D2 receptor stimulation.J Pharmacol Exp Ther.200330738633894
- BeuckmannCT.YanagisawaM.Orexins: from neuropeptides to energy homeostasis and sleep/wake regulation.J Mol Med.20028032934212072908
- TaheriS.ZeitzerJM.MignotE.The role of hypocretins (orexins) in sleep regulation and narcolepsy.Annu Rev Neurosci.20022528331312052911
- NishinoS.RipleyB.OvereemS.LammersGJ.MignotE.Hypocretion (orexin) deficiency in human narcolepsy.Lancet.2000355394010615891
- PeyronC.FarcoJ.RogersW.et al.A mutation in a rare case of early onset narcolepsy and a generalised absence of hypocretin peptides in human narcoleptic brains.Nat Med.2000699199710973318
- ThannickalTC.MooreRY.NïenhuisR.et al.Reduced number of hypocretins in human narcolepsy.Neuron.20002746947411055430
- SaperCB.ChouTC.ScammellTE.The sleep switch: hypothalamic control of sleep and wakefulness.Trends Neurosci.20012472673111718878
- ChouTC.BjorkumAA.GaussSE.et al.Afférents to the ventrolateral preoptic nucleus.J Neurosci.20022297799011826126
- BasheerR.StreckerRE.ThakkarMM.Mc CarleyRW.Adenosine and sleep-wake regulation.Progr Neurobiol.200473379396
- ChamberlainNL.ArrigoniE.ChouTC.et al.Effects of adenosine on GABAergic synaptic inputs to identified ventrolateral preoptic neurons.Neuroscience.200311991391812831851
- MorairtryS.RainnieD.McCarleyR.GreeneR.Dishinbition of ventrolateral preoptic area sleep-active neurons by adenosine: a new mechanism for sleep promotion.Neuroscience.200412345145714698752
- ThakkarMM.WinstonS.McCarleyRW.Orexin neurons of the hypothalamus express adenosine receptors.Brain Res.200294419019412106679
- ChouTC.ScammellTE.GooleyJJ.GausSE.SaperCB.LuJ.Critical role of dorsomedial hypothalamic nucleus in a wide range of behavioral circadian rhythms.J Neurosci.200323106911070214627654
- DeboerT.OvereemS.VisserNA.et al.Convergence of circadian and sleep regulatory mechanisms on hypocretin-1.Neuroscience.200412972773215541893
- SackRL.HughesRJ.EdgarDM.LewyAJ.Sleep-promoting effects of melatonin: at what dose, in whom, under what conditions, and by what mechanisms?Sleep.1997209089159415954
- MendelsonWB.Melatonin microinjection into the medial preoptic area increases sleep in the ratLife Sci.2002712067207012175899
- DorfmanLJ.JarvickME.Comparative stimulant and diuretic actions of caffeine and theobromine in man.Clin Pharmacol Ther.1970118698725481573
- LiebermanHR.WurtmanRJ.EmdeGG.RobertsC.CoviellaIL.The effects of low dose of caffeine on human performance and mood.Psychopharmacology.1987923083123114783
- RibeiroJA.SebastiaoAM.de MendonçaA.Adenosine receptors in the nervous system: pathophysiological implications.Prog Neurobiol.20026837739212576292
- FredholmBB.BattigK.HolmenJ.NehiigA.ZwartauEE.Actions of caffeine in the brain with special reference to factors that contribute to its widespread use.Pharmacol Rev.1999518313310049999
- HoppenbrowersML.BusscheGV.Mioflazine, a nucleoside transport inhibitor is it effective as a sleep promoter in humans? In: Wauqier A, Dugovïc C, Radulovacki M, et al, eds.Slow Wave Sleep: Pathophysiological and Functional Aspects. New York, NY: Raven Press;1989301309
- RadekRJ.DeckerMW.JarvisMF.The adenosine kinase inhibitor ABT702 augments EEG slow waves in rats.Brain Res.20041026748315476699
- TaylorD.HoBT.Comparison of inhibition of monoamine uptake by cocaine, methylphenidate and amphetamine.Res Commun Chem Pathol Pharmacol.1978216575
- NishinoS.MaoJ.SampathkumaranR.SheltonJ.MignotE.Increased dopaminergic transmission mediates the wake-promoting effects of CNS stimulants.Sleep Res Online.19981496111382857
- WisorJP.NishinoS.SoraI.UhlGH.MignotE.EdgarDM.Dopaminergic role in stimulant-induced wakefulness.J Neurosci.2001211787179411222668
- LinJS.HouY.JouvetM.Pontential brain neuronal targets for amphetamine-, methylpenidate-, and modafinil-induced wakefulness, evidenced by c-fos immunocytochemistry in the cat.Proc Natl Acad Sci USA.19969314128141338943072
- EngberTM.KouryEJ.DennisSA.MillerMS.ContrerasPC.BhatRV.Differential patterns of regional c-Fos induction in the rat brain by amphetamine and the novel wakefulness-promoting agent modafinil.Neurosci Lett.199824195989507929
- ScammellTE.EstabrookeIV.McCarthyMT.et al.Hypothalamic arousal regions are activated during modafinil-induced wakefulness.J Neurosci.2000208620862811069971
- AkaokaH.RousselB.LinJS.ChouvetG.JouvetM.Effect of modafinil and amphetamine on the rat catecholaminergic neuron activity.Neurosci Lett.199112320241676498
- LinJS.RousselB.AkaokaH.FortP.DebillyG.JouvetM.Role of catecholamines in the modafinil- and amphetamine-induced wakefulness, a comparative pharmacological study in the cat.Brain Res.19925913193261359924
- de Saint HilaireZ.OroscoM.RouchC.BlancG.NicolaidisS.Variations in extracellular monoamines in the prefrontal cortex and medial hypothalamus after modafinil administration: a microdialysis study in rats.Neuroreport.2001123533353711733706
- BrownRE.StevensDR.HaasHL.The physiology of brain histamine.Prog Neurobiol.20016363767211164999
- PassaniMB.LinJS.HancockA.CrochetS.BlandinaP.The histamine H3 receptor as a novel therapeutic target for cognitive and sleep disorders.TIPS.20042561862515530639
- LigneauX.LinJ.Vanni-MercierG.et al.Neurochemical and behavioral effects of ciproxifan, a potent histamine H3-receptor antagonist.J Pharmacol Exp Ther.19982876586669808693
- ParmentierR.OhtsuH.Djebbara-HannasZ.ValatxJL.WatanabeT.LinJS.Anatomical, physiological, and pharmacological characteristics of histidine decarboxylase knock-out mice: evidence for the role of brain histamine in behavioral and sleep-wake control.J Neurosci.2002227695771112196593
- KomaterVA.BrowmanKE.CurzonP.HancockAA.DeckerMW.FoxGB.H3 receptor blockade by thioperamide enhances cognition in rats without inducing locomotor sensitization.Psychopharmacology.200316736337212682709
- SakuraiT.AmemiyaA.IshiiM.et al.Orexins and orexin receptors: a family of hypothalamic neuropeptides and G proteincoupled receptors that regulate feeding.Cell.1998925735789491897
- TrivediP.YuH.MacNeilDJ.Van der PloegLH.GuanXM.Distribution of orexin receptor mRNA in the rat brain.FEBS Lett.199843871759821961
- MarcusJN.AschkenasiCJ.LeeCE.et al.Differential expression of orexin receptors 1 and 2 in the rat brain.J Comp Neurol.200143562511370008
- LinL.FaracaoJ.LiR.et al.The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene.Cell.19999836537610458611
- AkanmuMA.HondaK.Selective stimulation of orexin receptor type 2 promotes wakefulness in freely behaving rats.Brain Res.2005104813814515919057
- MontiJM.JantosH.PonzoniA.MontiD.Sleep and waking during acute histamine H3 agonist BP 2.94 or H3 antagonist carboperamide (MR 16155) administration in rats.Neuropsychopharmacology.19961531358797189
- LinJS.Brain structure and mechanisms involved in the control of cortical activation and wakefulness, with emphasis on the posterior hypothalamus and histaminergic neurons.Sleep Med Rev.2000447150317210278
- EspanaRA.ValentinoRJ.BerridgeCW.Fos immunoreactivity in hypocretin-synthesizing and hypocretin-1 receptor-expressing neurons: effects of diurnal and nocturnal spontaneous waking, stress and hypocretin-1 administration.Neuroscience.200312120121712946712
- Winsky-SommererR.YamanakaA.DïanoS.et al.Interaction between the corticotropin-releasing factor system and hypocretins (orexins): a novel circuit mediating stress response.J Neurosci.200424114391144815601950
- Schneider-HelmertD.Twenty-four hour sleep-wake function and personality pattern in chronic insomniacs and healthy controls.Sleep.1987104524623685753
- StepanskiE.ZorickF.RoehrsT.YoungD.RothT.Daytime alertness in patients with chronic insomnia compared with asymptomatic control subjects.Sleep.19881154603363270
- RegensteinQH.DambrosiaJ.HallettM.MurawskiB.PaineM.Daytime alertness in patients with primary insomnia.Am J Psychiatry.1993150152915348379559
- StanerL.CornetteF.MauriceD.et al.Sleep microstructure around sleep onset differentiates major depressive insomnia from primary insomnia.J Sleep Res.20031231933014633244
- VgontzasAN.BixlerEO.LinHM.et al.Chronic insomnia is associated with nyctohemeral activation of the hypothalamic-pituitary-adrenal axis: clinical implications.J Clin Endocr Metab.2001863787379411502812
- BonnetMH.ArandDL.Heart rate variability in insomniacs and matched normal sleepers.Psychosom Med.1998606106159773766
- MileykovskiyBY.KiyashchenkoLI.SiegelJM.Behavioral correlates of activity in identified hypocretin/orexin neurons.Neuron.20054678779815924864
- ProsiseGL.BonnetMH.BerryRB.DickelMJ.Effects of abstinence from smoking on sleep and daytime sleepiness.Chest.1994105113611418162739
- WetterDW.FioreMC.BakerTB.YoungTB.Tobacco withdrawal and nicotine replacement influence objective measures of sleep.J Consult Clin Psychology.199563658667
- JorenbyDE.SmithSS.FioreMC.et al.Varying nicotine patch dose and type of smoking cessation counselling.JAMA.1995274134713527563558
- GreenlandS.SatterfieldMH.LanesSF.A meta-analysis to assess the incidence of adverse effects associated with the transdermal nicotine patch.Drug Safety.1998182973089565740
- GourlaySG.ForbesA.MarrïnerT.McNeilJJ.Predictors of adverse experiences during transdermal nicotine therapy.Drug Safety.19992054555510392670
- Salin-PascualRJ.Moro-LopezML.Gonzalez-SanchezH.Bianco-CenturionC.Changes in sleep after acute and repeated administration of nicotine in the rat.Psychopharmacology.199914513313810463313
- LenaC.PopaD.GrailheR.EscourrouP.ChangeuxJP.AdrienJ.p2-containing nicotinic receptors contribute to the organization of sleep and regulate putative microarousals in mice.J Neurosci.2004245711571815215293
- GillinJC.LardonM.RuizC.GolshanS.SalinpascualR.Dose-dependent effects of transdermal nicotine on early morning awakening and rapid eye movement sleep time in non-smoking normal volunteers.J Clin Psychopharmacol.1994142642677962682
- DavilaDG.HurtRD.OffordKP.HarrisCD.ShepardJW.Acute effects of transdermal nicotine on sleep architecture, snoring, and sleep-disordered breathing in nonsmokers.Am J Resp Critic Care Med.1994150469471
- StanerL.LuthringerR.DupontC.AubinHJ.LagrueG.Sleep effects of a 24-hour versus a 16-hour nicotine patch: a polysomnographic study during smoking cessation.Sleep Med. 2005. In press.
- BrowerKJ.Alcohol's effects on sleep in alcoholics.Alcohol Res Health.20012511012511584550
- LittleHJ.The contribution of electrophysiology to knowledge of acute and chronic effects of ethanoi.Pharmacol Ther.19998433335310665833
- KrystalJH.PetrakisIL.MasonG.TrevisanL.D'SouzaDC.N-Methyl-Daspartate glutamate receptors and alcoholism: reward, dependence, treatment and vulnerability.Pharmacol Ther.200399799412804700
- GrantKA.LovingerDM.Cellular and behavioural neurobiology of alcohol: receptor-mediated neuronal processes.Clin Neurosci.199531551648612060
- WoodwardJJ.lonotropic glutamate receptors as sites of action for ethanoi in the brain.Neurochem Int.19993510711310405994
- HolterSM.DanyszW.SpanagelR.Evidence for alcohol anti-craving properties of memantine.Eur J Pharmacol.1996314R1R28957265
- HolterSM.DanyszW.SpanagelR.Novel uncompetitive A/-methyl-Daspartate (NMDA)-receptor antagonist MRZ 2/579 suppresses ethanoi intake in long-term ethanol-experienced rats and generalizes to ethanoi cue in drug discrimination procedure.J Pharmacol Exp Ther.200029254555210640291
- MasonBJ.Treatment of alcohol dependent outpatients with acamprosate: a clinical review.J Clin Psychiatry.200162424811584875
- BobN.NedelecJF.MuzetM.et al.Central effects of acamprosate: part 2. Acamprosate modifies the brain in-vivo proton magnetic resonance spectrum in healthy young male volunteers.Psychiatry Res.1998821151279754454
- SchneiderU.WohlfartK.Schulze-BonhageA.et al.Lack of psychotomimetic or impairing effect on psychomotor performance of acamprosate.Pharmacopsychiatry.199831110113
- BoeijingaPH.ParotP.SouffletL.et al.Pharmacodynamic effects of acamprosate on markers of cerebral function in alcohol-dependent subjects administered as pretreatment and during alcohol abstinence.Neuropsychobiology.200450717715179024
- MöhlerH.FritschyJM.RudolphU.A new benzodiazepine pharmacology.J Pharmacol Exp Ther.20023002811752090
- SteriadeM.Corticothalamic resonance, states of vigilance and mentation.Neuroscience.200010124327611074149
- ToblerI.KoppC.DeboerT.RudolphU.Diazepam-induced changes in sleep: role of α1 GABAA receptor subtype.Proc Natl Acad Sci USA.2001986646664911371608
- KoppC.RudolphU.KeistR.ToblerI.Diazepam-induced changes on sleep and the EEG spectrum in mice: role of the α3 GABAA receptor subtype.Eur J Neurosci.2003172226223012786990
- KoppC.RudolphU.LowK.ToblerI.Modulation of rhythmic brain activity by diazepam: GABAA receptor subtype and state specificity.Proc Natl Acad Sci US A.200410136743679
- LancelM.Role of GABAA receptors in the regulation of sleep: initial sleep responses to peripherally administered modulators and agonists.Sleep.19992233429989364
- GottesmanC.GABA mechanisms and sleep.Neuroscience.200211123123911983310
- MendelsonWB.GillinJC.WyattRD.Human Sleep and its Disorders. New York, NY: Plenum;1977
- ReynoldsCF III.KupferDJ.Sleep research in affective illness: state of the art circa 1987.Sleep.1987101992153306874
- BuysseDJ.KupferDJ.Diagnostic and research applications of electroencephalographic sleep studies in depression: conceptual and methodological issues.J Nerv Ment Dis.19901784054142195133
- InselTR.GillinJC.MooreA.et al.The sleep of patients with obsessivecompulsive disorder.Arch Gen Psychiatry.198239137213777149896
- UhdeTW.Roy-ByrneP.GillinJC.et al.The sleep of patients with panic disorder. A preliminary report.Psychiatry Res.1984122512596593756
- RosaRR.BonnetMH.KramerM.The relationship of sleep and anxiety in anxious subjects.Biol Psychol.1983161191266850022
- ReynoldsCF 3rd.SoloffPH.KupferDJ.et al.Depression in borderline patients: a prospective EEG sleep study.Psychiatry Res.1985141153857645
- KatzJL.KuperbergA.PollackCP.et al.Is there a relationship between eating disorder and affective disorder? New evidence from sleep recordings.AmJ Psychiatry.19841417537596731616
- MoellerFG.GillinJC.IrwinM.et al.A comparison of sleep EEGs in patients with primary major depression and major depression secondary to alcoholism.J Affect Disord.19932739428432959
- McCarleyRW.REM sleep and depression: common neurobiological control mechanisms.Am J Psychiatry.19821395655706122380
- StanerL.LuthringerR.MâcherJP.Effects of antidepressant drugs on sleep EEG in patients with major depression. Mechanisms and therapeutic implications.CNS Drugs.1999114960
- NofzingerEA.BuysseDJ.GermainA.et al.Increased activation of anterior paralimbic and executive cortex from waking to rapid eye movement sleep in depression.Arch Gen Psychiatry.20046169567215237081
- RiemannD.BergerM.VoderholzerU.Sleep and depression - results from psychobiological studies: an overview.Biol Psychol.2001576710311454435
- GillinJC.SitaramN.WehrT.et al.Sleep and affective illness. In, Post RM, Ballenger JC, eds.Neurobiology of Mood Disorders. Baltimore, Md: Williams SWilkins;1984157188
- MooreP.GillinJC.BhattiT.et al.Rapid tryptophan depletion, sleep electroencephalogram, and mood in men with remitted depression on serotonin reuptake inhibitors.Arch Gen Psychiatry.1998555345399633672
- EvansL.GolshanS.KelsoeJ.et al.Effects of rapid tryptophan depletion on sleep electroencephalogram and mood in subjects with partially remitted depression on bupropion.Neuropsychopharmacology.2002271016102612464458
- LandoltHP.KelsoeJR.RapaportMH.GillinJC.Rapid tryptophan depletion reverses phenelzine-induced suppression of REM sleep.J Sleep Res.200312131812603782
- HaynesPL.McQuaidJR.KelsoeJ.et al.Affective state and EEG sleep profile in response to rapid tryptophan depletion in recently recovered nonmedicated depressed individuals.J Affect Disord.20048325326215555723
- BhattiT.GillinJC.SeifritzE.et al.Effects of a tryptophan-free amino acid drink challenge on normal human sleep electroencephalogram and sleep.Biol Psychiatry.19984352599442344
- VoderholzerU.HornyakM.ThielB.et al.Impact of experimentally induced serotonin deficiency by tryptophan depletion on sleep EEG in healthy subjects.Neuropsychopharmacology.1998181121249430135