Abstract
The term “epilepsy” describes a heterogeneous group of disorders, most of them caused by interactions between several or even many genes and environmental factors. Much rarer are the genetic epilepsies that are due to single-gene mutations or defined structural chromosomal aberrations, such as microdeletions. The discovery of several of the genes underlying these rare genetic epilepsies has already considerably contributed to our understanding of the basic mechanisms epileptogenesis. The progress made in the last 15 years in the genetics of epilepsy is providing new possibilities for diagnosis and therapy. Here, different genetic epilepsies are reviewed as examples, to demonstrate the various pathways that can lead from genes to seizures.
El término “epilepsia” describe un grupo heterogéneo de trastornos, la mayoria de los cuales son causados por interacciones entre algunos o incluso muchos genes, y factores ambientales. Son bastante más raras las epilepsias genéticas que se deben a mutaciones de un gen único o aberraciones cromosómicas estructurales definidas, como las microdeleciones, El descubrimiento de algunos de los genes que están a la base de estas raras epilepsias genéticas ya ha contribuido considerablemente a la comprensión de los mecanismos básicos en la epileptogénesis. El progreso que se ha llevado a cabo en los últimos 15 años en la genética de la epilepsia está aportando nuevas posibilidades diagnósticas y terapéuticas. En este artículo se revisan, a modo de ejemplo, diferentes epilepsias genéticas para demostrar las diversas vías que pueden llevar desde los genes a las convulsiones.
Le terme «épilepsie» décrit un groupe hétérogène de troubles dont la plupart sont dus à des interactions entre quelques, ou même de nombreux gènes et des facteurs environnementaux. Plus rares sont les épilepsies génétiques qui sont dues à des mutations d'un seul gène ou à des aberrations chromosomiques de structure définies comme les microdélétions. Notre compréhension des mécanismes de base de l'épileptogenèse a déjà été considérablement améliorée par la découverte de plusieurs de ces gènes impliqués dans ces épilepsies génétiques peu répandues. Les progrès effectués ces 15 dernières années dans la génétique de l'épilepsie ouvrent de nouvelles perspectives de diagnostic et de traitement. Dans cet article, nous passons en revue différentes épilepsies génétiques comme exemples illustrant les différentes voies qui mènent des gènes à l'apparition d'une crise comitiale.
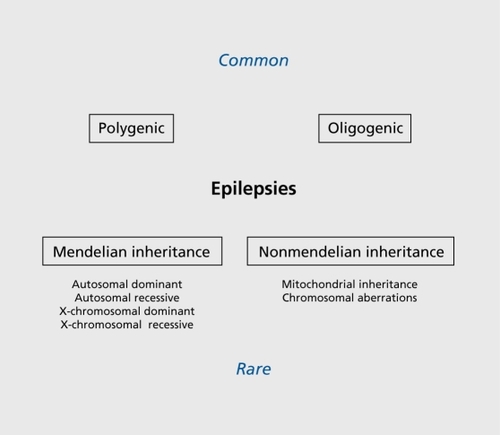
Epilepsy is one of the most common and heterogeneous neurological conditions, and the molecular pathomechanisms underlying the different seizure disorders have now been studied intensively for more than two decades.Citation1 There exists a large group of epilepsies that are often labeled as symptomatic, in order to distinguish them from the idiopathic epilepsies that are believed to be mainly of genetic origin. However, with the progress in genetic analysis, it has become more and more obvious that no clear division exists between the two groups of epilepsies. Findings such as SCN1A mutations underlying severe myoclonic epilepsy of infancy (SMEI), an epilepsy syndrome that was formerly labeled as symptomatic, as well as the failure to find major genes for common idiopathic epilepsies such as juvenile myoclonic epilepsy, raise questions about the usefulness of a strict classification that distinguishes between symptomatic and idiopathic cases. In the etiology of most epilepsies, a combination of acquired and genetic factors is involved, while predominantly genetic epilepsies constitute only a minority of all seizure disorders ().Citation2 The latter nevertheless serve as an important source for our increasing knowledge about the genes and gene families that can be involved in epileptogenesis, and help us gain insight into the pathomechanisms underlying the more common forms of epilepsy. In the following paragraphs, examples of seizure syndromes are described that are representative of the different genetic mechanisms in epileptogenesis known today. These include ion-channel disorders, examples of the different mechanisms underlying progressive myoclonus epilepsies, a group of neurogenetic disorders that can be due to either developmental abnormality, and defects in energy metabolism or metabolic disturbances, as well as neuronal migration disorders.
lon-channel mutations in epilepsy
Dysfunctions of mutated voltage- or ligand-gated ion channels have been shown to be a major cause of idiopathic epilepsies, at least in the rare genetic forms (Table I). The detailed genetic and electrophysiological analyses of different ion channels have provided significant knowledge on the pathophysiological pathways leading from mutation to seizures.Citation3 Ion-channel mutations arc a known cause of rare monogenic idiopathic epilepsies, but are also suspected to play a major role in more common epilepsies such as juvenile myoclonic epilepsy or childhood and juvenile absence epilepsies. In the following paragraphs, some monogenic epilepsies have been selected as examples, to illustrate the importance of ion channels in epileptogenesis.
Table I. Genes in idiopathic epilepsy. AD, autosomal dominant; OG, oligogen; (AD), rare families with monogenic inheritance have been described
Familial nocturnal frontal lobe epilepsy
The gene for autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) was the first one described for an inherited form of idiopathic epilepsy. The mean age of onset in ADNFLE is during adolescence or young adult-hood, with considerable intra- and intcrfamilial variance. Most mutations (with the exception of low penetrance mutation I312M) demonstrate a penetrance of 70 % to 80 % . The clusters of brief motor seizures that are typical of ADNFLE occur mostly out of nonREM (rapid eye movement) sleep. Patients often have recurrent paroxysmal awakenings associated with stereotyped movements. Most seizure episodes are of 2 to 20 s duration, but some of them might last considerably longer. In sleep-walking periods, patients might move around, speak unintelligibly, or scream for 3 min or more. Patients report aura phenomena including epigastric, sensory, or psychic symptoms that often precede the seizures. A typical seizure starts with gasps, grunts, or vocalizations followed by thrashing hyperkinetic activity or tonic stiffening of the limbs, and superimposed clonic jerking. Many patients are, at least initially, not aware of their seizures, or they underestimate their number of seizures per night. Typical compiaints are tiredness during daytime and poor sleep quality. Treatment with antiepileptic drugs such as carbamazepine is effective with respect to seizure control or reduction in many, but not all, patients.Citation4
In 1995 a first mutation was identified in the CHRNA4 gene as the underlying cause in a large Australian ADNFLE family that previously helped to map the disorder to chromosome 20q13.3.Citation5 Since then, additional CHRNA4 mutations, as well as mutations in two additional gènes (CHRNB2, CHRNA2), hâve been found in sleep-relatcd frontal lobe epilcpsy.Citation6-Citation8 CHRNA4, CHRNA2, and CHRNB2 encode the α4- and β2-subunits of the neuronal nicotinic acetyleholine receptor (nAChR), respectively. The nAChRs are members of the large family of lig-and-gated ion channels. They are characterized by five (identical or different) homologous subunits that assemble around a central axis and form a cation-selective ion channel. With the exception of CHRNB2-I312M the known ADNFLE mutations in either CHRNA4 or CHRNB2 are located within the second transmembrane domain. The second transmembrane (and in parts the third) domain mainly builds the walls of the ion channel; thus, it seems that ADNFLE mutations specifically target the channel's gating structure. Until now, four CHRNA4 and three CHRNB2 mutations have been described in ADNFLE families from different parts of the world. Most interesting arc those mutations that have occurred independently in different families, because they offer the opportunity to study the effects genetic backgrounds might have on the clinical expression of the disorder.Citation9
Not surprisingly in view of the important role of the cholinergic System in higher brain functions, there has been evidence presented that at least some ADNFLE mutations not only cause epilepsy but are also associated with other neurological disorders or cognitive deficits. A good example is presented by the Norwegian ADNFLE family carrying the CHRNA4-776ins3 mutation.Citation10,Citation11 More than half of the 11 mutation carriers are affected by either schizophrenia, negative symptoms of schizophrenia, or severe apathy. Another ADNFLE mutation, CHRNA4-S252L, is associated with mental retardation and/or behavioral problems in two families of different geographic origin. In the latter families the differences in geographic origin strongly suggest that the cognitive deficits are caused by the mutation rather than by unrelated factors.Citation8 Most of the known ADNFLE mutations have already been studied in different expression systems. They typically display an increased sensitivity for the natural agonist acetylcholine, demonstrated by a shift of the agonist response curve to the left. Thus, the pathomechanism caused by these mutations seems to be based on a gain-of-function effect that is likely to increase ion flow through the channel pore. Furthermore, comparison between the mutations showed that they are characterized by different biopharmacological profiles. For example, reduced calcium permeability was observed for the mutants CHRNA4-S248F and CHRNA4-776ins3 but not for the CHRNA4-S252L mutation. It is tempting to speculate that the particular functional signatures of each mutant contribute to the abovementioned observations of associated neurological features or cognitive defects in ADNFLE, while the gain-of-function effect might be responsible for the epilepsy phenotype itself.Citation12
It has been hypothized that in presynaptically located nAChRs the gain-of-function effect might activate inhibitory γ-aminobutyric acid (GABA)ergic interneurons. Such interneurons have an important role in controlling the activity of neuronal networks in brain structures such as neocortex and hippocampus by synchronizing the firing of the participating neurons. An enhanced GABA release would first inhibit a larger number of pyramidal cells than usual. After recovery from inhibition, the sudden enhancement in network synchrony could eventually cause the pathological hypersynchronization that might give rise to a seizure.
Benign familial neonatal convulsions
Benign familial neonatal convulsions (BFNC) is an autosomal dominantly inherited seizure disorder of the new-born. BFNC is characterized by an age of onset between the first day and, at latest, the fourth month of life. The seizures are mostly unprovoked, generalized, or multifocal, and of the tonic and/or clonic type. They are often accompanied by dyspnea, ocular symptoms, or other autonomic signs. The course of the disorder is usually benign and self-limiting, and, with or without pharmacotherapy, in the majority of patients the seizures remit spontaneously within a few days or weeks. Most patients are seizure-free by the age of 6 months.Citation13 Later in life seizures can reoccur in about 10 % to 15 % of the patients, starting mostly at school age or in young adulthood. These late-onset seizures are often provoked, for example, by lack of sleep. Recently, it has become a point of discussion as to whether the term “benign” should be used to describe the course of the disorder, since several BFNC families have come to attention in which some or all of the patients had a less than benign outcome. These patients often show a higher frequency of seizures and are often not seizure-free after the age of 4 months. Two BFNC families have been described in which a mutation carrier developed drug-resistant seizures and/or epileptic encephalopathy shortly after birth, resulting in severe psychomotor retardation.Citation14,Citation15 Follow-up studies showed that even patients that have formerly been believed to have a benign course of the disorder later often showed moderate delays of psychomotor development.
Mutations in the voltage gated potassium channel genes KCNQ2 (chromosome 20q13.3) and (more rarely) KCNQ3 (chromosome 8q24) have been described in different BFNC families.Citation16-Citation18 Both genes encode channel subunits with identical structures including six transmembrane regions (TM), a voltage sensor in TM4, a loop between TM5 and TM6 that builds the ion channel pore, and a long C-terminal region that contains sequence motifs for subunit assembling. So far more than 40 mutations have been reported for KCNQ2 and three for KCNQ3. The BFNC mutations are either missense mutations located in one of the TMs, truncating mutations (nonsense, insertion/deletions or splice site mutations), or large deletions. The majority of mutations are private, ie, they have not been found in other BFNC families.
The ion channel encoded by KCNQ2 and KCNQ3 provides one of the major physical equivalents of the socalled M-current, a potassium current which is known to be a powerful controller of neuronal firing. M-currents regulate the frequency with which action potentials are built by opposing sustained membrane depolarization. They have a pivotal role in the stabilization of membrane potentials, and are therefore in a powerful position to control excess neuronal excitability and prevent seizures.Citation19 Given this important role in brain excitability, it is not surprising that BFNC mutations were shown to cause only modest reductions (20 % to 30 %) in potassium currents in reconstitution experiments. Even such slight alterations of M-channel activity are obviously sufficient to increase seizure susceptibility in affected newborns.Citation20 Only a few KCNQ2 mutations have been identified that, at least in reconstitution experiments, had a dominant negative effect on channel function, ie, reduced the current by more than 50 %. One of these mutations is the R207W amino acid exchange in KCNQ2 that causes both BFNC and myokymia, a spontaneous and repetitive involuntary contraction of muscle fiber groups. In the BFNC/myokymia syndrome the occurrence of both central and peripheral neurological symptoms might be explained by the unusual electrophysiological profile of the underlying mutation. The magnitude in loss of current observed for R207W depends strongly on the pattern and time course of depolarization. It is therefore possible that the unusual dominant negative effect of R207W establishes itself only in the peripheral nervous system, causing symptoms like myokymia.Citation21
Infantile convulsion syndromes
Benign familial infantile convulsions (BFIC) is an autosomal dominantly inherited partial epilepsy syndrome of early childhood.Citation22 Seizures usually start between months 4 and 6, with remission before the age of 3 years. The partial seizures occur in clusters and usually respond well to antiepileptic drug treatment. BFIC is genetically heterogeneous, and has been linked to loci on chromosomes 1q23, 2q24, 19q, and 16p12-q12 in various families. Interestingly, three other disorders with overlapping neurological features map to the candidate region on chromosome 16p12-q12, including the infantile convulsions and choreoathetosis syndrome (ICCA; OMIM 602066), paroxysmal kinesigenic choreoathetosis (PKC; OMIM 128200), and a syndrome comprising rolandic epilepsy, paroxysmal exercise-induced dystonia, and writer's cramp (EPRPDC; OMIM 608105). Another rare seizure disorder with an age of onset intermediate between BFNC and BFIC is benign familial neonatal/infantile convulsions (BFNIC). In BFNIC both neonatal and early infantile onsets of the seizures can be present in the same family. BFNIC has been shown to be caused by mutations in the voltagegated sodium channel subunit gene SCN2A, a gene that is also discussed as a minor gene for generalized epilepsy with febrile seizures plus (GEFS+).Citation23
Febrile seizures, generalized epilepsy with febrile seizures plus, and Dravet syndrome
Febrile seizures are the most common seizure type in humans; they affect 5 % to 10 % of children under the age of 6 years. In most patients an oligo- or polygenic background rather than a monogenic cause of the seizures is assumed. Rare families with an apparent autosomal dominant mode of inheritance have been identified, and several putative gene loci described. These tentative gene locations include FEB1 on chromosome 8q13-q21, FEB2 on 19p, FEB3 on 2q23-q24, FEB4 on 5q14-q15, FEB5 on 6q22-q24, and FEB6 on 18p11.2.
In some families febrile seizures may persist beyond the age of 6 years and/or may be associated with variable afebrile seizures (febrile seizures plus). This probably not so rare syndrome has been named generalized epilepsy with febrile seizures plus (GEFS+). Afebrile seizure types in GEFS+ individuals include generalized tonoclonic seizures, myoclonic, absence, and atonic seizures, and in some patients also partial seizures.Citation24 The mode of inheritance underlying GEFS+ is still a matter of debate. Although in some families the trait is likely to be autosomal dominant, in others it is probably better described as oligogenic or as a major gene effect. A genetic concept involving more than one gene would also better fit to the observed clinical variability in GEFS+. Several different ion channel genes have been implicated in GEFS+ - SCN1B, SCN1A, SCN2A, GABRG2 - but in most families the underlying mutation(s) remain elusive. The first GEFS+ mutation was identified in the SCN1B gene on chromosome 19q13.1, a gene that encodes an accessory subunit of the voltage gated sodium channel.Citation25 Nevertheless, most of the mutations identified since then were found in the SCN1A gene, one of the genes coding for the major, pore-forming α-subunit of the voltage gated sodium channel.Citation25 These α-subunits are composed of four domains each containing sixTM (TM1-TM6), and the SCN1A mutations arc distributed over the whole length of this large gene.Citation26
Mutations in the SCN1A gene have also been identified in patients with SMEI (also known as Dravet syndrome) and in the phenotypically overlapping syndromes of borderline SMEI (SMEB) or intractable childhood epilepsy with generalized tonic-clonic seizures (ICEGTC).Citation27 The clinical phenotype of SMEI is characterized by an initially normal psychomotor development, onset of febrile seizures within the first year of life, and subsequent manifestation of various afebrile seizure types including absence, myoclonic, and partial seizures. Pharmacoresistant seizures and developmental delay become manifest during the second year of life. Nearly all reported SMEI mutations are de novo, and they are often truncating and/or located in functionally critical parts of the gene, while GEFS+ mutations are mostly found in less highly conserved parts of SCN1A. It is therefore likely that SMEI and GEFS+ mutations differ with respect to their predicted impact on ion channel function, explaining the differences in clinical severity between both syndromes.
Expression experiments with different SMEI and GEFS+ mutations have shown, probably due to different experimental setups, a variety of biophysical aberrations, including complete loss-of-function, altered gating properties, or even gain-of-function effects. Thus, the exact functional effects of SMEI and GEFS+ mutations and their correlations to clinical phenotypes are still unclear.Citation28
Non-ion channel genes in idiopathic epilepsy
Most of the epilepsy genes mentioned so far code for various ion channels, or for proteins that modulate ion-channel function (eg, accessory channel subunits). This has led to the assumption that idiopathic epilepsies are a group of channelopathies. However, in 2001 it was demonstrated that non-ion channel genes play at least a minor role in the etiology of idiopathic epilepsies. Mutations in the LG11 gene (leucine-rich glioma inactivated gene 1) have been shown to be a cause of autosomal dominant partial epilepsy with auditory features (AD-PEAF), also called autosomal dominant lateral temporal lobe epilepsy.Citation29 This rare syndrome is characterized by simple partial seizures with mainly acoustic and sometimes also visual hallucinations.Citation30 Some families have been described in which the seizures tend to start with a brief sensory aphasia without reduced consciousness.Citation31 The mutational spectrum known for the LG11 gene on chromosome 10q24 includes both missense mutations and truncating mutations. So far, not much is known about the function of the LG11 gene, which codes for a protein characterized by a leucine-rich repeat motif (LRR) in its N-terminal end and seven so-called epilepsy-associated repeats (EARs) in the C-terminal half. The LRR motif is found mostly in proteins that participate in some kind of protein-protein interaction or receptor function.Citation32,Citation33 Two recently published articles have shed some light on the possible function of LG11 protein in human brain. They were able to show that two different isoforms of LG11 protein are expressed in brain, with a long isoform that is secreted and a short isoform that is retained in the intracellular compartment. In the experimental setup the secreted isoform of LG11 binds to differentiated PC12 cells, suggesting that a LG11-specific receptor complex is present on these cells. A putative receptor for LG11 has been identified in ADAM22, a trans-membrane protein anchored to the postsynaptic density-95 (PSD-95)-associated protein complex by stargazin.Citation34 Both ADAM22 and stargazin are genetically linked to epilepsy, at least in knockout animal models. The PSD-95 protein complex has a scaffolding function at excitatory synapses and is involved in both synaptogcncsis and synaptic plasticity. It controls synaptic AMPA receptor surface expression, and the number of expressed receptors could be significantly increased by LG11 expression in cultured hippocampal neurons. Thus, ADPEAF mutations might cause epilepsy by interfering with the protein-protein interaction between LG11 and ADAM22, causing a dysregulation of synaptic transmission.Citation34 Additional non-ion channel genes have been described in epilepsy families since LG11 was first reported several years ago. Examples are the EFHC1 gene and the MASS1/VLGR1 gene, but so far the respective initial reports have not been confirmed in independent studies.Citation35
Progressive myoclonus epilepsies
Numerous neurogenetic syndromes arc known in which seizures are a predominant feature but are usually accompanied by other neurological or non-neurological symptoms. The genetic causes (and therefore the pathways leading to epilepsy) found in these disorders are more diverse than those described above for idiopathic epilepsies. Mutated genes might be involved in many different functions, such as metabolic disturbances, mitochondrial dysfunction, or aberrant neuronal (precursor) cell migration. The heterogeneous group of progressive myoclonus epilepsies that includes Unverricht-Lundborg disease (Baltic myoclonus), myoclonic epilepsy and ragged-red fiber disease (MERRF), neuronal ceroid lipofuscinosis (CLN), dentatorubropallidoluysian atrophy, Gaucher disease, Lafora disease, and sialidosis, is representative of the various mechanisms that might underlie different neurogenetic syndromes characterized predominantly by seizures and cognitive decline. Some of these syndromes are therefore discussed in more detail in the following sections.
Unverricht-Lundborg disease
Unverricht-Lundborg disease (EPM1, also known as Baltic or Mediterranean myoclonus epilepsy) is a typical example of a progressive myoclonus epilepsy characterized by generalized epileptic seizures, myoclonus (brief contraction of a muscle or a group of muscles), and progressive neurological deterioration including ataxia and dementia. EPM1, the most common form of progressive myoclonus epilepsy, is an autosomal recessive neurodegenerative disorder with an age of onset between 6 and 18 years. Severe stimulus-sensitive myoclonus and generalized tonic-clonic seizures are the main features; additionally, mental deterioration, intention tremor, dysarthria, and mild ataxia may develop in later stages of the disorder. So far a few point mutations in the CSTB gene (cystatin B, also stefin B) on chromosome 21q22.3 have been identified in different EPM1 patients (Table II). However, in the majority of patients the disorder is caused by an unstable expansion of a dodecamer repeat located in the 5' flanking region of the CSTB gene.Citation36 Expansion of the repeat causes absence of, or greatly reduced, CSTB expression; thus, a loss-of-function effect is a kev event in EPM1 pathogenesis. CSTB encodes the cystatin B protein, a widely expressed reversible inhibitor of cysteine protease that is thought to have lysosome-associated physiological functions. Cysteine protease inhibitors play an important role in controlling endogenous and exogenous protease activities, protecting organisms from protein degradation. Cystatin B is thought to be involved in the maintenance of normal neuronal structure, and loss of its expression in mouse models caused increased expression of genes involved in proteolysis, apoptosis, and glial activation. Thus neuronal loss by apoptosis and gliosis seems to be an important mechanism in the pathogenesis of EPM1.Citation37,Citation38
Table II. Cystatin B mutations in Unverricht-Lundborg disease. *different nucleotide numbering
There is also mounting evidence that Cystatin B might have an additional function in the cerebellum unrelated to its role in protease inhibition. Coprecipitation experiments showed that cystatin B interacts with different cerebellumexpressed proteins that are not functioning as proteases. What this additional role of cystatin B might be remains unknown so far.Citation39
Myoclonic epilepsy and ragged-red fiber disease
Mitochondria are small intracellular organelles that possess their own circular DNA molecules (mtDNA). Mutations in mtDNA that interfere with or even abolish the ability of mitochondria to perform their role in aerobic respiration are known to cause a wide spectrum of different disorders. They all have in common a pattern of symptoms that predominantly affect tissues with a high dependence on oxidative metabolism, such as brain, muscle, and heart. The clinical manifestations of mitochondrial disorders are extremely heterogeneous; they range from lesions of single tissues to severe impairments including myopathics, encephalomyopathies, cardiomyopathies, or complex multisystem syndromes. Citation40,Citation41 A typical example of such a mitochondrial disorder is MERRF, another member of the group of progressive myoclonus epilepsies. MERRF is characterized by myoclonus, generalized epileptic seizures, myopathy, and slowly progressive dementia. Additional symptoms can be hearing loss, ataxia, and lipomatosis. Histopathological analysis of muscle bioptic material typically shows ragged-red fibers and abnormal mitochondria with concentric cristae. Age of onset and clinical severity differ widely from patient to patient, even between siblings. This is mainly explained by heteroplasmy, ie, each patient inherits a different mixture of normal and mutated mitochondria from his or her mother (there is no paternal inheritance of mitochondria since sperm mitochondria that enter the oocyte are “digested”). The most commonly found MERRF mutation is mt8344A>G that affects the tRNALysine gene within the mtDNA.Citation42 Due to the heteroplasmatic nature of the disorder the molecular diagnosis from blood not always reliably detects the underlying mutation. Skeletal muscle frequently has the highest percentage of mutated mtDNA molecules in MERRF patients, and this percentage correlates best with the clinical severity of the disorder.Citation43 Muscle biopsy is therefore the first choice to obtain material for diagnostic mutation analysis. Close to 150 pathogenic mutations in 21 of the 22 mitochondrial tRNA genes have been described up to now, and 50 % of these mutations are located in either one of three tRNA genes, t.RNALeur (UUR), tRNALYS, or tRNAIle. Mutations in tRNA genes might have different pathological effects, including structural perturbanccs of the three-dimensional tRNA structure, reduced or abolished binding capacity to translation factors such as the elongation factor EF-Tu, or impairment of tRNA maturation. All of these mutations affect the essential role tRNA genes have in the synthesis of proteins involved in energy metabolism, causing various neuromuscular and neurodegenerative disorders.
Neuronal ceroid lipofuscinoses
The different subtypes of neuronal ceroid lipofuscinoses (CLN, or NCL) are (with one exception) autosomal recessively inherited neurodegenerative disorders belonging to the group of lysosomal storage diseases.Citation44 The childhood forms of CLN are characterized by mental and motor deterioration, as well as progressive loss of vision, myoclonic, tonic-clonic, and atypical absence seizures, and premature death, while dementia presents as the main feature in the rare adult forms of CLN. The human CLNs are presently classified into eight main genetic forms (CLN1-8). Tlic infantile subtype of CLN, Santavuori-Haltia-Hagberg disease (CLN1), occurs primarily in the Finish population. The classical late-infantile form of CLN, Jansky-Bielschowsky disease (CLN2), starts at age 2 to 4 years with myoclonus and seizures. There are at least three additional subtypes that are classified as variants of late infantile CLN. These include the Finnish variant (CLN5), the CLN6 variant of late infantile CLN, and the Turkish variant of CLN (CLN8). The CLN8 gene also causes Northern epilepsy or progressive epilepsy with mental retardation, an autosomal recessive epilepsy of childhood onset that is only found in parts of northern Finland. Juvenile-onset CLN (Batten disease, Vogt-Spielmeyer disease) is the most common neurodegenerative disorder of childhood, with an age of onset at 5 to 10 years. Kufs disease or adult CLN (CLN4) is distinguished clinically from the infantile and juvenile subtypes by onset of progressive myoclonus epilepsy in adulthood and by the absence of ocular involvement. Additionally, a few rare forms exist that have not been genetically assigned yet, including congenital NCL and dominantly inherited adult onset NCL (Parry disease, Table III).Citation45-Citation48
Table III. Ceroid lipofuscinosis subtypes in humans. AR, autosomal recessive; AD autosomal dominant; INCL, infantile CLN; LINCL, late infantile CLN; ANCL, adult NCL; vLINCL, variant late infantile CLN; NK, not known
Characteristic features of CLN are an accumulation of autofluoresccnt, periodic acid-Schiff- and Sudan black B-positive granules in the cytoplasm of most nerve cells, astrocytic proliferation and hypertrophy, and progressive and remarkably selective neuronal degeneration and loss.Citation49 The storage cytosomes characteristic for CLN mainly contain of two hydrophobic proteins, the sphingolipid activator proteins A and D (infantile form of CLN) or the subunit c of mitochondrial ATP synthase (late infantile and juvenile CLN).Citation50 The CLN1 and CLN2 genes code for the soluble lysosomal enzymes PPT1 and tripeptidyl peptidase 1 (TPP1), whereas CLN3, CLN6, CLN8 and, possibly, CLN5 are transmembrane proteins of largely unknown functions. It is still unclear how a group of genes as heterogeneous as the CLN genes can cause such a remarkably uniform morphological phenotype characterized by intraneuronal accumulation of hydrophobic proteins. Different mechanisms including both apoptotic and excitotoxic processes are discussed, but the exact nature of the pathophysiological pathways underlying the different CLN subtypes remain to be elucidated.Citation51
Neuronal migration disorders
The migration and maturation of neurons, synapses, and cortical neuronal networks during embryonal and fetal development is a sequential process composed of different steps that are regulated by genetic and environmental factors.Citation52 The cortical neurons are formed in the neuroepithelium of the ventricular zone, and then migrate considerable distances to reach their final position in the cortex. In humans, neuronal migration in the cortex starts at approximately 7 weeks of gestation from the proliferative ventricular zone. The radially migrating neurons as well as the nonradially (tangential) migrating future interneurons are guided by glial fibers through an interaction of adhesion molecules, trophic factors, and guidance molecules. Any disturbances (genetic or environmental) of these complicated migration and matu-rating processes have the potential to cause severe neurological disorders with various symptoms, including mental retardation and epilepsy. Genetic neuronal migration disorders include different lissencephaly syndromes and subcortial band heterotopia, cobblestone dysplasia (a term describing the bumpy surface of the brain that is caused by ectopic neurons and gliovascular proliferation), and different gray matter heterotopia disorders (see also the article by Le venter et al in this issue, p 47).
Conclusion
Although the number of already known epilepsy genes is impressive, they probably represent only the tip of the proverbial iceberg. A roughly estimated 50 % of all genes are, at least during fetal development, expressed in brain and might therefore be regarded as candidates for seizure disorders. Furthermore, recent research has shown that alterations of genomic DNA copy number and gene regulatory elements are likely to be as important for human disorders as mutations that directly affect genes.Citation53 In the future, whole-genome screening methods such as array-based comparative genomic hybridization (aCGH) or genome-wide single nucleotide polymorphism (SNP) analysis will become important tools for the identification of genetic alterations with potential application to common forms of human epilepsy.
Selected abbreviations and acronyms
| ADNFLE | = | autosomal dominant nocturnal frontal lobe epilepsy |
| BFIC | = | benign familial infantile convulsions |
| BFISC | = | benign familial neonatal convulsions |
| CLN | = | neuronal ceroid lipofuscinoses |
| EPM1 | = | epilepsy progressive myoclonus (Unverricht-Lundborg disease) |
| GEFS+ | = | generalized epilepsy with febrile seizures plus |
| MERRF | = | myoclonic epilepsy and ragged-red fiber disease |
| nAChR | = | nicotinic acetylcholine receptor |
| SMEI | = | severe myoclonic epilepsy of infancy |
| TM | = | transmembrane regions |
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG-STE 1651/1-1)
REFERENCES
- GreenbergDA.PalDK.The state of the art in the genetic analysis of the epilepsies.Curr Neurol Neurosci Rep.2007732032817618539
- AndradeDM.MinassianBA.Genetics of epilepsies.Exp Rev Neurother.20077727734
- AvanziniG.FranceschettiS.MantegazzaM.Epileptogenic channelopathies: experimental models of human pathologies.Epilepsia.200748 (suppl 2)516417571353
- SchefferIE.BhatiaKP.Lopes-CendesI.et al.Autosomal dominant nocturnal frontal lobe epilepsy: a distinctive clinical disorder.Brain.199511861737895015
- SteinleinO.MulleyJC.ProppingP.et al.A missense mutation in the neuronal nico-tinic acetylcholine receptor 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy.Nat Genet.1995112012037550350
- FuscoMD.BecchettiA.PatrignaniA.et al.The nicotinic receptor beta2 subunit is mutant in nocturnal frontal lobe epilepsy.Nat Genet.20002627527611062464
- PhillipsHA.FavreI.KirkpatrickM.et al.CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy.Am J Hum Genet.20016822523111104662
- AridonP.MariniC.Di RestaC.et al.Increased sensitivity of the neuronal nicotinic receptor alpha 2 subunit causes familial epilepsy with nocturnal wandering and ictal fear.Am J Hum Genet.2006734235016826524
- SteinleinOK.Genetic disorders caused by mutated acetylcholine receptors.Life Sci.2007802186219017434185
- MagnussonA.StordalE.BrodtkorbE.SteinleinO.Schizophrenia, psychotic illness and other psychiatric symptoms in families with autosomal dominant nocturnal frontal lobe epilepsy.Psychiat Genet.2003139195
- SteinleinOK.MagnussonA.StoodtJ.et al.An insertion mutation of the CHRNA4 gene in a family with autosomal dominant nocturnal frontal lobe epilepsy.Hum Mol Genet.199769439479175743
- BertrandD.PicardF.Le HellardS.et al.How mutations in the nAChRs can cause ADNFLE epilepsy.Epilepsia.200243 (suppl 5)11212212121305
- RonenGM.RosalesTO.ConnollyM.AndersonVE.LeppertM.Seizure characteristics in chromosome 20 benign familial neonatal convulsions.Neurology.199343135513608327138
- DedekK.FuscoL.TeloyN.SteinleinOK.Neonatal convulsions and epileptic encephalopathy in an Italian family with a missense mutation in the fifth transmembrane region of KCNQ2.Epilepsy Res.200354212712742592
- SchmittB.WohlrabG.SanderT.SteinleinOK.HajnalBL.Neonatal seizures with tonic clonic sequences and poor developmental outcome.Epilepsy Res.20056516116816039833
- BiervertC.SchroederBC.KubischC.et al.A potassium channel mutation in neonatal human epilepsy.Science.19982794034069430594
- CharlierC.SinghNA.RyanSG.et al.A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family.Nat Genet.19981853559425900
- SinghNA.CharlierC.StaufferD.et al.A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns.Nat Genet.19981825299425895
- WangHS.OanZ.ShiW.et al.KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel.Science.1998282189018939836639
- WatanabeH.NagataE.KosakaiA.et al.Disruption of the epilepsy KCNQ2 gene results in neural hyperexcitability.J Neurochem.200075283310854243
- DedekK.KunathB.KananuraC.ReunerU.JentschTJ.SteinleinOK.Myokymia and neonatal epilepsy caused by a mutation in the voltage sensor of the KCNQ2 K+ channel.Proc Natl Acad Sci USA.200198122721227711572947
- VigevanoF.FuscoL.Di CapuaM.RicciS.SebastianelliR.LucchiniP.Benign infantile familial convulsions.Europ J Pediat.19921516086121505581
- HeronSE.CrosslandKM.AndermannE.et al.Sodium-channel defects in benign familial neonatal-infantile seizures.Lancet.200236085185212243921
- SchefferIE.BerkovicSF.Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes.Brain.19971204794909126059
- WallaceRH.WangDW.SinghR.et al.Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel betal subunit gene SCN1B.Nat Genet.1998193663709697698
- EscaygA.MacDonaldBT.MeislerMH.et al.Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2.Nat Genet.20002434334510742094
- ClaesL.CeulemansB.AudenaertD.et al.De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy.Hum Mutat.20032161562112754708
- RhodesTH.LossinC.VanoyeCG.WangDW.GeorgeAL. Jr.Noninactivating voltage-gated sodium channels in severe myoclonic epilepsy of infancy.Proc Natl Acad Sci USA.2004101111471115215263074
- KalachikovS.EvgrafovO.RossB.et al.Mutations in LG11 cause autosomal-dominant partial epilepsy with auditory features.Nat Genet.20023033534111810107
- WinawerMR.OttmanR.HauserWA.PedleyTA.Autosomal dominant partial epilepsy with auditory features: defining the phenotype.Neurology.2000542173217610851389
- GuW.BrodtkorbE.SteinleinOK.LG11 is mutated in familial temporal lobe epilepsy characterized by aphasie seizures.Ann Neurol.20025236436712205652
- StaubE.Perez-TurJ.SiebertR.et al.The novel EPTP repeat defines a superfamily of proteins implicated in epileptic disorders.Trends Biochem Sci.20022744144412217514
- GuW.WeversA.SchröderH.et al.The LG11 gene involved in lateral temporal lobe epilepsy belongs to a new subfamily of leucine-rich repeat proteins. .FEBS Lett.2002519717612023020
- FukataY.AdesnikH.IwanagaT.BredtDS.NicollRA.FukataM.Epilepsy-related ligand/receptor complex LG11 and ADAM22 regulate synaptic transmission.Science.20063131792179516990550
- NakayamaJ.FuYH.ClarkAM.et al.A nonsense mutation of the MASS1 gene in a family with febrile and afebrile seizures.Ann Neurol.20025265465712402266
- LafreniereRG.RochefortDL.ChretienN.et al.Unstable insertion in the 5' flanking region of the cystatin B gene is the most common mutation in progressive myoclonus epilepsy type 1, EPM1.Nat Genet.1997152983029054946
- PennacchioLA.BouleyDM.HigginsKM.ScottMP.NoebelsJL.MyersRM.Progressive ataxia, myoclonic epilepsy and cerebellar apoptosis in cystatin B-deficient mice.Nat Genet.1998202512589806543
- LieuallenK.PennacchioLA.ParkM.MyersRM.LennonGG.Cystatin B-deficient mice have increased expression of apoptosis and glial activation genes.Hum Mol Genet.2001101867187111555622
- AlakurttiK.WeberE.RinneR.et al.Loss of lysosomal association of cystatin B proteins representing progressive myoclonus epilepsy, EPM1, mutations.Eur J Hum Genet.20051320821515483648
- DiMauroS.DavidzonG.Mitochondrial DNA and disease.Ann Med.20053722223216019721
- SchonEA.BonillaE.DiMauroS.Mitochondrial DNA mutations and pathogenesis.J Bioenerg Biomembr.1997291311499239539
- ShoffnerJM.LottMT.LezzaAM.SeibelP.BallingerSW.WallaceDC.Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation.Cell.1990619319372112427
- BouletL.KarpatiG.ShoubridgeEA.Distribution and threshold expression of the tRNA (Lys) mutation in skeletal muscle of patients with myoclonic epilepsy and ragged-red fibers (MERRF).Am J Hum Genet.199251118712001334369
- HaltiaM.The neuronal ceroid-lipofuscinoses: from past to present.Biochim Biophys Acta.2006176285085616908122
- VesaJ.HellstenE.VerkruyseLA.et al.Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis.Nature.19953765845877637805
- van DiggelenOP.ThoboisS.TiliketeC.et al.Adult neuronal ceroid lipofuscinosis with palmitoyl-protein thioesterase deficiency: first adultonset patients of a childhood disease.Ann Neurol.20015026927211506414
- SleatDE.DonnellyRJ.LacklandH.et al.Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis.Science.1997277180218059295267
- SavukoskiM.KlockarsT.HolmbergV.SantavuoriP.LanderES.PeltonenL.CLN5, a novel gene encoding a putative transmembrane protein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis.Nat Genet.1998192862889662406
- MoleSE.WilliamsRE.GoebelHH.Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses.Neurogenetics.2005610712615965709
- EzakiJ.KominamiE.The intracellular location and function of proteins of neuronal ceroid lipofuscinoses.Brain Path.200414778514997940
- KyttäläA.LahtinenU.BraulkeT.HofmannSL.Functional biology of the neuronal ceroid lipofuscinoses (NCL) proteins.Biochim Biophys Acta.2006176292093316839750
- GressensP.Pathogenesis of migration disorders.Curr Opinion Neurol.200619135140
- OoiL.WoodIC.Chromatin crosstalk in development and disease: lessons from REST.Nat Rev Genet.2007854455417572692