Abstract
Malformations of cortical development (MCDs) are macroscopic or microscopic abnormalities of the cerebral cortex that arise as a consequence of an interruption to the normal steps of formation of the cortical plate. The human cortex develops its basic structure during the first two trimesters of pregnancy as a series of overlapping steps, beginning with proliferation and differentiation of neurons, which then migrate before finally organizing themselves in the developing cortex. Abnormalities at any of these stages, be they environmental or genetic in origin, may cause disruption of neuronal circuitry and predispose to a variety of clinical consequences, the most common of which is epileptic seizures, A large number of MCDs have now been described, each with characteristic pathological, clinical, and imaging features. The causes of many of these MCDs have been determined through the study of affected individuals, with many MCDs now established as being secondary to mutations in cortical development genes. This review will highlight the best-known of the human cortical malformations associated with epilepsy. The pathological, clinical, imaging, and etioiogic features of each MCD will be summarized, with representative magnetic resonance imaging (MRI) images shown for each MCD, The malformations tuberous sclerosis, focal cortical dysplasia, hemimegalencephaiy, classical iissencephaly, subcortical band heterotopia, periventricular nodular heterotopia, polymicrogyria, and schizencephaly will be presented.
Las malformaciones del desarrollo cortical (MDC) son anormalidades macro o microscópicas de la corteza cerebral que surgen como consecuencia de una interrupción de las etapas normales en la formación de la placa cortical, La corteza cerebral desarrolla su estructura básica durante los dos primeros trimestres del embarazo como una serie de etapas sobrepuesitas, que se inician con la proliferación y diferenciación de neuronas las cuales luego migran antes de organizarse finalmente en la corteza desarrollada. Las anormalidades en cualquiera de estas etapas, sean ellas de origen ambiental o genético, pueden causar interrupción de los circuitos neuronales y predisponer a una variedad de consecuencias clínicas, siendo las más comunes las convulsiones epilépticas, Actualmente se ha descrito un gran número de MDC, cada una con sus características patológicas, clínicas y de imágenes. Las causas de gran parte de estas MDC se han determinado mediante el estudio de sujetos afectados, y actualmente se ha establecido que muchas de ellas son secundarias a mutaciones en genes del desarrollo cortical. Esta revisión destaca lo mejor conocido de las malformaciones corticales humanas asociadas con la epilepsia. Se resumen las características patológicas, clínicas, de imágenes y etiológicas de cada MDC, con imágenes representativas de resonancia nuclear magnética para cada una de ellas. Se presentan las malformaciones de la esclerosis tuberosa, displasia cortical focal, hemimegalencefalia, lisencefalia clásica, heterotopia subcortical en banda, heterotopia nodular periventricular, polimicrogiria y esquizencefalia.
Les malformations du développement cortical (MDC) sont des anomalies macroscopiques ou microscopiques du cortex cérébral qui surviennent comme conséquence d'une interruption des étapes normales de formation de la lame corticale. Le cortex humain développe ses structures de base pendant les deux premiers trimestres de la grossesse, par le biais d'une série d'étapes chevauchantes, commençant par la prolifération et la différenciation des neurones qui migrent ensuite avant de s'organiser finalement dans le cortex en développement. Les anomalies survenant à chacune de ces étapes, qu'elles soient d'origine environnementale ou génétique, peuvent interrompre le circuit neuronal et prédisposer à des conséquences cliniques variées, la plus fréquente étant les crises épileptiques. Un grand nombre de MDC ont été décrites à ce jour, chacune avec ses caractéristiques pathologiques, cliniques et d'imagerie propres. Les causes de la plupart de ces MDC ont été déterminées en étudiant les sujets atteints, et de nombreuses MDC seraient secondaires des mutations affectant des gènes du développement cortical. Cet article mettra en évidence les malformations corticales humaines les plus connues associées à l'épilepsie. Les caractéristiques pathologiques, cliniques, d'imagerie et étiologiques de chaque MDC seront résumées, des images d'IRM (imagerie par résonance magnétique) illustrant chacune d'entre elles. Les malformations telles la sclérose tubéreuse de Bourneville, la dysplasie corticale focale, l'hémiencéphalomégalie, la lissencéphalie classique, l'hétérotopie en bandes sous-corticale, l'hétérotopie nodulaire périventriculaire, la polymicrogyrie et la schizencéphalie seront présentées.
Malformations of cortical development (MCDs) are brain malformations that result from abnormalities affecting the normal processes of cortical development and involving cells that under normal circumstances would participate in formation of the cerebral cortex. Epileptic seizures result from paroxysmal, uncontrolled discharges of electricity from the brain that arise predominantly from the cerebral cortex. It is not surprising therefore that MCDs are often associated with recurrent, seizures, and that these seizures may be difficult, to control. The seizures in MCDs arise as a consequence of either malpositioning of normal cortical neurons or the presence of abnormal cortical neurons which results in abnormal cortical circuitry and a subsequent imbalance between the excitatory (glutaminergic) and inhibitory (y-aminobutyric acid [GABA]ergic) systems which would normally control electrical discharges and prevent, spontaneous abnormal electrical discharges and seizures.
The precise incidence of MCDs is not known; however, they have been diagnosed with increased frequency since the use of magnetic resonance imaging (MRI) to investigate patients with epilepsy, mental retardation, and congenital neurological deficits. It is estimated that 25% to 40% of intractable or medication-resistant childhood epilepsy is attributable to MCDs,Citation1,Citation2 and that at least 75% of patients with MCDs will have epilepsy.Citation3 A large number of MCDs have now been identified and classified using embryologie, genetic, and imaging criteria.Citation4 Contrary to previous assumptions, the majority of these disorders are now thought to have a genetic basis, although environmental causes such as in utero infection or ischemia are still possible. At the time of preparation of this manuscript, mutations in over 30 genes have been identified as causes of MCDs. MCD syndromes with specific clinical, imaging, and genetic criteria are being defined and delineated.
The aim of this review is to discuss the main types of MCDs encountered in clinical practice, highlighting those MCDs in which epilepsy is a frequent accompaniment. The different. MCDs shall be discussed in the order in which they arc currently classified, based on the presumed timing of the “insult,” be it genetic or environmental, within the overlapping stages of cortical development. Each MCD shall be discussed in terms of its pathological, clinical, imaging, and etiological features.
MCDs as a consequence of abnormal neuronal and glial proliferation or differentiation
Tuberous sclerosis
Tuberous sclerosis complex (TSC) is a multisystem syndrome characterized by hamartomata in multiple organ systems, including abnormal proliferation of neurons and glia in the central nervous system. The brain is the most frequently affected organ, but other organs including skin, eyes, heart, and kidneys may be involved.Citation5 Typical brain abnormalities include cortical tubers, subependymal nodules, and subependymal giant cell astrocytoma. 'ITtic pathological features of cortical tubers may be indistinguishable from those of some forms of focal cortical dysplasia (FCD), showing large bizarre neurons, atypical astrocytes, and subpial fibrillary gliosis.
TSC is one of the most, common causes of MCDs, with a birth incidence of 1/6000.Citation6 The clinical features of TSC are highly variable, depending on what organ systems are involved and the location of and severity of involvement within the affected organs. Neurological, symptoms include seizures, intellectual disability, and behavioral problems. Some patients may have minimal or no neurological features despite showing abnormalities in other organ systems or carrying a mutation in one of the two known TSC genes, whilst others may be neurologically asymptomatic despite known cerebral lesions. Seizures may commence at any age and are usually partial seizures originating in cortical tubers. Infantile spasms are common, with seizures arising in infancy. Hie severity of neurological symptoms in TSC generally correlates with the patient's tuber count,Citation7 although this may not hold true for an individual patient. Evidence suggests that the presence and severity of epilepsy is the most important variable associated with intellectual disability.Citation8,Citation9 Overall, approximately 80% of patients with TSC have epilepsy, whilst approximately 65% have intellectual disability of some degree.Citation5
MRI may show cortical tubers, subependymal nodules, giant, cell astrocytoma, and linear white matter abnormalities, as shown in . Computerized tomography (CT) scanning may be required to adequately show calcifications, which are most commonly seen in subependymal nodules. In addition to these typical findings, MRI may also detect cerebellar tubers, subtle cortical dysplasia, transmantle dysplasia,Citation10 hemimegalencephaly (HMEG),Citation11,Citation12 focal megalencephaly, and cortical infoldings.Citation13
TSC is an autosomal dominant syndrome with high penetrance. Based on the study of affected families, two genes have been identified; TSC1 on 9q34 which codes for hamartin,Citation14-Citation14 and TSC2 on 16pl3 which codes for tuberin.Citation15 Ninety percent of patients with TSC will have mutations in one of these genes.Citation16,Citation17 Hamartin and tuberin cooperate in pathways that control cell growth and thus are associated with defective control of neuronal and glial proliferation or differentiation.
Focal cortical dysplasia
The term “focal cortical dysplasia” (FCD) was first used by Taylor et al in 1971 to describe a histological abnormality seen in surgical specimens from 10 patients with epilepsy.Citation18 The abnormality was described as a “malformation,” visible by histology and characterized by the “congregations of large, bizarre neurons...(and) in most ... cases, grotesque cells ... present in the depths of affected cortex and in the subadjacent white matter.” Most, methods of classification divide FCD according to both the degree of dysplasia (architectural or cytoarchitecturai dysplasia) and the presence or absence of abnormal cells, primarily balloon cells or large dysmorphic neurons.Citation19-Citation21 FCD shows a spectrum of severity in terms of its gross morphology, topography, and microscopic features. At the mildest end of the spectrum is “microdysgenesis,” which is poorly defined and refers to subtle developmental cortical abnormalities including neuronal heterotopia, undulations of cortical layering, or neuronal clusters amongst, cell-sparse areas.Citation22 Microdysgenesis has been found at autopsy more commonly in those with epilepsy compared with controls without epilepsy or other neurological disorders,Citation23 as well as in surgical specimens from patients with medically intractable epilepsy.Citation22,Citation24 Despite this, it is still unclear what, degree of “microdysgenesis” may fall within the normal spectrum.Citation25 It has been suggested that the term FCD only be applied to lesions with architectural abnormalities such as dyslamination or the presence of abnormal cells within the cortex.Citation26 The extent of FCD may be highly variable, ranging from focal areas involving part of a gyrus, to involvement of one or more gyri to transmantle dysplasia, lobar dysplasia, hemispheric dysplasia, or multifocal bilateral dysplasia.
Apart from TSC, no particular dysmorphic, neurocutaneous, or multiple congenital anomaly syndromes have been described in which FCD is a feature. Hie most common clinical sequelae of FCD are seizures, developmental delay or intellectual disability, and focal neurological deficits.Citation27-Citation29 Seizures from FCD may arise at any age from in utero seizuresCitation30 until adulthood; however, patients usually present in childhood.Citation27 Extratemporal FCD is usually associated with an earlier age of seizure onset than temporal FCD.Citation27,Citation31,Citation32 Seizures may be simple partial, complex partial, or secondarily generalized, depending on the location of the FCD and the age of the patient. The seizure disorder may be intractable and life -threatening,Citation33 and surgical resection of the area of FCD may be required to control seizures, as they are often resistant to anticonvulsant medications. FCD has been shown to be intrinsically epileptogenic, both in vivo using corticography during epilepsy surgeryCitation34 and in vitro using cortex resected from patients with intractable epilepsy.Citation35,Citation36
FCD is rarely visible on CT, and may not be visible even with high-quality MRI. Subtle abnormalities in gyration, cortical thickness, and the gray-white junction may be a clue to underlying FCD.Citation37 Some forms of FCD may show increased signal on FLAIR and T2 -weighted images which has been thought to represent the presence of balloon cells.Citation20,Citation38,Citation39 White matter signal may be abnormal in the region of a FCD producing intractable seizures.Citation26,Citation40 It is not clear whether this represents dysplastic white matter, or an effect of continued seizure activity producing advanced myelination. Imaging examples of FCD with and without T2 signal increase are shown in .
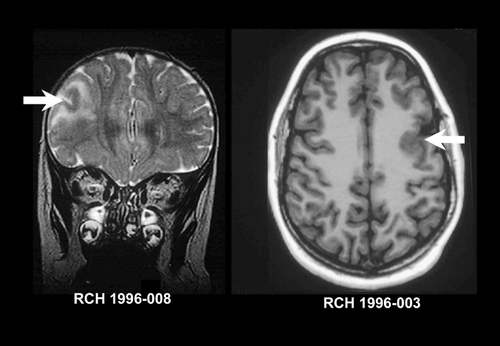
Barkovich and colleagues have described two forms of cortical dysplasia with characteristic imaging appearances. In focal transmantle dysplasia (FTD) there is a wedge of dysplastic tissue from the lateral ventricle to the cortical surface. Histology showed the features of FCD with balloon cells as well as white -matter astrogliosis, and MRI shows a wedge of disorganized tissue with increased T2 signal.Citation41 FTD may also be seen in patients with TSC. Sublobar dysplasia is characterized by a deep infolding of the cortex with a thickened cortex and possible poor gray-white differentiation in the malformed egion. There arc associated brain abnormalities including ventricular dysmorphism and callosal and cerebellar dysgenesis. Tissue was not available for pathological examination.Citation42 Another form of FCD affecting one or other posterior quadrant, of the brain has also been described as “posterior quadrantic dysplasia.” Citation43 This form of FCD is alternately known by the clumsy term “hemihemimegalen cephaly.”
Apart from FCD due to TSC, the etiology of FCD remains unknown. There is no good evidence for environmental causes. There are no published multiplex pedigrees for typical forms of FCD other than families with TSC. However homozygous mutations in the gene CNTNAP2 were recently identified in Amish children with cortical dysplasia, macrocephaly, and intractable seizures with subsequent language regression.Citation44
Hemimegalencephaly
HMEG is a brain malformation characterized by the presence of an abnormally enlarged and dysplastic cerebral hemisphere. The contralateral cerebral hemisphere usually appears normal, except for being compressed or distorted, although a recent, study demonstrated reduced size.Citation45 Macroscopically, one hemisphere is enlarged and there is usually cortical dysgenesis, white-matter hypertrophy, and a dilated and dysmorphic lateral ventricle. The majority of the cerebral hemisphere is affected, with no clear predilection for right or left hemisphere.Citation46 The microscopic features of HMEG can vary significantly. These may include polymicrogyria (PMG), heterotopic grey matter, cortical dyslamination, bizarre enlarged neurons, balloon cells, blurring of the gray-white junction, and an increase in the number of both neurons and astrocytes.Citation47-Citation49
The clinical triad of HMEG is typically: (i) intractable partial seizures from the neonatal period or early infancy, (ii) hemiparesis, and (iii) developmental delay.Citation50 Although the seizures are partial in origin, children may present with tonic seizures, or infantile spasms and the electroclinical features of Ohtahara syndromeCitation51 or West, syndrome. With the exception of the hemiparesis, the developmental abnormalities may in part be the consequence of intractable seizures, and the developmental outcome may be more favorable if seizures arc well controlled from an early age.Citation52,Citation53 Seizures are usually resistant to medical therapy and control may only be achieved by surgery such as anatomical or functional hemispherectomy.Citation53-Citation55
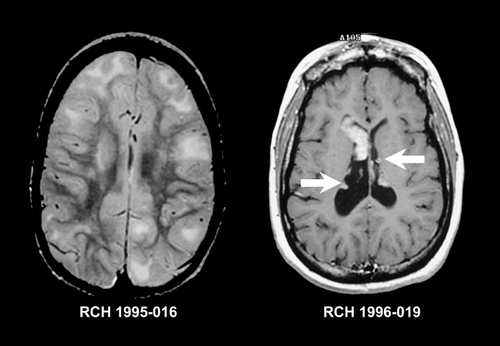
HMEG has been seen in association with both neurocutaneous and overgrowth syndromes. Neurocutaneous associations include the linear nevus sebaceous syndrome,Citation56 hypomclanosis of Ito,Citation57 tuberous sclerosis,Citation11 and neurofibromatosis.Citation58 Approximately 50% of cases of linear nevus sebaceous syndrome have associated HMEG.Citation59 On MRI the cortical gray matter is almost uniformly abnormal, showing areas of thickening and gyral simplification similar to pachygyria or overfolding that resembles polymicrogyria (PMG). In both cases the gray-white junction appears indistinct. White matter is generally markedly increased in volume, and often contains tissue isointense to cortical gray matter, consistent with graymatter heterotopia. There may be white-matter signal change consistent with either dysmyelination or advanced myclination.Citation37,Citation46 The ipsilatcral ventricle is usually enlarged and dysmorphic, often with extension of the posterior horn of the lateral ventricle across the midline.Citation46,Citation60,Citation61 There may be enlargement of the ipsilatcral cerebellar hemisphere and brain stem, an appearance which has been named “total hemimegalencephaly.” Citation62 The typical imaging features of HMEG are shown in .
The etiology of HMEG remains unknown. There are no clear environmental causes or associations with known chromosomal abnormalities. It is generally assumed that HMEG results from a defect leading to excessive proliferation of both neurons and astrocytes and the known association of HMEG with other disorders of cellular proliferation such as TSC and neurofibromatosis supports this hypothesis. One study has shown the abnormal expression of the L1 neural cell adhesion molecule (L1CAM) in 10 children with HMEG compared with 23 controls.Citation63 L1CAM is known to be involved in regulation of neuroblast migration and axonal development.
MCDs as a consequence of abnormal neuronal migration
Classical lissencephaly
The term lissencephaly (LIS) has generally been used to describe disorders in which the mature brain is deficient in gyration. Classical LIS was previously known as “type I” US.Citation64 Classical LIS is a different malformation to cobblestone LIS (or cobblestone dysplasia), previously referred to as “type II LIS.” Citation64 The terms classical LIS, type I LIS, and agyria/pachygyria all still appear in the current literature and all refer to the same malformation. The macroscopic hallmarks of classical LIS are reduced or absent gyration combined with thickening of the cerebral cortex. Most, cases arc a combination of agyria (absent gyration) and pachygyria (broad, simplified gyration), with total agyria or total pachygyria being unusual. On macroscopic inspection the brain shows poorly developed Sylvian and Rolandic fissures and failure of opercularization of the insular areas.Citation65 The brain size and weight are usually at the lower range of normal. Associated abnormalities may include enlarged lateral ventricles, absence of the claustra and external capsules, abnormalities of the corpus callosum, persistent cavum septum pellucidum, hypoplasia of the pyramidal tracts, heterotopia of the inferior olives, and less often abnormalities of the cerebellum. Microscopic examination shows a thick and poorly organized cortex with four rather than the normal six layers.Citation65-Citation67 From the cortical surface inwards, these consist of: (i) a poorly defined marginal zone with increased ccllularity; (ii) a superficial cortical gray zone with diffusely scattered neurons; (iii) a relatively neuron-sparse zone; and (iv) a deep cortical gray zone with neurons often oriented in columns.Citation68 Hie deep cortical gray zone is much thicker than the superficial cellular layer, and consists of large numbers of neurons presumed to have arrested their migration prematurely. Other forms of LIS have recently been described, including LIS associated with cerebellar hypoplasia and RELN mutations,Citation69 and LIS associated with agenesis of the corpus callosum and ARX mutations.Citation70 The pathological findings in these rarer forms of LIS may be somewhat different to those described above.Citation68
The clinical manifestations of LIS> are variable depending on: (i) the severity and topography of the malformation; (ii) associated congenital brain abnormalities; and (iii) congenital abnormalities in other organ systems. Intractable epilepsy may be an independent contributor to intellectual disability and developmental delay. The common clinical features of classical LIS include severe or profound intellectual disability, early hypotonia (which may persist or evolve to mixed axial hypotonia and limb spasticity), epileptic seizures (usually presenting as infantile spasms) and feeding problems.Citation71-Citation75 Hie Miller-Dieker syndrome (MDS) is a contiguous gene deletion syndrome with the deletion of multiple genes at the tip of the short arm of chromosome 17, including both the LISI and YWHAE (14-3-3ε) genes which are both required for normal brain development.Citation76 Children with MDS have a severe form of LIS associated with facial dysmorphism and occasionally other congenital abnormalities, and have a severely shortened life expectancy.
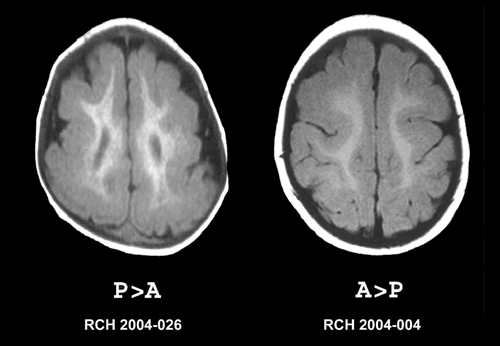
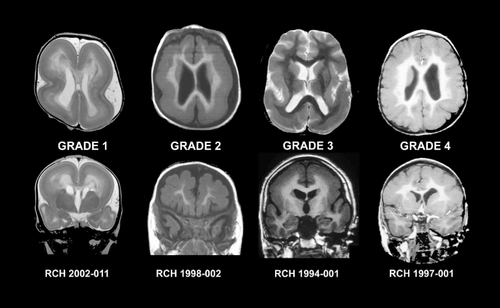
Moderate and severe forms of LIS can usually be diagnosed using CT scanning. The cerebral surface appears smooth with absent opercularization and a characteristic “figure eight” appearance.Citation77 Milder forms of LIS and accompanying brain malformations such as cerebellar abnormalities may be missed using CT scanning. Using 1.5T MRI the gyral pattern (agyria or pachygyria), thickened cortex and other brain abnormalities can readily be appreciated.Citation75 Several different patterns of LIS arc recognized using MRI, which led to development of a detailed grading systemCitation78,Citation79 which considers both the severity of the gyral pattern simplification and the gradient along the anterior to posterior axis. Most patients have a posterior to anterior (P>A) gradient in which the gyral malformation is more severe posteriorly than anteriorly. This pattern is seen most, often as a consequence of a mutation in the LIS1 gene, but may also occur with mutations of TUBA1A. Citation80 Others have the reverse anterior to posterior (A>P) gradient, which is seen most commonly as a consequence of mutations in the DCX gene. shows the imaging features of the two main gradients of LIS, and shows different grades of LIS severity.
Six genes associated with LIS syndromes have been identified, and in approximately 80% of cases, a genetic cause can be found, usually an abnormality of the LIS1 or DCX genes.Citation78,Citation81 The six known genes associated with causation of LIS are LIS1,Citation82 DCX,Citation83 ARX,Citation70 RELN,Citation69 YWHAE,Citation76 and TUBA1A Citation84 These genes are all known to be required for optimal migration of neurons during brain development. All but the ARX gene are required for normal radial migration of neurons whereas the ARX gene is required for normal tangential migration.Citation85 Mutations in the LIS1 gene are the most common cause of LIS.Citation81 The LIS1 protein is not only required for neuronal migration, but. it is also required for cellular proliferation and intracellular transport (reviewed in ref 86).
Subcortical band heterotopia
Subcortical band heterotopia (SBH) is alternately known as double cortexCitation87 or subcortical laminar heterotopia.Citation88 The term SBH is preferable to double cortex, as the heterotopic gray matter lacks cortical lamination and organization, and does not resemble a cerebral cortex other than being composed of gray matter. SBH is characterized microscopically by bilateral bands of heterotopic gray matter located in the white matter between the lateral ventricular walls and the cortex.Citation65 The overlying cortex appears normal with the exception of mildly shallow sulci. In the most typical forms, the bands are bilateral and symmetric and slightly more prominent anteriorly.Citation89 Occasionally the bands are seen restricted to the frontal lobes (partial frontal) or rarely the occipitoparietal regions (partial posterior) and unilateral and asymmetric bands have also been reported.Citation89,Citation90 There are usually no associated brain anomalies or other congenital malformations, although occasionally the SBH can merge anteriorly with pachygyric cortex which has been described as a “pachy-band.”Citation79 Microscopic examination of SBH shows the band to consist of a superficial zone of disorganized neurons, an intermediate zone of small neurons with some columnar organization and a deeper zone where the heterotopia may break into nodules. Trie overlying cortex has a normal histological appearance.Citation65 All forms of SBH are thought to be a defect of neuroblast migration with neurons that fail to migrate completely forming the heterotopic band.Citation91
Patients with SBH will usually have mild-to-moderate intellectual disability and a mixed seizure disorder with onset at any age, but occasionally delayed until the second or third decade.Citation87,Citation92,Citation93 The spectrum of epilepsy and intellectual disability is wide with severity roughly correlating with the thickness of the heterotopic band.Citation92 Typical SBH shows a striking skewing of sex ratio to females,Citation87,Citation91 although the malformation has rarely been reported in males as well.Citation94,Citation97 Occasional patients with mild partial forms of SBH may appear asymptomatic.Citation90 Patients with SBH usually have no dysmorphic features or other congenital anomalies.
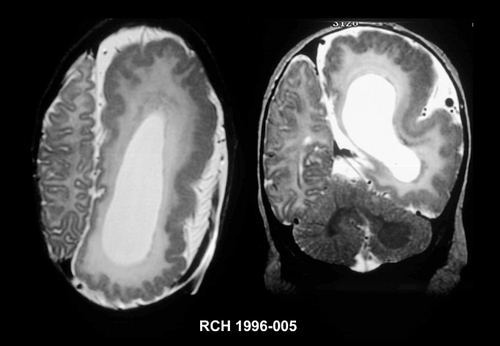
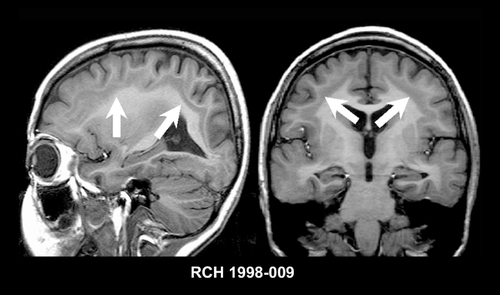
SBH is rarely recognized using CT scanning and when seen may be mistaken for lissencephaly, and partial forms may be difficult to appreciate, even using MRI. MRI will show a four-layered cerebral parenchyma composed of (from ventricle to cortex); (i) normal periventricular white matter; (ii) layer of heterotopic gray matter; (iii) thin layer of subcortical white matter; and (iv) normal cortical gray matter,Citation92 as shown in .
Mutations in two genes have been identified as causing SBH; the DCX gene and the LIS1 gene. The vast, majority of both sporadic and familial cases of the most common form of SBH (bilateral, symmetric, and with a frontal predominance) are due to mutations of DCX. Citation89,Citation98 As DCX is carried on the X chromosome males with mutations in DCX will usually have classical lissencephaly whereas females will have SBH. It is assumed that females with SBH secondary to DCX mutations have two populations of neurons; those with the mutant gene inactive that migrate normally and form the cortex, and those with the normal gene inactivated that migrate abnormally and form the heterotopic band. Carriers of mild DCX mutations may show no evidence of SBH on MRI, but may have intellectual disability or epilepsy.Citation99 A few cases of posterior SBH have been found to be due to somatic mutations of the LIS1 gene.Citation100 SBH has also been reported in association with trisomy 9pCitation101 and in a family without mutations of either the DCX or LIS1 genes,Citation102 suggesting that mutations in other genes may also result in SBH phenotypes.
Periventricular nodular heterotopia
Heterotopia are defined as groups of cells found in an inappropriate location in the correct, tissue of origin. Nodular gray matter heterotopia are relatively common in the brain, most, often found in the periventricular or subcortical white matter, suggesting a failure of migration of neurons normally destined for the cerebral cortex. They are thus correctly defined as MCDs. Heterotopia may occur in isolation, in association with other developmental anomalies of the brain or as part of a multiple congenital anomaly syndrome. Macroscopically, periventricular or subependymal heterotopia are nodular masses of gray matter adjacent to or protruding into the walls of the lateral ventricles. They may be single, multiple, and separated or contiguous. Microscopically, the heterotopic gray matter forms clusters of rounded, irregular nodules separated from each other by layers of myelinated fibers. Both neurons and glia may be present with a pattern ranging from apparent, disorganization to one with rudimentary lamination.Citation103
The most, frequent manifestation of periventricular nodular heterotopia (PNH) is epilepsy, occurring in 80% to 90% of patients with most, having various types of partial seizures, which are usually intractable.Citation104 Studies using depth electrodes in patients with PNH and epilepsy have shown the nodules to be intrinsically epileptogenicCitation105 and temporal lobe surgery for patients with PNH and associated hippocampal sclerosis has generally been unsuccessful.Citation106 Most patients with PNH have normal intelligence, although the curve may be shifted slightly to the left, with an average IQ of approximately 85. This data applies best, to the more common forms of PNH, with manifestations of the variant syndromes generally being more severe. There is a skewed sex ratio towards females among patients with bilateral PNH.
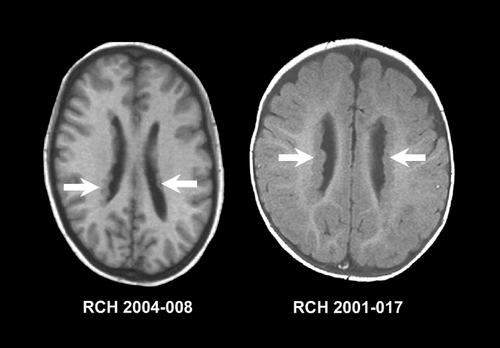
In typical PNH, MRI will show nodular masses of gray matter, lying adjacent, to the lateral ventricles and often protruding into the lumen, as seen in . The signal intensity is identical to that of cortical gray matter. Functional studies using fluorodcoxyglucosc positron emission tomography (FDG-PET) and hexamethylpropyleneamine oxime single positron emission computed tomography (HMPAO-SPECT) have shown changes in metabolic activity and perfusion to be almost identical in the heterotopic nodules and normal overlying cortex.Citation107 Most are located along the lateral ventricular walls, although they may occasionally be seen posteriorly or medially. The nodules may be single or multiple, unilateral or bilateral, large or small, and symmetric or asymmetric. They may be contiguous or separated to resemble “pearls on a string.” PNH differ from the subependymal nodules of TSC, which are usually smaller, fewer, inhomogeneous, and calcified, and have signal intensity resembling white matter. PNH may be associated with additional brain anomalies such as cerebellar vermis hypoplasia, and is the most common MCD found in association with hippocampal sclerosis.Citation24 Unilateral or focal PNH may occur in combination with subcortical nodular heterotopia (SNH) or in association with other MCDs such as PMG.Citation3,Citation108,Citation109 Typical bilateral PNH may be associated with mild-to-moderate hypoplasia of the corpus callosum or cerebellum, the latter primarily involving the vermis. Usually, PNH is limited to the periventricular region but may occasionally form a larger mass that may deform or displace the lateral ventricle.
Mutations in the FLNA gene were identified in families with multiple affected members with bilateral periventricular nodular heterotopia.Citation110 FLNA is located on the long arm of the X-chromosome, and mutations in males are thought to be lethal, thus explaining the female predominance of PNH. FLNA may be necessary for efficient cell motility,Citation111 possibly by promoting actin networks at the leading edge of motile cells or by keeping cells attached to supporting cells until the necessary signal for cell locomotion. Defects in these functions may account for defective initiation of neuronal migration in bilateral PNH.Citation110 Although approximately 80% of familial cases of PNH have FLNA mutations, mutations have been detected in only approximately 20% of sporadic PNH patients.Citation112 Those with mutations usually have a typical bilateral PNH pattern,Citation113 with most patients with atypical PNH not having FLNA mutations.Citation112,Citation114 An autosomal recessive form of PNH with microcephaly has been found to be due to mutations in the ARFGEF2 gene in a small number of children from consanguineous parents.Citation115
Bilateral PNH is also described in association with structural abnormalities of chromosome 5p.Citation116 It is likely that PNH is a genetically heterogeneous disorder secondary to abnormalities of genes involved in neuroblast proliferation or initiation of neuroblast, migration.
MCDs as a consequence of abnormal cortical organization
Polymicrogyria
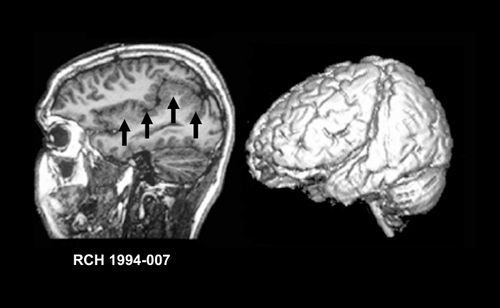
Polymicrogyria (PMG) refers to a cerebral cortex with excessive microscopic gyration, and is probably one of the most common of the MCDs. Macroscopically PMG appears as an irregular cortical surface. The distribution of PMG varies significantly from unilateral forms, to bilateral symmetric and asymmetric forms. The perisylvian cortex is the most frequently affected area and the affected Sylvian fissures may appear extended and superiorly orientated posteriorly as shown in PMG is reported to occur at the periphery of many porencephalic or hydranencephalic defects.Citation117,Citation118 There may be a variety of associated brain malformations, including ventriculomegaly and abnormalities of the corpus callosum, brain stem, and cerebellum, although PMG is usually the isolated brain malformation. PMG may show a variety of histological patterns, but all show abnormal cortical lamination, excessive folding and fusion of adjacent gyri.Citation65 Two main forms of PMG are described; unlayered and layered, the latter of which has been described as the “true” or “structured” PMG.Citation119 Occasionally, both forms are found in the same patient, suggesting that they may be variations of the same malformation.Citation120
The clinical sequelae of PMG are highly variable depending on the extent and location of the PMG, the presence of other brain malformations, and the influence of complications such as epilepsy. In addition, PMG is reported as an occasional component, in multiple different syndromes or disorders including metabolic disorders, chromosome deletion syndromes, and multiple congenital anomaly syndromes. These patients may have a wide spectrum of clinical problems other than those attributable to the PMG. Some patients with PMG have fewer clinical problems than would be expected for the location and extent of cortex involved. Trie most, common form of PMG involves the perisylvian regions in a bilateral and symmetric pattern. The combination of bilateral perisylvian PMG (BPP) associated with oromotor dysfunction and a seizure disorder has been called the “congenital bilateral perisylvian syndrome,” and is the best described syndrome with PMG. Detailed clinical data is published in over 50 patients with this distribution of PMG,Citation121,Citation122 with the first, description appearing in the German pathological literature in 1905.Citation123 Patients with BPP typically have oromotor dysfunction including difficulties with tongue (tongue protrusion and side to side movement), facial and pharyngeal motor function resulting in problems with speech production, sucking, and swallowing, excessive drooling, and facial diplegia. They may also have an expressive dysphasia in addition to dysarthria. More severely affected patients have minimal or no expressive speech, necessitating the use of alternate methods of communication such as signing. On examination there is facial diplegia, limited tongue movement, a brisk jaw jerk, and frequent, absence of the gag reflex.Citation121 In patients presenting in childhood there may be other abnormalities including arthrogryposis, hemiplegia, and hearing loss, although there is limited pediatric data available.Citation124 There may be mild-to-moderate intellectual disability in up to 75% of cases.Citation121 Motor dysfunction may include limb spasticity, although this is rarely severe if present. Other patterns of PMG have been described including unilateral perisylvian PMG,Citation125 bilateral frontal PMG,Citation126 bilateral frontoparietal PMG,Citation127 bilateral parasagittal parietooccipital PMG,Citation128 bilateral parietooccipital PMG,Citation129 multilobar PMG,Citation130 and bilateral generalized PMG.Citation131 The clinical features of these rarer forms of PMG vary from those seen in BPP, although epilepsy and some degree of developmental delay are common accompaniments.
The frequency of epilepsy in PMG is 60% to 85 %,Citation121,Citation122,Citation132 aithough seizure onset may not occur until the second decade, however usually between the ages of 4 and 12.Citation133 Seizure types include atypical absence (62%), atonic and tonic drop attacks (73%), generalized tonic-clonic (35%) and partial (26%). Citation133 It is rare for the partial seizures to secondarily generalize. Occasionally patients develop bilateral facial motor seizures with retained awareness. A small number of patients may present, with infantile spasmsCitation122,Citation133,Citation134 in contrast to patients with LIS, TSC, or FCD, in which the frequency of spasms is higher. Electroencephalography (EEG) typically shows generalized spike and wave or multifocal discharges with a centroparietal emphasis.Citation133 Seizures may be daily and intractable in at least 50% of patients.Citation133
Using CT and low field strength MRI, PMG is difficult to discern and may only appear as thickened cortex.Citation135-Citation138 The only role for CT in the evaluation of PMG is to assess for evidence of calcification which is seen in PMG resulting from congenital CMV infection. Using high-quality 1.5T MRI with appropriate age-specific protocols, it is now possible to reliably differentiate PMG from other MCDs.Citation139 Polymicrogyric cortex often appears mildly thickened (6 to 10 mm) on imaging due to cortical overfolding rather than true cortical thickening. With better imaging (such as inversion recovery) using thin contiguous slices, microgyri and microsulci may be appreciated as shown in figure 8 T2 signal within the cortex is usually normal, although there may be delayed myelination or high T2 signal in the underlying white matter.Citation140 Diffusely abnormal white matter signal should raise the question of an in utero infection (such as cytomegalovirus [CMV]) or a peroxisomal disorder.Citation141-Citation143 There may be an expansion of the subarachnoid space over PMG, and this may contain excessive or anomalous venous drainage, especially in the Sylvian fissures.Citation140 Other developmental anomalies may also be seen including ventricular enlargement or dysmorphism and abnormalities of the corpus callosum and cerebellum, although the patterns and prevalence of these associated brain malformations are poorly documented.
Few topics in the field of MCDs have generated as much discussion as the etiology and pathogenesis of PMG. Initial theories of PMG suggested that it was the result of a vascular defect such as arterial ischemia. Numerous etiologies, both genetic and nongenetic, have since been reported in association with PMG. Nongenetic causes other than hypoxia or hypoperfusion mainly relate to congenital infections including CMV.Citation141,Citation144-Citation146 There are a multitude of reports of PM'G in association with genetic factors, either as part of a known genetic disease or a multiple congenital anomaly syndrome, in association with a structural chromosomal abnormality, or in families with multiple affected members and/or consanguinity. There is an association of PMG with some metabolic diseases including Zellweger syndrome, although the pathological changes differ from typical PMG.Citation143,Citation147,Citation148
Zellweger syndrome has been found to be due to mutations in the PEX family of genes.Citation149,Citation150 Despite the longheld assumption that, most forms of PMG are the result of a nongenetic insult, familial cases and examples of PMG occurring in other genetic syndromes and structural chromosomal abnormalities are now abundant in the literature, as reviewed in Jansen and Andermann.Citation151 All modes of inheritance have been suggested although an X-linked inheritance pattern appears most, frequent.Citation152 The gene for bilateral frontoparietal PMG has been identified as GPR56, yet the function of this gene in cortical development is unclear.Citation153 Our experience and recent data from the mouse suggest that the pathological changes have features in common with cobblestone cortical malformation rather than typical PMG.Citation154,Citation155
Mutations in the gene SRPX2 have been found in one family with BPP,Citation156 but. thus far mutations in this gene have not been reported in other patients with BPP. PMG is also reported as a component, of several chromosomal deletion syndromes, particularly the 22q11.2 deletion syndromes such as the DiGeorge and velocardiofacial syndromes.Citation157
Schizencephaly
“Schizencephaly” (SCZ) is a term first used by Yakovlev and Wadsworth in 1946 to describe “true clefts formed in the brain as the result of failure of development of the cerebral mantle in the zones of cleavage of the primary cerebral fissures.”Citation158,Citation159 SCZ is differentiated from clefts in the cerebral mantle that arise as a consequence of destructive lesions, which Yakovlev and Wadsworth call “encephaloclastic porencephalies,” now known simply as porencephaly. As part, of the definition of SCZ, the clefts must, be lined by abnormal gray matter described as “microgyria,” a term now synonymous with PMG. Macroscopically, the clefts of SCZ can be unilateral or bilateral and “openlipped” or “closed-lipped,” as shown in In openlipped clefts, the walls of the clefts do not appose each other. In closed-lipped clefts the walls of the cleft are apposed and often fused, although a line of continuity between the lateral ventricle and subarachnoid space is usually visible (the “pia epcndymal seamCitation158”). Clefts are frontal or parietal in approximately 65%, and temporal or occipital in approximately 35%.Citation160 Other brain malformations may accompany SCZ. Most, are rare, with the exception of agenesis of the septum pellucidum which is present in approximately 70% of cases.Citation161 Microscopically, the gray matter lining the clefts of SCZ is consistent with PMG, often indistinguishable from other forms of PMG.
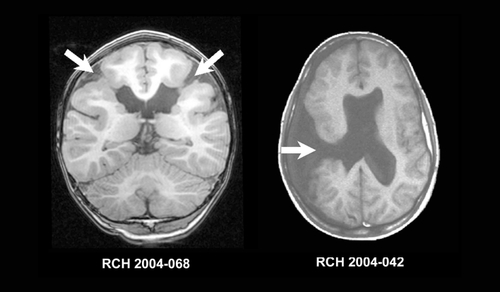
The clinical features of SCZ are well described in the literature, and depend on two factors: (i) unilateral vs bilateral SCZ and (ii) open vs. closed-lipped SCZ. Patients with closed-lipped SCZ typically present with hemiparesis or motor delay whereas patients with open-lipped SCZ typically present with hydrocephalus or seizures.Citation162 In a large series of 47 children with different, types of SCZ, Packard et al found a prevalence of epilepsy in 57% and moderateto-sevcre developmental delay in 83%. The median age for seizure onset was 13 months, although those with openlipped SCZ generally had seizure onset at an earlier age than those with closed-lipped SCZ. The most common seizure type was complex partial, although infantile spasms, tonic, atonic, and tonic-clonic seizures were also reported. The severity and type of seizures does not. appear to correlate with the topography of the SCZ.Citation162,Citation163 Outcome is worst, for those with bilateral open-lipped SCZ and best for those with unilateral closed-lip SCZ.Citation162,Citation164 A large number of patients have associated brain abnormalities which may account for the severity of some cases. These included agenesis of the septum pellucidum, focal cortical dysplasia, and dysgenesis of the corpus callosum.Citation162-Citation165 An interesting finding is that some patients with SCZ have relatively minor clinical problems relative to the appearance of their malformation. Citation166-Citation169
Routine structural MRI scanning is usually sufficient to diagnose SCZ and determine whether the SCZ is open- or closed-lipped. Subtle SCZ may recognizable by a “puckering” or “dimple” outwards of the lateral ventricle at the point at which the cleft reached the ventricular margin (seen in the left, image in figure 9). The cleft, is lined by gray matter. The presence of white matter or T2 signal increase suggestive of gliosis lining the cleft suggests that the lesion is porencephaly rather than SCZ. The gray matter lining the cleft has the imaging appearance of PMG with apparent, cortical thickening, an irregular surface, and stippling of the gray-white interface. SCZ may be asymmetric, and the contralateral hemisphere should be closely evaluated for the presence of a milder SCZ or PMG of another form. Agenesis of the septum pellucidum is a common finding and hypoplasia of the optic nerves may be present, in up to 30% of cases, placing some forms of SCZ in the septo-optic dysplasia spectrum.Citation136,Citation170
The etiology of SCZ remains highly controversial, and there are likely both genetic and non-genetic causes. In SCZ, there is definite evidence for nongenetic causes such as congenital CMV infectionCitation144,Citation171 and in utero ischemic insults.Citation172 There is also now ample evidence supportive of a genetic etiology for some cases of SCZ, including reports of a number of familial cases.Citation173-Citation175 A few patients with both familial and nonfamilial SCZ were found to have mutations in the homeobox gene EMX2. Citation176,Citation177 Unfortunately, other researchers have failed to reproduce these results, raising the question as to the true role of EMX2 in SCZ.Citation174
Conclusion
MCDs are significant causes of neurological and developmental disability and epileptic seizures are an associated symptom in over three quarters of patients. The seizures may arise at any age, but epilepsy will usually commence in childhood and is often resistant to anticonvulsant medications. Surgery may have a role in the treatment of seizures caused by these malformations. Discrete cortical malformation syndromes with specific pathological, clinical, imaging, and genetic syndromes are being defined, and this knowledge has improved the clinician 's ability to provide more accurate prognostic and genetic counseling to affected families, including prenatal testing for certain disorders. The study of these disorders has provided researchers with a unique opportunity to investigate the mechanisms of epileptogenesis. In addition, MCDs have provided molecular biologists and developmental neurobiologists with another method by which to identify new genes and mechanisms for the normal development of the human cerebral cortex.
Selected abbreviations and acronyms
| FCD | = | focal cortical dysplasia |
| HMEG | = | hemimegencephay |
| LIS | = | lissencephaly |
| MCD | = | malformation of cortical development |
| MRI | = | magnetic resonance imaging |
| PMG | = | polymicrogyria |
| PNH | = | periventricular nodular heterotopia |
| SBH | = | subcortical band heterotopia |
| SCZ | = | schizencephaly |
| TSC | = | tuberous sclerosis |
REFERENCES
- KuznieckyRI.JacksonGD.Magnetic Resonance in Epilepsy. New York, NY: Raven Press;1995183202
- GuerriniR.HolthausenH.ParmeggianiL.et al.Epilepsy and malformations of the cerebral cortex. In: Roger J, Bureau M, Dravet C, eds.Epileptic Syndromes in Infancy, Childhood and Adolescence. 3rd ed. London, UK: John Libbey;2002457479
- LeventerRJ.PhelanEM.ColemanLT.et al.Clinical and imaging features of cortical malformations in childhood.Neurology19995371572210489031
- BarkovichAJ.KuznieckyRI.JacksonGD.et al.A developmental and genetic classification for malformations of cortical development.Neurology2005651873188716192428
- GomezMR.Tuberous Sclerosis 2nd ed. New York, NY: Raven Press;1988
- WebbDW.OsborneJP.Tuberous sclerosis.Arch Dis Child1995724714747618927
- GoodmanM.LammSH.EngelA.et al.Cortical tuber count: a biomarker indicating neurologic severity of tuberous sclerosis complex.J Child Neurol19971285909075016
- WebbDW.FryerAE.OsborneJP.On the incidence of fits and mental retardation in tuberous sclerosis.J Med Genet1991283953971870096
- ShepherdCW.StephensonJBP.Seizures and intellectual disability associated with tuberous sclerosis complex in the west of Scotland.DevMed Child Neurol199234766774
- ViglianoP.CanaveseC.BobbaB.et al.Transmantle dysplasia in tuberous sclerosis: clinical features and surgical outcome in four children.J Child Neurol20021775275812546430
- MaloofJ.SledzK.HoggJP.et al.Unilateral megalencephaly and tuberous sclerosis: related disorders?J Child Neurol199494434467822741
- GriffithsPD.GardnerSA.SmithM.et al.Hemimegalencephaly and focal megalencephaly in tuberous sclerosis complex.AJNR Am J Neuroradiol199819193519389874550
- ChristopheC.SekharaT.RypensF.et al.MRI spectrum of cortical malformations in tuberous sclerosis complex.Brain Dev20002248749311111062
- van SlegtenhorstM.de HoogtR.HermansC.et al.Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34.Science19972778058089242607
- European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. The European Chromosome 16 Tuberous Sclerosis Consortium.Cell199375130513158269512
- DaboraSL.JozwiakS.FranzDN.et al.Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs.Ami J Hum Genet2001686480
- van SlegtenhorstM.VerhoefS.TempelaarsA.et al..Mutational spectrum of the TSC1 gene in a cohort of 225 tuberous sclerosis complex patients: no evidence for genotype-phenotype correlation.J Med Genet19993628528910227394
- TaylorDC.FalconerMA.BrutonCJ.et al.Focal dysplasia of the cerebral cortex in epilepsy.J Neurol Neurosurg Psychiatry1971343693875096551
- TassiL.ColomboN.GarbelliR.et al.Focal cortical dysplasia: neuropathological subtypes, EEG, neuroimaging and surgical outcome.Brain20021251719173212135964
- ColomboN.TassiL.GalliC.et al.Focal cortical dysplasias: MR imaging, histopathologic, and clinical correlations in surgically treated patients with epilepsy.AJNR Am J Neuroradiol20032472473312695213
- KuznieckyRI.AndermannF.Non-lissencephalic cortical dysplasias. In: Barth PG, ed.Disorders of Neuronal Migration. 1st ed. London, UK: MacKeith Press;20035871
- HardimanO.BurkeT.PhillipsJ.et al.Microdysgenesis in resected temporal neocortex: incidence and clinical significance in focal epilepsy.Neurology198838104110473386820
- MeenckeHJ.VeithG.Migration disturbances in epilepsy.Epilepsy Res1992Suppl 93140
- RaymondAA.FishDR.SisodiyaSM.et al.Abnormalities of gyration, heterotopias, tuberous sclerosis, focal cortical dysplasia, microdysgenesis, dysembryoplastic neuroepithelial tumour and dysgenesis of the archicortex in epilepsy. Clinical, EEG and neuroimaging features in 100 adult patients.Brain1995118(Pt 3)6296607600083
- MeenckeHJ.VeithG.The relevance of slight migrational disturbances (microdysgenesis) to the etiology of the epilepsies.Adv Neurol19997912313110514809
- PalminiA.NajmI.AvanziniG.et al.Terminology and classification of the cortical dysplasias.Neurology200462S2S815037671
- WyllieE.BaumgartnerC.PraysonR.et al.The clinical spectrum of focal cortical dysplasia and epilepsy.J Epilepsy19947303312
- MackayMT.BeckerLE.ChuangSH.et al.Malformations of cortical development with balloon cells: Clinical and radiologic correlates.Neurology20036058058712601096
- BarkovichAJ.KjosBO.Nonlissencephalic cortical dysplasias: correlation of imaging findings with clinical deficits.AJNR Am J Neuroradiol199213951031375803
- du PlessisAJ.KaufmannWE.KupskyWJ.Intrauterine-onset myoclonic encephalopathy asociated with cerebral cortical dysgenesis.J Child Neurol199381641708505480
- PraysonRA.FraterJL.Cortical dysplasia in extratemporal lobe intractable epilepsy: a study of 52 cases.Ann Diagn Pathol2003713914612808564
- BautistaJF.Foldvary-SchaeferN.BingamanWE.et al.Focal cortical dysplasia and intractable epilepsy in adults: clinical, EEG, imaging, and surgical features.Epilepsy Res20035513113612948622
- DesbiensR.BerkovicSF.DubeauF.et al.Life-threatening focal status epilepticus due to occult cortical dysplasia.Arch Neurol1993506957008323470
- PalminiA.GambardellaA.AndermannF.et al.Instrinsic epileptogenicity of human dysplastic cortex as suggested by corticography and surgical results.Ann Neurol1995374764877717684
- MattiaD.OlivierA.AvoliM.Seizure-like discharges recorded in human dysplastic neocortex maintained in vitro.Neurology199545139113957617202
- AvoliM.LouvelJ.MattiaD.et al.Epileptiform synchronization in the human dysplastic cortex.Epileptic Disord20035 (suppl 2)S45S5014617420
- YagishitaA.AraiN.TamagawaK.et al.Hemimegalencephaly: signal changes suggesting abnormal myelination on MRI.Neuroradiology1998407347389860124
- BronenRA.VivesKP.KimJH.et al.Focal cortical dysplasia of Taylor, balloon cell subtype: MR differentiation from low grade tumors.AJNR Am J Neuroradiol199718114111519194442
- ColomboN.CitterioA.GalliC.et al.Neuroimaging of focal cortical dysplasia: neuropathological correlations.Epileptic Disord20035 (suppl 2)S67S7214617423
- EltzeCM.ChongWK.BhateS.et al.Taylor-type focal cortical dysplasia in infants: some MRI lesions almost disappear with maturation of myelination.Epilepsia2005461988199216393166
- BarkovichAJ.KuznieckyRI.BollenAW.et al.Focal transmantle dysplasia: a specific malformation of cortical development.Neurology199749114811539339707
- BarkovichAJ.PeacockW.Sublobar dysplasia: a new malformation of cortical development.Neurology199850138313879595991
- D'AgostinoMD.BastosA.PirasC.et al.Posterior quadrantic dysplasia or hemi-hemimegalencephaly: A characteristic brain malformation.Neurology200462221415210885
- StraussKA.PuffenbergerEG.HuentelmanMJ.et al.Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2.N Engl J Med20063541370137716571880
- SalamonN.AndresM.ChuteDJ.et al.Contralateral hemimicrencephaly and clinical-pathological correlations in children with hemimegalencephaly.Brain200612935236516291806
- BarkovichAJ.ChuangSH.Unilateral megalencephaly: correlation of MR imaging and pathologic characteristics.AJNR Am J Neuroradiol1990115235311693466
- RobainO.FloquetC.HeldtN.et al.Hemimegalencephaly: a clinicopathological study of four cases.Neuropathol Appl Neurobiol1988141251353399023
- De RosaMJ.SecorDL.BarsomM.et al.Neuropathologic findings in surgically treated hemimegalencephaly: immunohistochemical, morphometry, and ultrastructural study.Acta Neuropathol (fieri)199284250260
- BosmanC.BoldriniR.DimitriL.et al.Hemimegalencephaly. Histological, immunohistochemical, ultrastructural and cytofluorimetric study of six patients.Childs NervSyst199612765775
- TrounceJQ.RutterN.MellorDH.Hemimegalencephaly: diagnosis and treatment.Dev Med Child Neurol1991332612661902803
- OhtsukaY.OhnoS.OkaE.Electroclinical characteristics of hemimegalencephaly.Pediatr Neurol19992039039310371388
- FuscoL.FerracutiS.FarielloG.et al.Hemimegalencephaly and normal intellectual development.J Neurol Neurosurg Psychiatry1992557207221326602
- HumbertclaudeVT.CoubesPA.RobainO.et al.Early hemispherectorny in a case of hemimegalencephaly.Pediatr Neurosurg1997272682719620005
- BonioliE.PalmieriA.BelliniC.Surgical vs. medical treatment of seizures in hemimegalencephaly.Brain Dev1994161698048711
- BattagliaD.Di RoccoC.luvoneL.et al.Neuro-cognitive development and epilepsy outcome in children with surgically treated hemimegalencephaly.Neuropediatrics19993030731310706025
- DobynsWB.GargBP.Vascular abnormalities in epidermal nevus syndrome.Neurology1991412762781992375
- PesericoA.BattistellaPA.BertoliP.et al.Unilateral hypomelanosis of Ito with hemimegalencephaly.Acta Paediatr Scand1988774464473389141
- RossGW.MillerJQ.PersingJA.et al.Hemimegalencephaly, hemifacial hypertrophy and intracranial lipoma: a variant of neurofibromatosis.Neurofibromatosis1989269772516459
- PavoneL.CuratoloP.RizzoR.et al.Epidermal nevus syndrome: a neurologic variant with hemimegalencephaly, gyral malformation, mental retardation, seizures, and facial hemihypertrophy.Neurology1991412662711992373
- KalifaGL.ChironC.SellierN.et al.Hemimegalencephaly: MR imaging in five children.Radiology198716529333628788
- Flores-SarnatL.Hemimegalencephaly: part 1. Genetic, clinical, and imaging aspects.J Child Neurol20021737338412150586
- SenerRN.MR demonstration of cerebral hemimegalencephaly associated with cerebellar involvement (total hemimegalencephaly).Comput Med Imaging Graph1997212012049258598
- TsuruA.MizuguchiM.UyemuraK.et al.Immunohistochemical expression of cell adhesion molecule L1 in hemimegalencephaly.Pediatr Neurol19971645499044401
- DambskaM.WisniewskiK.SherJH.Lissencephaly: two distinct clinicopathological types.Brain Dev198353023106614389
- HardingB.CoppAJ.Malformations. In: Greenfield JD, Lantos PL, Graham DI, eds.Greenfield's Neuropathology. Vol. 1. 7th ed. London, UK: Arnold2002
- CromeL.Pachygyria.J Pathol Bacteriol19567133535213398879
- NormanMG.McGillivrayBC.KalousekDK.et al.Congenital Malformations of the Brain: Pathological, Embryological, Clinical, Radiologic and Genetic Aspects. New York, NY: Oxford University Press;1995
- FormanMS.SquierW.DobynsWB.et al.Genotypically defined lissencephalies show distinct pathologies.J Neuropathol Exp Neurol20056484785716215456
- HongSE.ShugartYY.HuangDT.et al.Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations.Nat Genet200026939610973257
- KitamuraK.YanazawaM.SugiyamaN.et al.Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans.Nat Genet20023235936912379852
- DobynsWB.EliasER.NewlinAC.et al.Causal heterogeneity in isolated lissencephaly.Neurology199242137513881620349
- DobynsWB.CurryCJ.HoyrneHE.et al.Clinical and molecular diagnosis of Miller-Dieker syndrome.Am J Hum Genet1991485845941671808
- de Rijk-van AndelJF.ArtsWFM.BarthPG.et al.Diagnostic features and clinical signs of 21 patients with lissencephaly type 1.Dev Med Child Neurol1990327077172210085
- DobynsWB.ReinerO.CarrozzoR.et al.Lissencephaly: a human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13.JAMA199323283828427907669
- BarkovichAJ.KochTK.CarrolCL.The spectrum of lissencephaly: report of ten patients analyzed by magnetic resonance imaging.Ann Neurol1991301391461897907
- Toyo-OkaK.ShionoyaA.GambelloMJ.et al.14-3-3epsilon is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome.Nat Genet20033427428512796778
- DobynsWB.McCluggageCW.Computed tomographic appearance of lissencephaly syndromes.AJNR Am J Neuroradiol198565455503927671
- PilzDT.MatsumotoN.MinnerathSR.et al.LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation.Hum Mol Genet19987202920379817918
- DobynsWB.TruwitCL.RossME.et al.Differences in the gyral pattern distinguish chromosome 17- linked and X- linked lissencephaly.Neurology19995327027710430413
- PoirierK.KeaysDA.FrancisF.et al.Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A).Hum Mutat2007281055106417584854
- CardosoC.LeventerRJ.DowlingJJ.et al.Clinical and molecular basis of classical lissencephaly: Mutations in the LIS1 gene (PAFAH1B1).Hum Mutat20021941511754098
- ReinerO.CarrozzoR.ShenY.et al.Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats.Nature1993364717721 8355785
- GleesonJG.AllenKM.FoxJW.et al.Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein.Cell19989263729489700
- KeaysDA.TianG.PoirierK.et al.Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans.Cell2007128455717218254
- KatoM.DobynsWB.X-linked lissencephaly with abnormal genitalia as a tangential migration disorder causing intractable epilepsy: proposal for a new term, “interneuronopathy”.J Child Neurol20052039239715921244
- LeventerRJ.CardosoC.LedbetterDH.et al.LIS1: from cortical malformation to essential protein of cellular dynamics.Trends Neurosci20012448949211506866
- PalminiA.AndermannF.AicardiJ.et al.Diffuse cortical dysplasia, or the “double cortex” syndrome: the clinical and epileptic spectrum in 10 patients.Neurology199141165616621922811
- PinardJM.MotteJ.ChironC.et al.Subcortical laminar heterotopia and lissencephaly in two families: a single X-linked dominant gene.J Neurol Neurosurg Psychiatry1994579149208057113
- GleesonJG.LuoRF.GrantPE.et al.Genetic and neuroradiological heterogeneity of double cortex syndrome.Ann Neurol20004726526910665503
- DemelasL.SerraG.ContiM.et al.Incomplete penetrance with normal MRI in a woman with germline mutation of the DCX gene.Neurology20015732733011468322
- DobynsWB.AndermannE.AndermannF.et al.X-linked malformations of neuronal migration.Neurology1996473313398757001
- BarkovichAJ.GuerriniR.BattagliaA.et al.Band heterotopia: correlation of outcome with magnetic resonance imaging parameters.Ann Neurol1994366096177524438
- BarkovichAJ.JacksonDE.BoyerRS.Band heterotopias: a newly recognized neuronal migration anomaly.Radiology19891714554582468173
- PoolosNP.DasS.ClarkGD.et al.Males with epilepsy, complete subcortical band heterotopia, and somatic mosaicism for DCX.Neurology2002581559156212034802
- PilzDT.KucJ.MatsumotoN.et al.Subcortical band heterotopia in rare affected males can be caused by missense mutations in DCX (XLIS) or LIS1.Hum Mol Genet199981757176010441340
- OnoJ.ManoT.AndermannE.et al.Band heterotopia or double cortex in a male: bridging structures suggest abnormality of the radial glial guide system.Neurology199748170117039191790
- KatoM.KanaiM.SomaO.et al.Mutation of the doublecortin gene in male patients with double cortex syndrome: somatic mosaicism detected by hair root analysis.Ann Neurol20015054755111601509
- MatsumotoN.LeventerRJ.KucJA.et al.Mutation analysis of the DCX gene and genotype/phenotype correlation in subcortical band heterotopia.Eur J Hum Genet2001951211175293
- GuerriniR.MoroF.AndermannE.et al.Nonsyndromic mental retardation and cryptogenic epilepsy in women with Doublecortin gene mutations.Ann Neurol200354303712838518
- SiccaF.KelemenA.GentonP.et al.Mosaic mutations of the LIS1 gene cause subcortical band heterotopia.Neurology2003611042104614581661
- FedericoA.TomasettiP.ZollinoM.et al.Association of trisomy 9p and band heterotopia.Neurology19995343043210430446
- DeconinckN.DuprezT.des P.V.et al.Familial bilateral medial parietooccipital band heterotopia not related to DCX or LIS1 gene defects.Neuropediatrics20033414614812910438
- HardingBN.Gray matter heterotopia. In: Guerrini R, Andermann F, Canapicchi R, et al, eds.Dysplasias of Cerebral Cortex and Epilepsy. 1st ed. Philadelphia, Pa: Lippincott-Raven;19968188
- DubeauF.TampieriD.LeeN.et al.Periventricular and subcortical nodular heterotopia. A study of 33 patients.Brain1995118(Pt 5)127312877496786
- KothareSV.VanLandinghamK.ArmonC.et al.Seizure onset from periventricular nodular heterotopias: depth- electrode study.Neurology199851172317279855532
- LiLM.DubeauF.AndermannF.et al.Periventricular nodular heterotopia and intractable temporal lobe epilepsy: poor outcome after temporal lobe resection.Ann Neurol1997416626689153529
- MoriokaT.NishioS.SasakiM.et al.Functional imaging in periventricular nodular heterotopia with the use of FDG-PET and HMPAO-SPECT.Neurosurg Rev199922414410348206
- Soto AresG.Hamon-KerautretM.Houletteet al.Unusual MRI findings in grey matter heterotopia.Neuroradiology19984081879541917
- HannanAJ.ServotteS.KatsnelsonA.et al.Characterization of nodular neuronal heterotopia in children.Brain1999122(Pt 2)21923810071051
- FoxJW.LampertiED.EksiogluYZ.et al.Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia.Neuron199821131513259883725
- CunninghamCC.GorlinJB.KwiatkowskiDJ.et al.Actin-binding protein requirement for cortical stability and efficient locomotion.Science19922553253271549777
- SheenVL.DixonPH.FoxJW.et al.Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females.Hum Mol Genet2001101775178311532987
- PoussaintTY.FoxJW.DobynsWB.et al.Periventricular nodular heterotopia in patients with filamin-1 gene mutations: neuroimaging findings.Pediatr Radiol20003074875511100490
- ParriniE.RamazzottiA.DobynsWB.et al.Periventricular heterotopia: phenotypic heterogeneity and correlation with Filamin A mutations.Brain20061291892190616684786
- SheenVL.GaneshVS.TopcuM.et al.Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex.Nat Genet200436697614647276
- SheenVL.WhelessJW.BodellA.et al.Periventricular heterotopia associated with chromosome 5p anomalies.Neurology2003601033103612654978
- DekabanA.Large defects in cerebral hemispheres associated with cortical dysgenesis.J Neuropathol Exp Neurol196524512530
- FerrerI.CatalaI.Unlayered polymicrogyria: structural and developmental aspects.Anat Embryol (Berlj)1991184517528
- NiewhuijseP.Zur kenntnis der mikrogyrie.Psychiatr Neurol Bl191317953
- McBrideMC.KemperTL.Pathogenesis of four-layered m icrogyric cortex in man.Acta Neuropathol fieri)1982579398
- KuznieckyRl.AndermannF.GuerriniR.Congenital bilateral perisylvian syndrome: study of 31 patients.Lancet19933416086128094839
- GropmanAL.BarkovichAJ.VezinaLG.et al.Pediatric congenital bilateral perisylvian syndrome: clinical and MRI features in 12 patients.Neuropediatrics1997281982039309709
- 0ekonmakisM.Uber umschriebene, mikrogyrische Verbildungen an der Grosshirnoberflache und ihre Beziehung zur Porencephalie.Arch Psychiatrie Nervenkrank190539676
- MillerSP.ShevellM.RosenblattB.et al.Congenital bilateral perisylvian polymicrogyria presenting as congenital hemiplegia.Neurology199850186618699633745
- SebireG.HussonB.DusserA.et al.Congenital unilateral perisylvian syndrome: radiological basis and clinical correlations.J Neurol Neurosurg Psychiatry19966152568676160
- GuerriniR.BarkovichAJ.SztrihaL.et al.Bilateral frontal polymicrogyria: a newly recognized brain malformation syndrome.Neurology20005490991310690985
- ChangBS.PiaoX.BodellA.et al.Bilateral frontoparietal polymicrogyria: clinical and radiological features in 10 families with linkage to chromosome 16.Ann Neurol20035359660612730993
- GuerriniR.DubeauF.DulacO.et al.Bilateral parasagittal parietooccipital polymicrogyria and epilepsy.Ann Neurol19974165739005867
- FerrieCD.JacksonGD.GiannakodimosS.et al.Posterior agyria-pachygyria with polymicrogyria: evidence for an inherited neuronal migration disorder.Neurology1995451501537824106
- GuerriniR.GentonP.BureauM.et al.Multilobar polymicrogyria, intractable drop attack seizures, and sleep- related electrical status epilepticus.Neurology1998515045129710026
- ChangBS.PiaoX.GianniniC.et al.Bilateral generalized polymicrogyria (BGP): a distinct syndrome of cortical malformation.Neurology2004621722172815159468
- BarkovichAJ.HevnerR.GuerriniR.Syndromes of bilateral symmetrical polymicrogyria.AJNR Am J Neuroradiol1999201814182110588102
- KuznieckyRl.AndermannF.GuerriniR.et al.The epileptic spectrum in the congenital bilateral perisylvian syndrome.Neurology1994443793858145902
- KuznieckyR.AndermannF.GuerriniR.Infantile spasms: an early epileptic manifestation in some patients with the congenital bilateral perisylvian syndrome.J Child Neurol199494204237822736
- ByrdSE.OsbornRE.BohanTP.et al.The CT and MR evaluation of migrational disorders of the brain. Part II. Schizencephaly, heterotopia and polymicrogyria.Pediatr Radiol1989192192222748227
- BarkovichAJ.ChuangSH.NormanD.MR of neuronal migration anomalies.AJNR Am J. Neuroradiol1987810091017
- KuznieckyRl.AndermannF.CBPS Study GroupThe congenital bilateral perisylvian syndrome: imaging findings in a multicenter study.AJNR Am J Neuroradiol1994151391448141045
- BarkovichAJ.KuznieckyRl.Neuroimaging of focal malformations of cortical development.J Clin Neurophysiol1996134814948978620
- RaybaudC.GerardN.Canto-MoreiraN.et al.High-definition magnetic resonance imaging identification of cortical dysplasias: micropolygyria versus lissencphaly. In: Guerrini R, Andermann F, Canapicchi R, et al, eds.Dysplasias of Cerebral Cortex and Epilepsy. 1sted. Philadelphia, Pa: Lipincott -Raven.1996131143
- ThompsonJE.CastilloM.ThomasD.et al.Radiologie-pathologie correlation polymicrogyria.AJNR Am J Neuroradiol1997183073129111668
- BarkovichAJ.LindanCE.Congenital cytomegalovirus infection of the brain: imaging analysis and embryologie considerations.AJNR Am J Neuroradiol199415703715 8010273
- van der KnaapMS.ValkJ.The MR spectrum of peroxisomal disorders.Neuroradiology1991333037 2027442
- BarkovichAJ.PeckWW.MR of Zellweger syndrome.AJNR Am J Neuroradiol199718116311709194444
- lannettiP.NigroG.SpaliceA.et al.Cytomegalovirus infection and schizencephaly: case reports.Ann Neurol1998431231279450779
- HaywardJC.TitelbaumDS.ClancyRR.et al.Lissencephaly-pachygyria associated with congenital cytomegalovirus infection.J Child Neurol199161091141646253
- CromeL.FranceNE.Microgyria and cytomegalic inclusion disease in infancy.J Clin Pathol19591242713812952
- ZellwegerH.The cerebro-hepato-renal (Zellweger) syndrome and other peroxisomal disorders.Dev Med Child Neurol1987298218293319743
- KaufmannWE.ThedaC.NaiduS.et al.Neuronal migration abnormality in peroxisomal bifunctional enzyme defect.Ann Neurol1996392682718967760
- ShimozawaN.SuzukiY.ZhangZ.et al.Identification of PEX3 as the gene mutated in a Zellweger syndrome patient lacking peroxisomal remnant structures.Hum Mol Genet200091995199910942428
- RaymondGV.Peroxisomal disorders.CurrOpin Neurol200114783787
- JansenA.AndermannE.Genetics of the polymicrogyria syndromes.J Med Genet20054236937815863665
- GuerreiroMM.AndermannE.GuerriniR.et al.Familial perisylvian polymicrogyria: a new familial syndrome of cortical maldevelopment.Ann Neurol200048394810894214
- PiaoX.HillRS.BodellA.et al.G protein-coupled receptor-dependent development of human frontal cortex.Science20043032033203615044805
- DobynsWB.PattonMA.StrattonRF.et al.Cobblestone lissencephaly with normal eyes and muscle.Neuropediatrics19962770758737821
- JinZ.TietjenI.BuL.et al.Disease-associated mutations affect GPR56 protein trafficking and cell surface expression.Hum Mol Genet2007161972198517576745
- RollP.RudolfG.PereiraS.et al.SRPX2 mutations in disorders of language cortex and cognition.Hum Mol Genet2006151195120716497722
- RobinNH.TaylorCJ.Donald-McGinnDM.et al.Polymicrogyria and deletion 22q11 .2 syndrome: window to the etiology of a common cortical malformation.Am J Med Genet A20061402416242517036343
- YakovlevPI.WadsworthRC.Schizencephalies: a study of the congenital clefts in the cerebral mantle. I. Clefts with fused lips.J Neuropathol Exp Neurol1946511613021026933
- YakovlevPI.WadsworthRC.Schizencephalies: a study of the congenital clefts in the cerebral mantle II. Clefts with hydrocephalus and lips separated.J Neuropathol Exp Neurol1946516920620993391
- BarkovichAJ.Paediatric Neuroimaging. 3rd ed. Philadelphia, Pa: Lippincott Williams and Wilkins;2000
- BarkovichAJ.NormanD.Absence of the septum pellucidum: a useful sign in the diagnosis of congenital brain malformations.AIR Am J Roentgenol1989152353360
- PackardAM.MillerVS.DelgadoMR.Schizencephaly: correlations of clinical and radiologic features.Neurology199748142714349153485
- GranataT.BattagliaG.D'lncertiL.et al.Schizencephaly: neuroradiologic and epileptologic findings.Epilepsia199637118511938956850
- BarkovichAJ.KjosBO.Schizencephaly: correlation of clinical findings with MR characteristics.AJNR Am J Neuroradiol19921385941595498
- DenisD.ChateilJF.BrunM.et al.Schizencephaly: clinical and imaging features in 30 infantile cases.Brain Dev20002247548311111060
- BrownMC.LevinBE.RamsayRE.et al.Comprehensive evaluation of left hemisphere type I schizencephaly.Arch Neurol1993506676698503805
- BisgardC.HerningM.Severe schizencephaly without neurological abnormality.Seizure199321511538167968
- AvellanetM.MirapeixRM.EscuderoD.et al.An unusual clinical presentation of bilateral schizencephaly.Surg Radiol Anat1996182712738983105
- ChoWH.SeidenwurmD.BarkovichAJ.Adult-onset neurologic dysfunction associated with cortical malformations.AJNR Am J Neuroradiol1999201037104310445440
- BarkovichAJ.FramEK.NormanD.Septo-optic dysplasia: MR imaging.Radiology19891711891922928524
- SenerRN.Schizencephaly and congenital cytomegalovirus infection. J.Neuroradiol199825151152
- LandrieuP.LacroixC.Schizencephaly, consequence of a developmental vasculopathy? Aclinicopathological report.Clin Neuropathol1994131921967955664
- HilburgerAC.WillisJK.BouldinE.et al.Familial schizencephaly.Brain Dev1993152342368214352
- TietjenI.ErdoganF.CurrierS.et al.EMX2-independent familial schizencephaly: clinical and genetic analyses.Am J A/fed Genet A2005135166170
- GranataT.FarinaL.FaiellaA.et al.Familial schizencephaly associated with EMX2 mutation.Neurology199748140314069153481
- BrunelliS.FaiellaA.CapraA.et al.Germline mutations in the horneobox gene EMX2 in patients with severe schizencephaly.Nat Genet19961294968528262
- FaiellaA.BrunelliS.GranataT.et al.A number of schizencephaly patients including 2 brothers are heterozygous for germline mutations in the homeobox gene EMX2.Eur J Hum Genet199751861909359037