Abstract
Major depressive disorder (MDD) is associated with a high rate of developing serious medical comorbidities such as cardiovascular disease, stroke, dementia, osteoporosis, diabetes, and the metabolic syndrome. These are conditions that typically occur late in life, and it has been suggested that MDD may be associated with “accelerated aging.” We review several moderators and mediators that may accompany MDD and that may give rise to these comorbid medical conditions. We first review the moderating effects of psychological styles of coping, genetic predisposition, and epigenetic modifications (eg, secondary to childhood adversity). We then focus on several interlinked mediators occurring in MDD (or at least in subtypes of MDD) that may contribute to the medical comorbidity burden and to accelerated aging: limbic-hypothalamic-pituitary-adrenal axis alterations, diminution in glucocorticoid receptor function, altered glucose tolerance and insulin sensitivity, excitotoxicity, increases in intracellular calcium, oxidative stress, a proinflammatory milieu, lowered levels of “counter-regulatory” neurosteroids (such as allopregnanolone and dehydroepiandrosterone), diminished neurotrophic activity, and accelerated cell aging, manifest as alterations in telomerase activity and as shortening of telomeres, which can lead to apoptosis and cell death. In this model, MDD is characterized by a surfeit of potentially destructive mediators and an insufficiency of protective or restorative ones. These factors interact in increasing the likelihood of physical disease and of accelerated aging at the cellular level. We conclude with suggestions for novel mechanism-based therapeutics based on these mediators.
El trastorno depresivo mayor (TDM) tiene una alta frecuencia de asociación con el desarrollo de importantes comorbilidades médicas como enfermedad cardiovascular, accidentes vasculares, demencia, osteoporosis, diabetes y síndrome metabólico. Estas son patologías que de preferencia ocurren en la edad tardía de la vida y se ha propuesto que el TDM puede estar asociado con un “envejecimiento acelerado”. Se revisan algunos moderadores y mediadores que pueden acompañar al TDM y dar origen a estas condiciones médicas comórbidas. En primer lugar se revisan los efectos moderadores de los estilos psicológicos de adaptación, de la predisposición genética y de las modificaciones epigenéticas (por ejemplo, secundarias a la adversidad infantil). A continuación se revisan algunos mediadores interrelacionados que se presentan en el TDM (o al menos en algunos subtipos de TDM) que pueden incidir en la comorbilidad médica y en el envejecimiento acelerado: alteraciones del eje límbicohipotalámico-hipofisiario-adrenal, disminución de la función de los receptores de glucocorticoides, alteraciones en la tolerancia a la glucosa y en la sensibilidad a la insulina, excitotoxicidad, aumento del calcio intracelular, estrés oxidativo, un ambiente proinflamatorio, reducción de los niveles de neuroesteroides “contra-reguladores” (como alopregnanolona y dehidroepiandrosterona), disminución de la actividad neurotrófica y un envejecimiento celular acelerado que se manifiesta en alteraciones de la actividad de la telomerasa y acortamiento de los telómeros, lo que puede llevar a la apoptosis y la muerte celular. En este modelo, el TDM está caracterizado por un exceso de mediadores potencialmente destructores y una insuficiencia de los protectores o restauradores. Estos factores interactúan aumentando la posibilidad de enfermedad física y de un envejecimiento acelerado a nivel celular. Se concluye con propuestas de nuevas terapias basadas en los mecanismos que regulan estos mediadores.
Le trouble dépressif majeur (TDM) est associé à un taux élevé de comorbidités graves comme les pathologies card iovasculaires, les accidents vasculaires cérébrauxl (AVC), la démence, l'ostéoporose, le diabète et le syndrome métabolique. Ces pathologies surviennent habituellement tard dans la vie, et c'est pourquoi certains ont suggéré que le TDM pourrait être associé à un « vieillissement accéléré ». Dans cette revue, nous analysons plusieurs modérateurs et médiateurs pouvant accompagner le TDM et susceptibles de précipiter ces comorbidités. Tout d'abord, nous passons en revue les effets modérateurs des stratégies psychologiques d'adaptation (coping), des prédispositions génétiques et des modifications épigénétiques (par ex secondaires à des difficultés dans l'enfance). Nous nous consacrons ensuite à plusieurs médiateurs liés entre eux intervenant dans le TDM (ou au moins dans certains sous-types de TDM) qui pourraient contribuer à la charge comorbide et à l'accélération du vieillissement: modifications de l'axe limbo-hypophyso-hypothalamo-surrénalien, diminution de la fonction du récepteur des glucocorticoïdes, modification de la tolérance au glucose et de la sensibilité à l'insuline, excitotoxicité, augmentation du calcium intracellulaire, stress oxydatif, un milieu proinflammatoire, abaissement des taux des neurostéroïdes « contre-régulateurs » (comme l'alloprégnanolone et la déhydroépiandrostérone), diminution de l'activité neurotrophique et accélération du vieillissement cellulaire, se manifestant par des modifications de l'activité de la télomérase et par un raccourcissement des télomères, pouvant conduire à l'apoptose et à la mort cellulaire. Dans ce modèle, le TDM se caractérise par un excès de médiateurs potentiellement destructeurs et par une insuffisance de médiateurs protecteurs et restaurateurs. Ces facteurs interagissent en augmentant la probabilité de pathologie physique et de vieillissement accéléré à un niveau cellulaire. Nous concluons avec des suggestions concernant de mécanismes nouveaux pour des traitements s'appuyant sur ces médiateurs.
Major Depressive Disorder (MDD) is typically considered a mental illness, yet pathology associated with MDD is evident in cells and organs throughout the body. For example, MDD is associated with an increased risk of developing atherosclerosis, heart disease, hypertension, stroke, cognitive decline, and dementia (including Alzheimer's disease), osteoporosis, immune impairments (eg, “immunosenescence”), obesity, metabolic syndrome, insulin resistance, and type 2 diabetes,Citation1-Citation4 and individuals who are afflicted both by MDD and one of these diseases have a poorer prognosis than individuals afflicted by either alone.Citation3 This increased risk of serious medical diseases is not fully explained by lifestyle choices such as diet, exercise, and smoking, and the reasons for the heightened risk remain unknown.Citation4 Moreover, many of the medical comorbidities seen in MDD are diseases more commonly seen with advanced age, and MDD has even been characterized as a disease of “accelerated aging.”Citation1,Citation5,Citation6 In this review article, we explore certain biological mediators that are dysregulated in MDD and that may contribute to the depressed state itself, to the comorbid medical conditions, and to “accelerated aging.” Discovering novel pathological mediators in MDD could help identify new targets for treating depression and its comorbid medical conditions and could help reclassify MDD as a multisystem disorder rather than one confined to the brain.
Theoretical model
We propose a model of MDD comprised of certain pathogenic processes that are interlinked and often recursive, that occur in the brain and in the periphery, and that can culminate in cellular damage, cellular aging, and disease.Citation6-Citation10 This model is presented schematically in and is briefly described in this introduction; the individual moderators and mediators are described in greater detail in the remainder of this article. This model is not intended to be complete or all-encompassing but is meant to highlight and connect certain interesting findings in the study of depression. It does not propose that each component is necessary or sufficient, or that the specified mediators are the sole routes to MDD. It also does not speak to the directions of causality between depression and physical pathology. Further, many of the specified mediators may serve either protective or destructive functions depending on their context and chronicity.Citation11-Citation13 Nonetheless, the model presented here provides testable hypotheses for further investigation and provides rationales for considering novel treatment approaches. Earlier reviews of this model have been published elsewhere.Citation5,Citation6,Citation10
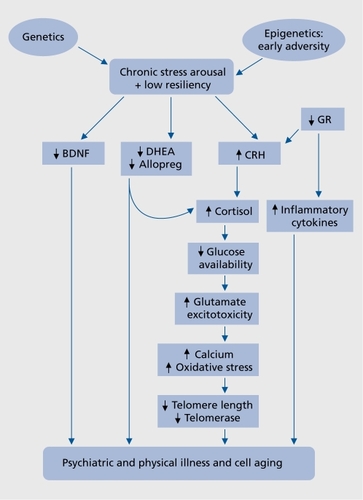
In brief, psychological and physical stressors trigger physiological responses that are acutely important for successful adaptation to the stress (“stress arousal”). However, when stress responses are disrupted or inappropriately prolonged, endangering effects may supersede the protective ones. The “cost” to the organism of maintaining these physiological responses over prolonged periods has been termed “allostatic load”Citation13 or “arousal pathology,”Citation14 and it has repeatedly been associated with poor medical outcomes.Citation12 In addition to chronicity of the stress response, certain psychological, environmental, genetic, and epigenetic circumstances (discussed below) favor dysregulation of two main stress response effectors, the limbic-hypothalamic -pituitaryadrenal (LHPA) axis and the locus coeruleus noradrenergic (NE) system.Citation15 A particular problem may arise when these two systems, which are generally counterregulatory, activate one another for prolonged periods of time (as may be seen in melancholic depression).Citation15 The failure of glucocorticoids (GCs) to effectively counter-regulate stress-induced NE and LHPA activity may underlie critical aspects of MDD.Citation15 Prolonged LHPA axis dysregulation can lead to neuroendangering or neurotoxic effects in vulnerable brain regions (eg, prefrontal cortex and hippocampus).Citation16 It can also lead to energetic disturbances (decreased intracellular glucose availability and insulin resistance), glutamatergic hyperactivity/excitotoxicity, increased intracellular calcium concentrations, mitochondrial damage, free radical generation and oxidative stress, immune alterations (leading to a proinflammatory milieu), and accelerated cell aging (via effects on the telomere/telomerase maintenance system). The nature of cortisol abnormalities in MDD is complex, however, and will be discussed below. Prolonged activation of central NE systems, as often seen in melancholic depression, may be associated with worsened outcome in cardiovascular diseases and with accelerated cell aging at the level of the telomere.Citation9,Citation15
In addition to increases in these destructive processes, normal compensatory or reparative processes may be diminished, eg, diminution of counter-regulatory neurosteroids, eg, dehydroepiandrosterone (DHEA),Citation17 and allopregnanolone,Citation18 decreased antioxidant compounds, diminished anti-inflammatory/immunomodulatory cytokines, decreased neurotrophic factor concentrations, eg, brainderived neurotrophic factor (BDNF), and altered telomerase activity. This juxtaposition of enhanced destructive processes with diminished (or inadequate) protective or restorative ones can culminate in cellular damage and physical disease (Table I). This model will be explored in greater depth in the following sections.
Table I. Possibly damaging and protective mediators in major depression LHPA, limbic-hypothalamic-pituitary-adrenal; DHEA, dehydroepiandrosterone; BDNF, brain-derived neurotrophic factor. * Evidence is mixed as to whether DHEA concentrations are elevated or lowered in depression. ** Evidence is mixed as to whether the anti-inflammatory/immunomodulatory cytokine, IL10, is elevated or lowered in depression. *** Evidence is mixed as to whether telomerase activity is elevated or lowered in states of chronic stress and depression.
Moderators
Psychological stress and individual differences
Psychological stress is frequently a precipitant of depressive episodes,Citation19 and under certain circumstances it can initiate the biochemical cascade described here.Citation7,Citation8,Citation10,Citation13,Citation16,Citation20 It is apparent, though, that individuals respond very differently to stress, due, in part, to differences in coping strategies, disposition, temperament, and cognitive attributional styles.Citation21-Citation23 These can moderate stress-associated biological changes such as LHPA axis arousal,Citation23 inflammation,Citation22,Citation24 neurogenesis,Citation25 amygdala arousal,Citation26 and cell aging. In the first study examining a personality trait and telomere length, O'Donovan et al found that pessimism was related to shorter telomere length, as well as higher IL-6 concentrations.Citation22 In a study of the effects of early-life parental loss on later-life depression, the quality of the family and home's adaptation to the loss was the single most power-ful predictor of adult psychopathology, and was more important than the loss itself.Citation27 Biochemical aspects of resilience vs stress vulnerability will not be covered here but have recently been reviewed.Citation28
Adverse childhood events
Alexander Pope noted in 1734, that “as the twig is bent, the tree is inclined.” A rapidly expanding body of evidence suggests that early-life adversity (such as parental loss, neglect, and abuse) predisposes to adult depressionCitation27,Citation29 as well as to LHPA axis hyper-reactivity to stress,Citation27,Citation30 increased allostatic load,Citation13,Citation31 diminished hippocampal volume (although this is controversial),Citation32 lower brain serotonin transporter binding potential,Citation33 and a myriad of adult physical diseases.Citation34 Childhood adversity also predisposes to alterations in many of the mediators presented in our model of stress/depression/illness/cell aging, such as: inflammation,Citation35,Citation36 oxidative stress,Citation37 neurotrophic factors,Citation38 neurosteroids,Citation39 glucose/insulin/ insulin-like growth factor (IGF-1) regulation,Citation40 telomerase activity,Citation41 and telomere length.Citation36,Citation42-Citation44 Alterations in LHPA axis activity (increased or decreased) have been well described in victims of childhood adversity, even when the individuals are not currently depressed.Citation27,Citation30 In fact, several instances of neurobiological changes reported in MDD may be more attributable to histories of early-life adversity,Citation30,Citation32 which are over-represented among individuals with MDD, than to the MDD itself. Thus, early-life adversity seems capable of “reprogramming” the individual to a certain lifetime repertoire of altered physiological responses to stress and to vulnerability to psychiatric and physical illness. This reprogramming toward stress arousal and preparedness may be adaptive when the individual is likely to be confronted with a lifetime of continuous adversity, but is clearly disadvantageous otherwise. The causes of early adversity-induced behavioral and biochemical changes, and the explanation for the very long-lasting effects of such adversity, are the subject of intense investigation. One explanation that has attracted much attention is epigenetic changes,Citation45,Citation46 discussed in the next section.
Genetic and epigenetic moderators
A number of variants in candidate genes have been implicated in contributing to maladaptive and resilient responses that underlie alterations in neuronal plasticity and subsequent behavioral depression.Citation47 Evidence is strongest for genes involved in HPA regulation and stress (corticotrophin-releasing hormone [CRH]1; glucocorticoid receptor [GR]), regulatory neurotransmitters, transporters, and receptors (serotonin (5-HT)1A, 5HT2, 5-HTTLPR, NET), neurotrophic factors, (brain-derived neurotrophic factor [BDNF], nuclear factor-kappaB, mitogen-activated protein kinase-1) and transcription factors (cAMP response element binding, Re-1 silencing transcription factor, delta FosB), but variations in other secondary modulatory factors (g-aminobutyric acid [GABA], catechol-O-methyl transferase, monoamine oxidase, dynorphin, neuropeptide-Y) have also been hypothesized to be important in determining individual differences in stress response.Citation48 Studies of the CRH-1 gene in humans, for example, have shown that specific variants are associated with differential hormonal responses to stress, and with differing rates of depression and suicidal behavior.Citation49 Increasingly, such genetic effects have themselves been found to be modulated by individual variation in environmental context and history (gene x environment, GxE).Citation45 Epigenetics, which focuses on nongenomic alterations of gene expression, provides a mechanism for understanding such findings, through alteration of DNA methylation and subsequent silencing of gene expression or through physical changes in DNA packaging into histones.Citation50 A comprehensive review of this literature is beyond the scope of this article, but the findings of selective recent studies in these areas are illustrative of the regulatory complexity that influences the possible translation of stressful experiences into depression. Long-lasting epigenetic effects of early life experience on hypothalamicpituitary-adrenal (HPA) responses have been demonstrated most clearly in animal models of differential maternal behavior and social isolation.Citation45,Citation46 Augmented maternal care was associated with reduced hypothalamic response to stress in rat pups and altered expression of CRH into adulthood.Citation51 Suggestive human data compatible with these mechanisms have been reported.Citation28,Citation52 Oberlander et al,Citation53 for example, found that prenatal exposure to third trimester maternal depression was associated with increased methylation of the glucocorticoid receptor gene at 3 months of age in the newborn child, while McGowanCitation45 reported decreased levels of GR expression in the hippocampus of suicide victims with a history of childhood abuse, in comparison with those without such history and to controls. Tyrka and colleaguesCitation54 have also shown that variants in the CRH1 receptor gene appear to interact with a history of childhood abuse in determining cortical response to CRH. A separate body of research has focused on genetic investigations in components of serotonergic function, most commonly on a variant in the serotonin promoter (5HTTLPR), and, to a lesser extent, on serotonin receptor genes.Citation55 In a small-scale study that remains controversial, Caspi et alCitation56 reported that the effect of a variant in 5HTTLPR on increasing risk of depression was dependent upon a history of previous life stresses; several large-scale attempts at replication failed to support these conclusions and subsequent meta-analyses have been both positive and negative.Citation57,Citation58 Ressler et alCitation59 have suggested that gene x gene x environment interactions may be involved, and reported that 5-HTTLPR alleles interacted with CRH1 haplotypes and child abuse history in predicting depressive symptoms. Others, however, have found it hard to demonstrate such effects.Citation55 Yet another example of a potential GxGxE interaction was found in a study by Kauffman et alCitation60 of child abuse victims, in whom BDNF and 5-HTTLPR genotypes interacted with maltreatment history in predicting depression, with social support showing some moderating influence. Despite the persuasive empirical animal data, the clinical relevance of epigenetic effects of stress on human emotional behavior is yet to be convincingly established.
Biochemical mediators
Glucocorticoids
Elevated circulating GC levels are often observed in depressed individuals (especially in those with severe, melancholic, psychotic, or inpatient depressions), although considerable variability exists between studies, between individuals, and even within individuals over time, and some individuals are even hypocortisolemic.Citation61,Citation62 The physiological significance of increased circulating GC levels remains unknown, and it is debatable whether hypercortisolemia results in hypercortisolism at the cellular level, or, rather, in hypocortisolism, perhaps due to downregulation of the GR (often referred to as “GC resistance”).Citation20 Thus, determination of “net” GC activity in depressed individuals at the intracellular level has remained elusive.Citation63,Citation64 In fact, different subclasses of depressed individuals may show opposite patterns of limbic-hypothalamic-pituitary-adrenal (LHPA) axis activity,Citation15 and levels of LHPA activation may be more related to individual depressive symptoms than to the depressive syndrome per se.Citation65 Further, it is possible that both hypo- and hypercortisolism are related to depression, in an inverted-U shaped manner.Citation62 Complicating our understanding of this issue, novel treatment strategies that decrease or increase GC activity may show antidepressant effects in certain patients.Citation66-Citation69 The “hypocortisolism” hypothesis is supported by findings that proinflammatory cytokine levels (eg, tumor necrosis factor [TNF]-a, interleukin [IL]-1ß and IL-6) tend to be increased in the serum of depressed patients, and that proinflammatory cytokines may contribute to depressive symptomatology. Since cortisol typically has anti-inflammatory actions and suppresses proinflammatory cytokines (although there are instances to the contrary [eg, ref 70]), the coexistence of elevated cortisol and elevated proinflammatory cytokine levels suggests an insensitivity to cortisol at the level of the lymphocyte GR.Citation20 Further supporting this notion, inflammatory cytokines downregulate GRs.Citation20 Also, antidepressants typically increase GR binding activity,Citation20 although in so doing, negative feedback onto the HPA axis is increased.Citation71 On the other hand, the “hypercortisolism” hypothesis is supported by certain phenotypic somatic features suggestive of cortisol excess and end-organ cortisol receptor overactivation in some individuals with depression, eg, osteoporosis, insulin resistance, type 2 diabetes, a relative hypokalemic alkalosis accompanied by neutrophilia and lymphocytosis, hypertension, metabolic syndrome, and visceral/intra-abdominal adiposity.Citation72,Citation73 Further support of net GC overactivation is provided by evidence of altered expression of target genes such as BDNF, which are believed to be under negative regulatory control by cortisol.Citation74
Pathologically elevated or diminished GC activity might have adverse neurobehavioral and physical health sequellae.Citation72,Citation75 Chronic hypercortisolemia, in particular, has been proposed by Sapolsky and othersCitation16 to result in a biochemical “cascade,” which can culminate in cell endangerment or cell death in certain cells, including cells in the hippocampus. In the simplest description of this model, GC excess engenders a state of intracellular glucoprivation (insufficient intracellular glucose energy stores) in certain cells, impairing the ability of glia and other cells to clear synaptic glutamate. The resulting excitotoxicity results in excessive influx and release of calcium into the cytoplasm, which contributes to oxidative damage, proteolysis, and cytoskeletal damage. Unchecked, these processes can culminate in diminished cell viability or cell death.
Neurosteroids
Although cortisol concentrations are often reported as elevated in depression, CSF concentrations of the potent GABA-A receptor agonist neurosteroid, allopregnanolone, are decreased in unmedicated depressives, and CSF levels of allopregnanolone increase with treatment in direct proportion to the antidepressant effect.Citation76 Selective serotonin reuptake inhibitor (SSRI) antidepressants rapidly increase allopregnanolone synthesis, and this may contribute to their anxiolytic effects.Citation77,Citation78 Another neurosteroid, DHEA, which may have “anticortisol” effects, has been reported to be both high and low in depression.Citation17 Notably, both of these neurosteroids modulate HPA axis activityCitation17,Citation18 and immune system activity,Citation17,Citation79 antagonize oxidative stressCitation17,Citation80 and have certain neuroprotective effects.Citation17,Citation81 Depressed patients entering remission show decreases in plasma cortisol concentrations along with increases in plasma allopregnanolone concentrations.Citation82 Endogenous decreases in this neurosteroid concentrations or exogenously produced increases in their concentrations might be expected to have damaging or beneficial effects, respectively, in the context of depression,Citation17,Citation78,Citation83,Citation84 and treatment trials have demonstrated significant antidepressant effects of exogenously administered DHEA.Citation17 Animal models suggest that 3 a hydroxy-5 a reduced steroids (allopregnanolone and allotetrahydrodeoxycorticosterone) are responsive to stressCitation85 and may function to restore normal g-aminobutyric acid (GABA)-ergic and hypothalamicpituitary-adrenal function following stress.Citation18,Citation85 In vitro, allopregnanolone suppresses release of gonadotropinreleasing hormoneCitation86 or CRHCitation87 via a GABA-A mediated mechanism. Allopregnanolone or allotetrahydrodeoxycorticosterone can also attenuate stress-induced increases in plasma ACTH and corticosterone and can affect arginine vasopression transcription in the hypothalamus (paraventricular nucleus).Citation18 Under chronic stress or in psychiatric disorders, dysregulation of the HPA axis could be exacerbated if there is insufficient activity of these “counter-regulatory” neurosteroids. In addition to protection against acute or chronic stress, neurosteroids such as allopregnanolone and allotetrahydrodeoxycorticosterone may be neuroprotective against early life stressorsCitation88 or against deleterious effects of social isolation.Citation89 In this way, these neurosteroids may be neuroprotective during development and may affect future responsiveness to stress.
The detrimental effects of neurosteroid dysregulation on stress responses has been particularly documented in women with premenstrual dysphoric disorder (PMDD).Citation90,Citation91 PMDD is a depressive disorder that is characterized by cyclic recurrence, during the luteal phase of the menstrual cycle, of a variety of physical and emotional symptoms that are so severe as to interfere with daily activities. In these studies in women with PMDD, both high and low concentrations of allopregnanolone during the luteal phase of the menstrual have been reported. However, women with PMDD have reduced responsiveness to neurosteroids (on GABA-A receptors)Citation92 as well as a blunted stress response (failing to demonstrate an increase in allopregnanolone concentrations after acute stress) in women with PMDD and a prior history of depression.Citation90 Furthermore, women with PMDD who also had prior histories of depression showed significant decreases in allopregnanolone after acute stress.Citation90 These data highlight that long-term histories of depression may be associated with persistent, long-term effects on the responsivity of the neurosteroid system, as well as long-term effects on modulation of the HPA axis following stress.
Glucose and insulin regulation
Abnormalities of glucose homeostasis (eg, insulin resistance and impaired glucose tolerance) are seen in MDD, even in individuals who are nonobese and not diabetic.Citation93 These glucose and insulin abnormalities are most pronounced in hypercortisolemic depressed individuals,Citation94 as would be predicted based on cortisol's well-known antiinsulin effects. Hypercortisolemic depressives, compared with normocortisolemic ones, are also at increased risk of having increased abdominal (visceral) fat depositionCitation95 and the metabolic syndrome,Citation96 which are also risk factors for cardiovascular disease. Insulin resistance and diminished cellular glucose uptake can also lead to a dangerous “energetic crisis.”Citation7,Citation16 When this occurs in the hippocampus,Citation16 for example, hippocampal excitotoxicity may develop, since there is insufficient energy available to clear glutamate from the synapse. Thereafter, cytosolic calcium is mobilized, triggering oxygen free radical formation and cytoskeletal proteolysis. The relevance of this in humans was demonstrated in a PET scan study, in which cortisol administration to normal individuals resulted in significant reductions in hippocampal glucose utilization.Citation97 The importance of hippocampal insulin resistance for depression and cognitive disorders (eg, Alzheimer's disease) is the subject of active investigation.Citation98,Citation99
Over and above these direct effects on energy balance, prolonged exposure to glucose intolerance and insulin resistance is associated with accelerated biological agingCitation7,Citation100 including shortened telomere length,Citation101 and visceral adiposity is associated with increased inflammation and oxidation,Citation102,Citation103 both of which, themselves, promote accelerated biological aging.Citation7 These will be further discussed below in the sections on inflammation, oxidation, and cell aging.
Immune function
Dysregulation of the LHPA axis contributes to immune dysregulation in depression, and immune dysregulation, in turn, can activate the HPA axis and precipitate depressive symptoms.Citation20 Immune dysregulation may be an important pathway by which depression heightens the risk of serious medical comorbidity.Citation7,Citation104,Citation105 Several major proinflammatory cytokines, such as IL-1ß, IL-2, IL-6 and TNF-a, are elevated in depression, either basally or in response to mitogen stimulation or acute stress.Citation20,Citation106,Citation107 Conversely, certain anti-inflammatory or immunomodulatory cytokines, such as IL-1 receptor antagonist and IL10 may be decreased or dysregulated.Citation106 Indeed, the ratio of proinflammatory to anti-inflammatory/ immunomodulatory cytokines may be disturbed in depression and could result in net increased inflammatory activityCitation106 as well as in oxidative stress.Citation108 Converging findings suggest that high peripheral levels of inflammatory cytokines, such as IL-6, are associated with the activation of central inflammatory mechanisms that can adversely affect the hippocampus, where IL-6 receptors are abundantly expressed.Citation109 High proinflammatory cytokine levels, for example, may directly contribute to depression, decreased neurotrophic support, and altered glutamate release/reuptake and hippocampal neurodegeneration,Citation110 and, plasma IL-6 levels are inversely correlated with hippocampal gray matter in healthy humans.Citation111 Further, inappropriately and chronically elevated proinflammatory cytokines can contribute to accelerated biological aging (eg, premature shortening of immune cell telomeresCitation112). Interestingly, the development of immunosenescence (eg, the loss of the CD28 marker from CD8+ T cells), can further aggravate the proinflammatory milieu, since CD8+CD28- cells hypersecrete IL-6.Citation113 It should be noted, however, that due to the complexity of cytokine actions in neurons and glia, the end effect of individual cytokines may be either detrimental or protective, depending on the circumstances.Citation106
Oxidation
Stress and increased LHPA axis activity can also increase oxidative stress and decrease antioxidant defenses.Citation5 ,Citation7,Citation114 Oxidative stress often increases with aging and various disease states, while antioxidant and antiinflammatory activities decrease, resulting in a heightened likelihood of cellular damage and of a senescent phenotype.Citation7,Citation115 The co-occurrence of oxidative stress and inflammation (the so-called “evil twins” of brain agingCitation115), as may be seen in depression, post-traumatic stress disorder (PTSD), stroke, Alzheimer's disease, and others, can be especially detrimental. Oxidative stress occurs when the production of oxygen free radicals (and other oxidized molecules) exceeds the capacity of the body's antioxidants to neutralize them. Oxidative stress damages DNA, protein, lipids, and other macromolecules in many tissues, with telomeres (discussed below) and the brain being particularly sensitive. Elevated plasma and/or urine oxidative stress markers (eg, increased F2-isoprostanes and 8-hydroxydeoxyguanosine [8-OHdG], along with decreased antioxidant compounds, such as Vitamin C, Vitamin E, and Coenzyme Q) have been reported in individuals with depression and in those with chronic psychological stress, and the concentration of peripheral oxidative stress markers is positively correlated with the severity and chronicity of depression.Citation114,Citation116 Further, the ratio of serum oxidized lipids (F2-isoprostanes) to antioxidants (Vitamin E) is directly related to psychological stress.Citation8 Importantly, this ratio (and the ratio of F2-isoprostanes to another antioxidant, Vitamin C) is inversely related to telomere length in chronically stressed caregiversCitation8 and in individuals with major depression.Citation117 Oxidative stress markers are also correlated with decreased telomerase activity.Citation118 Further, diminished levels of antioxidants reportedly lower BDNF activity.Citation119 Interestingly, antidepressants decrease oxidative stress.Citation120 Since cellular oxidative damage may be an important component of the aging process, prolonged or repeated exposure to oxidative stress might accelerate aspects of biological aging and promote the development of aging-related diseases in depressed individuals.Citation114 It is unknown whether antioxidant treatment would retard stress- or depressionrelated aging; this is discussed below under “novel treatment implications.”
Brain-derived neurotrophic factor
The “neurotrophic model” of depressionCitation74 emphasizes the centrality of neurogenesis and neuronal plasticity in the pathophysiology of depression. It posits that diminished hippocampal BDNF activity, caused by stress or excessive GCs, impairs the ability of stem cells in the subgranular zone of the dentate gyrus (as well as cells in the subventricular zone, projecting to the prefrontal cortex) to remain viable and to proliferate into mature cells. It is not known whether such effects can cause depression, but they may be relevant to the mechanism of action of antidepressant treatments.Citation121 Unmedicated patients with depression have decreased hippocampal (at autopsy) and serum concentrations of BDNFCitation121,Citation122 Over 20 studies have documented decreased serum concentrations of BDNF in unmedicated depressed individuals; this is now one of the most consistently replicated biochemical findings in major depression.Citation121,Citation123 Further, serum BDNF concentrations increase with antidepressant treatment.Citation121,Citation123 The relationship of peripheral BDNF concentrations to central ones is not known, but even peripherally administered BDNF abrogates depressive and anxiety-like behaviors and increases hippocampal neurogenesis in mice, suggesting that serum BDNF concentrations are functionally significant for brain function and are more than merely a biomarker.Citation124 A role of BDNF in antidepressant mechanisms of action is supported by findings that hippocampal neurogenesis (in animals) and serum BDNF concentrations (in depressed humans) increase with antidepressant treatment,Citation121,Citation123 and that hippocampal neurogenesis and intact BDNF expression are required for behavioral effects of antidepressants in animals.Citation125,Citation126
Apart from its direct neurotrophic actions, BDNF also has anti-inflammatory and antioxidant effects that may contribute to its neuroprotective efficacy,Citation127 and BDNF, in concert with telomerase (discussed below) promotes the growth of developing neurons.Citation128 In addition, BDNF (despite its name) has significant peripheral actions that are important for physical health, and the low levels of BDNF seen in MDD may be involved in certain comorbid illnesses such as cardiovascular disease, diabetes, obesity, and metabolic syndrome.Citation129 For example, BDNF improves glucose and lipid profiles, enhances glucose utilization, suppresses food intake, has an insulinotropic effect and protects cells in the islets of Langerhans (reviewed in ref 129). Plasma levels of BDNF are low in type 2 diabetes and are inversely correlated with fasting glucose levels.Citation129 Indeed, BDNF is increasingly considered not only a neurotrophin but a metabotrophin,Citation129 and its dysregulation has been proposed as a unifying feature of several clustered conditions, such as MDD, Alzheimer's disease, and diabetes.Citation130
Cell aging: telomeres and telomerase
Telomeres are DNA-protein complexes that cap the ends of linear DNA strands, protecting DNA from damage.Citation131 When telomeres reach a critically short length, as may happen when cells undergo repeated mitotic divisions in the absence of adequate telomerase (eg, immune cells and stem cells, including neurogenic stem cells in the hippocampus), cells become susceptible to apoptosis and death. Even in nondividing cells, such as mature neurons, telomeres can become shortened by oxidative stress, which preferentially damages telomeres to a greater extent than nontelomeric DNA. This nonmitotic type of telomere shortening also increases susceptibly to apoptosis and cell death. Telomere length is a robust indicator of “biological age” (as opposed to just chronological age) and may represent a cumulative log of the number of cell divisions and a cumulative record of exposure to genotoxic and cytotoxic processes such as oxidation.Citation7-Citation9,Citation113,Citation131,Citation132 Telomere length may also represent a biomarker for assessing an individual's cumulative exposure to, or ability to cope with, depression or stressful conditions. For example, chronically stressedCitation8,Citation9 or depressedCitation133-Citation135 individuals show premature leukocyte telomere shortening, a sign of cellular aging. In the former study, telomere length was inversely correlated with perceived stress and with cumulative duration of caregiving stress.Citation8 The estimated magnitude of the acceleration of biological aging in these studies was not trivial; it was estimated as approximately 9 to 17 additional years of chronological aging in the stressed caregivers and approximately 6 to 10 years in the depressed individuals. Preliminary data from our group suggest that telomere loss in MDD is most apparent in those individuals with more chronic courses of depression,Citation117 but another study did not observe that.Citation135 Interestingly, individuals with histories of early-life adversity or abuse also have shortened leukocyte telomeres.Citation36,Citation42,Citation43 Since individuals with MDD are more likely to have experienced earlylife adversity, it remains to be determined how much of the telomere shortening seen in studies of MDD relate to the MDD per se vs the histories of early-life adversity. In individuals with post-traumatic stress disorder (PTSD), telomere shortening was more closely linked to adverse childhood events than to the PTSD per se.Citation44
The importance of accelerated telomere shortening for understanding comorbid medical illnesses and premature mortality in depressed individuals is highlighted by multiple studies in nondepressed populations showing significantly increased medical morbidity and earlier mortality in those with shortened telomeres.Citation7,Citation136 For example, shortened leukocyte telomeres are associated with a greater than 3-fold increase in the risk of myocardial infarction and stroke and with a greater than 8-fold increase in the risk of death from infectious disease.Citation137 Thus, cell aging (as manifest by shortened telomeres), may provide a conceptual link between depression and its associated medical comorbidities and shortened life span.Citation7,Citation104,Citation132 The causes of accelerated telomere loss in MDD are not known, but they may include chronic exposure to inflammation and oxidation, both of which are commonly seen in MDD and both of which are associated with telomere shortening. In our own studies, telomere length in MDD was inversely correlated with inflammation (IL-6 concentrations) and oxidative stress (the F2-isoprostane/ Vitamin C ratio).Citation117
Telomere length is determined by the balance between telomere shortening stimuli (eg, mitotic divisions and exposure to inflammation and oxidation) and telomere lengthening or reparative stimuli. A major enzyme responsible for protecting, repairing, and lengthening telomeres is telomerase, a ribonucleoproptein enzyme that elongates telomeres, thereby counteracting telomere shortening and maintaining cellular viability.Citation131 Telomerase may also have antiaging or cell survival-promoting effects independent of its effects on telomere length by regulating transcription of growth factors, synergizing with the neurotrophic effects of BDNF, having antioxidant effects and intrinsic antiapoptotic effects, protecting cells from necrosis, and stimulating cell growth in adverse conditions (eg, ref 128). In one study in which telomere shortening was observed, telomerase activity
was significantly diminished in stressed (generally nondepressed) caregiversCitation8 but, in another caregiver study (in which caregivers were more depressed than controls), telomerase activity was significantly increased.Citation138 We recently found that telomerase activity was significantly increased in unmedicated depressed individuals.Citation139 It is possible that increased telomerase activity, in the face of shortened telomeres, is an attempted compensatory response to telomere shortening.Citation138,Citation139
Pointing to the inter-relatedness of several of the mediators considered in this review, telomerase activity can be down-regulated by cortisol,Citation140 tumor necrosis factor (TNF)-a and certain growth factors, and upregulated by IL-6 and certain other inflammatory cytokines, insulinlike growth factor-1, fibroblast growth factor-2, vascular endothelial growth factor, estrogen, and others.Citation141
Novel treatment implications
To the extent the biochemical mediators we have described are pathophysiologically involved in MDD and its medical comorbidities, new classes of treatments should be considered, and certain noncanonical mechanisms of action of traditional antidepressants should be emphasized in new drug development. Some of these novel approaches are already under investigation, while others remain to be tested. In Table II, we list certain traditional and nontraditional, but mechanism-based, interventions that may ameliorate the biochemical mediators we have discussed. These interventions range from purely behavioral (eg, exercise and improved fitness, environmental enrichment, yoga and meditation, dietary macronutrient modifications and calorie restriction) (see refs 7,142-144 for description of these behavioral approaches) to more purely medication-based (see ref 145 for additional descriptions of novel biological mechanism-based therapeutics). For example, early work suggests the promise, at least in certain patients, of antiglucocorticoids,Citation67-Citation69 DHEA supplementation,Citation17 insulin receptor sensitizers,Citation99,Citation146 glutamate antagonists,Citation147 calcium blockers,Citation148 anti-inflammatories,Citation149 antioxidants,Citation150 increased BDNF delivery to the brain, Citation124,Citation151 and, most speculatively, telomerase enhancers.Citation152,Citation153
Table II. Potential mechanism-based therapeutic interventions. LHPA, limbic-hypothalamic-pituitary-adrenal; GC, glucocorticoid; GR, glucocorticoid receptor; CRH, corticotrophin-releasing hormone; DHEA, dehydroepiandrosterone; BDNF brain-derived neurotrophic factor; TNF, tumor necrosis factor; SSRI, selective serotonin reuptake inhibitor
Summary: is depression accompanied by accelerated aging?
We began this review article by noting that depressed individuals are at increased risk of developing physical illnesses more commonly seen with aging. It remains unknown whether MDD and these medical conditions are causally related. This determination will be important in considering whether primary treatment of the depression (eg, with antidepressant medications or psychotherapy) should additionally treat some of the medical comorbidities (and vice versa) or whether the biochemical mediators that are common to both conditions (eg, inflammation and oxidation) should be a primary treatment focus. We also discussed the potent influence that early-life adversity can have on the subsequent development of depression and medical comorbidities. We noted that many of the biochemical mediators are linked to others, and that there are many examples of bidirectional influence. Finally, we postulated that certain of these mediators have the potential to accelerate cellular aging at the level of DNA. In any event, is important to recognize that MDD may be biologically heterogeneous, and this model may apply only to certain subsets of patients with MDD. This reconceptualization of MDD as a constellation of biochemical features conducive to physical as well as mental distress places MDD firmly in the taxonomy of physical disease and points to new types of treatment.
Selected abbreviations and acronyms
| 5-HT | = | serotonin |
| BDNF | = | brain-derived neurotrophic factor |
| CRH | = | corticotrophin-releasing hormone |
| DHEA | = | dehydroepiandrosterone |
| GC | = | glucocorticoid |
| GR | = | glucocorticoid receptor |
| IL | = | interleukin |
| LHPA | = | limbic-hypothalamic-pituitary-adrenal axis |
| MDD | = | major depressive disorder |
| PMDD | = | premenstrual dysphoric disorder |
We acknowledge the valuable intellectual input into this manuscript by Dr Elissa S. Epel, who coauthored previous reviews of an earlier model. We also thank her and Drs Elizabeth H. Blackburn, Jue Lin, Firdaus S. Dhabhar, Yali Su, Steve Hamilton, and J. Craig Nelson for their valued collaboration on our studies of cell aging in depression. This study was funded by an NIMH R01 grant (R01 MH083784), a grant from the O'Shaughnessy Foundation and grants from the UCSF Academic Senate and the UCSF Research Evaluation and Allocation Committee (REAC). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. None of the granting or funding agencies had a role in the preparation, review, or approval of the manuscript.
Portions of this paper are based on a prior review article: Wolkowitz O, Epel ES, Reus VI, Mellon S. Depression gets old fast: do stress and depression accelerate cell aging? Depress Anxiety. 2010;27:327-338.
Financial disclosures: Drs Owen Wolkowitz and Synthia Mellon, along with Drs Elizabeth Blackburn, Elissa Epel, and Jue Lin, on behalf of the Regents of the University of California (who will be assignees of the patent), have applied for a patent covering the use of cell aging markers (including telomerase activity) as a biomarker of depression.
Related Research Data
REFERENCES
- HeuserI.Depression, endocrinologically a syndrome of premature aging?Maturitas.200241(suppl 1)S19S2311955792
- McIntyreRS.SoczynskaJK.KonarskiJZ.et al.Should depressive syndromes be reclassified as "metabolic syndrome type II"?Ann Clin Psychiatry.20071925726418058283
- EvansDL.CharneyDS.LewisL.et al.Mood disorders in the medically ill: scientific review and recommendations.Biol Psychiatry.20055817518916084838
- SchulzR.BeachSR.IvesDG.MartireLM.AriyoAA.KopWJ.Association between depression and mortality in older adults: the Cardiovascular Health Study.Arch Intern Med.20001601761176810871968
- WolkowitzOM.EpelES.MellonS.When blue turns to grey: do stress and depression accelerate cell aging?World J Biol Psychiatry.200892518273736
- WolkowitzOM.EpelES.ReusVI.MellonSH.Depression gets old fast: do stress and depression accelerate cell aging?Depression and Anxiety.20102732733820376837
- EpelES.Psychological and metabolic stress: a recipe for accelerated cellular aging?Hormones (Athens).2009872219269917
- EpelES.BlackburnEH.LinJ.et al.Accelerated telomere shortening in response to life stress.Proc Natl Acad Sci USA.2004101173121731515574496
- EpelES.LinJ.WilhelmFH.et al.Cell aging in relation to stress arousal and cardiovascular disease risk factors.Psychoneuroendocrinology.20063127728716298085
- WolkowitzOM.EpelES.ReusVI.Stress hormone-related psychopathology: pathophysiological and treatment implications.World J Biol Psychiat.20012115143
- SelyeH.The Stress of Life. New York, NY: McGraw-Hill.1956
- SeemanT.McEwenBS.RoweJ.SingerB.Allostatic load as a marker of cumulative biological risk: MacArthur studies of successful aging.Proc Natl Acad Sci USA.200194770477511287659
- McEwenB.Allostasis and allostatic load: implications for neuropsychopharmacology.Neuropsychopharmacology.20002210812410649824
- SchulkinJ.McEwenBS.GoldPW.Allostasis, amygdala, and anticipatory angst.Neurosci Biobehav Rev.1994183853967984356
- WongML.KlingMA.MunsonPJ.et al.Pronounced and sustained central hypernoradrenergic function in major depression with melancholic features: relation to hypercortisolism and corticotropin-releasing hormone.Proc Natl Acad Sci USA.20009732533010618417
- SapolskyRM.The possibility of neurotoxicity in the hippocampus in major depression: a primer on neuron death.Biol Psychiatry.20004875576511063972
- ManingerN.WolkowitzOM.ReusVI.EpelES.MellonSH.Neurobiological and neuropsychiatric effects of dehydroepiandrosterone (DHEA) and DHEA sulfate (DHEAS).Front Neuroendocrinol.200930659119063914
- PatchevVK.HassanAH.HolsboerDF.AlmeidaOF.The neurosteroid tetrahydroprogesterone attenuates the endocrine response to stress and exerts glucocorticoid-like effects on vasopressin gene transcription in the rat hypothalamus.Neuropsychopharmacology.1996155335408946427
- KendlerK.KarkowskiLM.PrescottCA.Causal relationship between stressful life events and the onset of major depression.Am J Psychiatry.199915683784110360120
- RaisonCL.MillerAH.When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders.Am J Psychiatry.20031601554156512944327
- FolkmanS.LazarusRS.Dunkel-SchetterC.DeLongisA.GruenRJ.Dynamics of a stressful encounter: cognitive appraisal, coping, and encounter outcomes.J Pers Soc Psychol.19865099210033712234
- O'DonovanA.LinJ.DhabharFS.et al.Pessimism correlates with leukocyte telomere shortness and elevated interleukin-6 in post-menopausal women.Brain Behav Immun.20092344644919111922
- DensonTF.SpanovicM.MillerN.Cognitive appraisals and emotions predict cortisol and immune responses: a meta-analysis of acute laboratory social stressors and emotion inductions.Psychol Bull.200913582385319883137
- StowellJR.Kiecolt-GlaserJK.GlaserR.Perceived stress and cellular immunity: when coping counts.J Behav Med.20012432333911523331
- LyonsDM.BuckmasterPS.LeeAG.et al.Stress coping stimulates hippocampal neurogenesis in adult monkeys.Proc Natl Acad Sci USA.2010107148231482720675584
- TaylorSE.BurklundLJ.EisenbergerNI.et al.Neural bases of moderation of cortisol stress responses by psychosocial resources.J Pers Soc Psychol.20089519721118605860
- BreierA.Kelsoe JRJr.KirwinPD.et al.Early parental loss and development of adult psychopathology.Arch Gen Psychiatry.1988459879932972265
- FederA.NestlerEJ.CharneyDS.Psychobiology and molecular genetics of resilience.Nat Rev Neurosci.20091044645719455174
- ChapmanDP.WhitfieldCL.FelittiVJ.et al.Adverse childhood experiences and the risk of depressive disorders in adulthood.J Affect Disord.20048221722515488250
- HeimC.PlotskyPM.NemeroffCB.Importance of studying the contributions of early adverse experience to neurobiological findings in depression.Neuropsychopharmacology.20042964164815034558
- Grassi-OliveiraR.AshyM.SteinLM.Psychobiology of childhood maltreatment: effects of allostatic load?Rev Bras Psiquiatr.200830606818373020
- VythilingamM.HeimC.NewportJ.et al.Childhood trauma associated with smaller hippocampal volume in women with major depression.Am J Psychiatry.20021592072208012450959
- MillerJM.KinnallyEL.OgdenRT.et al.Reported childhood abuse is associated with low serotonin transporter binding in vivo in major depressive disorder.Synapse.20096356557319288578
- AndaRF.FelittiVJ.BremnerJD.et al.The enduring effects of abuse and related adverse experiences in childhood: A convergence of evidence from neurobiology and epidemiology.Eur Arch Psychiatry Clin Neurosci.200625617418616311898
- PaceTW.MletzkoTC.AlagbeO.et al.Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress.Am J Psychiatry.20061631630163316946190
- Kiecolt-GlaserJK.GouinJP.WengNP.et al.Childhood adversity heightens the impact of later-life caregiving stress on telomere length and inflammation.Psychosom Med201073162221148804
- BarnesSK.OzanneSE.Pathways linking the early environment to longterm health and lifespan.Prog Biophys Mol Biol. In press.
- RothTL.LubinFD.FunkAJ.SweattJD.Lasting epigenetic influence of early-life adversity on the BDNF gene.Biol Psychiatry.20096576076919150054
- AvitalA.RamE.MaayanR.WeizmanA.Richter-LevinG.Effects of early-life stress on behavior and neurosteroid levels in the rat hypothalamus and entorhinal cortex.Brain Res Bull.20066841942416459196
- ThomasC.HypponenE.PowerC.Obesity and type 2 diabetes risk in midadult life: the role of childhood adversity.Pediatrics.2008121e1240e124918450866
- WolkowitzOM.EpelES.MellonSH.et al.Major depression and history of childhood sexual abuse are related to increased PBMC telomerase activity. in International Society of Psychoneuroendocrinology Annual Meeting. San Francisco, CA. 2009
- KananenL.SurakkaI.PirkolaS.et al.Childhood adversities are associated with shorter telomere length at adult age both in individuals with an anxiety disorder and controls.PLoS One.20105e1082620520834
- TyrkaAR.PriceLH.KaoHT.PortonB.MarsellaSA.CarpenterLL.Childhood maltreatment and telomere shortening: preliminary support for an effect of early stress on cellular aging.Biol Psychiatry.20106753153419828140
- O'DonovanAEpelES.LinJ.et al.Childhood trauma associated with short leukocyte telomere length in post-traumatic stress disorder.Biol Psychiatry. In press.2011
- McGowanPO.SasakiA.DAlessioAC.et al.Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse.Nat Neurosci.20091234234819234457
- WeaverIC.CervoniN.ChampagneFA.et al.Epigenetic programming by maternal behavior.Nat Neurosci.2004784785415220929
- KrishnanV.NestlerEJ.Linking molecules to mood: new insight into the biology of depression.Am J Psychiatry.20101671305132020843874
- DeRijkR.de KloetER.Corticosteroid receptor genetic polymorphisms and stress responsivity.Endocrine.20052826327016388115
- WassermanD.WassermanJ.SokolowskiM.Genetics of HPA-axis, depression and suicidality.Eur Psychiatry.20102527828020444578
- SchroederM.KrebsMO.BleichS.FrielingH.Epigenetics and depression: current challenges and new therapeutic options.Curr Opin Psychiatry.20102358859220644477
- KorosiA.ShanabroughM.McLellandS.et al.Early-life experience reduces excitation to stress-responsive hypothalamic neurons and reprograms the expression of corticotropin-releasing hormone.J Neurosci.20103070371320071535
- MurgatroydC.PatchevAV.WuY.et al.Dynamic DNA methylation programs persistent adverse effects of early-life stress.Nat Neurosci.2009121559156619898468
- OberlanderTF.WeinbergJ.PapsdorfM.GrunauR.MisriS.DevlinAM.Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses.Epigenetics.200839710618536531
- TyrkaAR.PriceLH.GelernterJ.SchepkerC.AndersonGM.CarpenterLL.Interaction of childhood maltreatment with the corticotropin-releasing hormone receptor gene: effects on hypothalamic-pituitary-adrenal axis reactivity.Biol Psychiatry.20096668168519596121
- ChipmanP.JormAF.TanXY.EastealS.No association between the serotonin-1A receptor gene single nucleotide polymorphism rs6295C/G and symptoms of anxiety or depression, and no interaction between the polymorphism and environmental stressors of childhood anxiety or recent stressful life events on anxiety or depression.Psychiatr Genet.201020813
- CaspiA.SugdenK.MoffittTE.et al.Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene.Science.200330138638912869766
- KargK.BurmeisterM.SheddenK.SenS.The Serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation.Arch Gen Psychiatry. In press.
- RischN.HerrellR.LehnerT.et al.Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis.JAMA.20093012462247119531786
- ResslerKJ.BradleyB.MercerKB.et al.Polymorphisms in CRHR1 and the serotonin transporter loci: gene x gene x environment interactions on depressive symptoms.Am J Med Genet B Neuropsychiatr Genet.2010153B81282420029939
- KaufmanJ.YangBZ.Douglas-PalumberiH.et al.Brain-derived neurotrophic factor-5-HTTLPR gene interactions and environmental modifiers of depression in children.Biol Psychiatry.2006596738016458264
- FriesE.HesseJ.HellhammerJ.HellhammerDH.A new view on hypocortisolism.Psychoneuroendocrinology.2005301010101615950390
- BremmerMA.DeekDJ.BeekmanAT.PenninxBW.LipsP.HoogendijkWJ.Major depression in late life is associated with both hypo- and hypercortisolemia.Biol Psychiatry.20076247948617481591
- MillerAH.Letter to the Editor Re: An inflammatory review of glucocorticoids in the CNS. Sorrells SF, Sapolsky RM.Brain Behavior Immun.200721259272988989; author reply 990.
- SorrellsSF.SapolskyRM.A pro-inflammatory review of glucocorticoid actions in the CNS.Brain Behav Immun.20072125927217194565
- MillerKB.NelsonJC.Does the dexamethasone suppression test relate to subtypes, factors, symptoms, or severity?Arch Gen Psychiatry.1987447697743632250
- AranaGW.SantosAB.LaraiaMT.et al.Dexamethasone for the treatment of depression: a randomized, placebo- controlled, double-blind trial.Am J Psychiatry.19951522652677840362
- BelanoffJ.SchatzbergAF.Glucocorticoid antagonists.Neuropsychopharmacology.200023S56
- WolkowitzOM.ReusVI.Treatment of depression with antiglucocorticoid drugs.Psychosomat Med.199961698711
- GallagherP.WatsonS.SmithMS.YoungAH.FerrierIN.Antiglucocorticoid treatments for mood disorders.Cochrane Database Syst Rev.2008CD005168
- FrankMG.MiguelZD.WatkinsLR.MaierSF.Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide.Brain Behav Immun.201024193019647070
- BardenN.ReulJM.HolsboerF.Do antidepressants stabilize mood through actions on the hypothalamic-pituitary-adrenocortical system?Trends Neurosci.1995186117535490
- WolkowitzOM.BurkeH.EpelES.ReusVI.Glucocorticoids: mood, memory and mechanisms.Ann NY Acad Sci.20091179194019906230
- ReusVI.MinerC.Evidence for physiological effects of hypercortisolemia in psychiatric patients.Psychiatry Res.19851447563857648
- DumanRS.MonteggiaLM.A neurotrophic model for stress-related mood disorders.Biol Psychiatry.2006591116112716631126
- HeimC.EhlertU.HellhammerDH.The potential role of hypocortisolism in the pathophysiology of stress-related bodily disorders.Psychoneuroendocrinology.20002513510633533
- UzunovaV.ShelineY.DavisJM.et al.Increase in the cerebrospinal fluid content of neurosteroids in patients with unipolar major depression who are receiving fluoxetine or fluvoxamine.Proc Natl Acad Sci USA.199895323932449501247
- GriffinLD.MellonSH.Selective serotonin reuptake inhibitors directly alter activity of neurosteroidogenic enzymes.Proc Natl Acad Sci.199996135121351710557352
- GuidottiA.CostaE.Can the antidysphoric and anxiolytic profiles of selective serotonin reuptake inhibitors be related to their ability to increase brain allopregnanolone availability?Biol Psychiatry.1998448658739807641
- BauerME.Chronic stress and immunosenescence: a review.Neuroimmunomodulation.20081524125019047801
- ZampieriS.MellonSH.ButtersTD.et al.Oxidative stress in NPC1 deficient cells: protective effect of allopregnanolone.J Cell Mol Med.200133786379618774957
- WangJM.IrwinRW.LiuL.ChenS.BrintonRD.Regeneration in a degenerating brain: potential of allopregnanolone as a neuroregenerative agent.Curr Alzheimer Res.2007451051718220513
- StrohleA.RomeoE.HermannB.et al.Concentrations of 3a-reduced neuroactive steroids and their precursors in plasma of patients with major depression and after clinical recovery.Biol Psychiatry.19994527427710023501
- RupprechtR.Neuroactive steroids: mechanisms of action and neuropsychopharmacological properties.Psychoneuroendocrinology.20032813916812510009
- WolkowitzOM.ReusVI.Neurotransmitters, neurosteroids and neurotrophins: new models of the pathophysiology and treatment of depression.World J Biol Psychiatry.200349810212872201
- BarbacciaML.SerraM.PurdyRH.BiggioG.Stress and neuroactive steroids.Int Rev Neurobiol.20014624327211599302
- CalogeroAE.PalumboMA.BosboomAM.et al.The neuroactive steroid allopregnanolone suppresses hypothalamic gonadotropin-releasing hormone release through a mechanism mediated by the gamma-aminobutyric acidA receptor.J Endocrinol.19981581211259713333
- PatchevVK.ShoaibM.HolsboerF.AlmeidaOF.The neurosteroid tetrahydroprogesterone counteracts corticotropin-releasing hormoneinduced anxiety and alters the release and gene expression of corticotropinreleasing hormone in the rat hypothalamus.Neuroscience.1994622652717816204
- PatchevVK.MontkowskiA.RouskovaD.et al.Neonatal treatment of rats with the neuroactive steroid tetrahydrodeoxycorticosterone (THDOC) abolishes the behavioral and neuroendocrine consequences of adverse early life events.J Clin Invest.1997999629669062354
- DongE.MatsumotoK.UzunovG.et al.Brain 5alpha-dihydroprogesterone and allopregnanolone synthesis in a mouse model of protracted social isolation.Proc Natl Acad Sci USA.2001982849285411226329
- KlatzkinRR.MorrowAL.LightKC.PedersenCA.GirdlerSS.Histories of depression, allopregnanolone responses to stress, and premenstrual symptoms in women.Biol Psychol.20067121115951099
- RapkinAJ.MorganM.GoldmanL.et al.Progesterone metabolite allopregnanolone in women with premenstrual syndrome.Obstet Gynecol.1997907097149351749
- SundströmI.AnderssonA.NybergS.et al.Patients with premenstrual syndrome have a different sensitivity to a neuroactive steroid during the menstrual cycle compared to control subjects.Neuroendocrinology.1998671261389508043
- HenningsJM.IsingM.GrautoffS.et al.Glucose tolerance in depressed inpatients, under treatment with mirtazapine and in healthy controls.Exp Clin Endocrinol Diabetes.20101189810019834872
- KopfD.WestphalS.LuleyCW.et al.Lipid metabolism and insulin resistance in depressed patients: significance of weight, hypercortisolism, and antidepressant treatment.J Clin Psychopharmacol.20042452753115349009
- Weber-HamannB.HentschelF.KniestA.et al.Hypercortisolemic depression is associated with increased intra-abdominal fat.Psychosom Med.20026427427711914443
- VogelzangsN.SuthersK.FerrucciL.et al.Hypercortisolemic depression is associated with the metabolic syndrome in late-life.Psychoneuroendocrinology.20073215115917224244
- de LeonMJ.McRaeT.RusinekH.et al.Cortisol reduces hippocampal glucose metabolism in normal elderly, but not in Alzheimer's disease.J Clin Endocrinol Metab.199782325132599329348
- McIntyreRS.SoczynskaJZ.LewisGF.et al.Managing psychiatric disorders with antidiabetic agents: translational research and treatment opportunities.Expert Opin Pharmacother.200671305132116805717
- RasgonNL.KennaHA.Insulin resistance in depressive disorders and Alzheimer's disease: revisiting the missing link hypothesis.Neurobiol Aging.200526 Suppl 110310716225963
- AvogaroA.de KreutzenbergSV.FadiniGP.Insulin signaling and life span.Pflugers Arch.201045930131419756720
- GardnerJP.SrinivasanSR.ChenW.et al.Rise in insulin resistance is associated with escalated telomere attrition.Circulation.20051112171217715851602
- AlvehusM.BurenJ.SjostromM.GoedeckeJ.OlssonT.The human visceral fat depot has a unique inflammatory profile.Obesity (Silver Spring).20101887988320186138
- PalmieriVO.GrattaglianoI.PortincasaP.PalascianoG.Systemic oxidative alterations are associated with visceral adiposity and liver steatosis in patients with metabolic syndrome.J Nutr.20061363022302617116714
- BauerME.JeckelCM.LuzC.The role of stress factors during aging of the immune system.Ann N Y Acad Sci.2009115313915219236337
- Kiecolt-GlaserJ.K.GlaserR.Depression and immune function: central pathways to morbidity and mortality.J Psychosom Res.20025387387612377296
- DhabharFS.BurkeHM.EpelES.et al.Low serum IL-10 concentrations and loss of regulatory association between IL-6 and IL-10 in adults with major depression.J Psychiatr Res.20094396296919552919
- DowlatiY.HermannN.SwardfagerW.et al.A meta-analysis of cytokines in major depression.Biol Psychiatry.20106744645720015486
- KaurK.SharmaAK.DhingraS.SingalPK.Interplay of TNF-alpha and IL-10 in regulating oxidative stress in isolated adult cardiac myocytes.J Mol Cell Cardiol.2006411023103017045606
- PickeringM.O'ConnorJ.J.Pro-inflammatory cytokines and their effects in the dentate gyrus.Prog Brain Res.200716333935417765728
- MillerAH.MaleticV.RaisonCL.Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression.Biol Psychiatry.20096573274119150053
- MarslandAL.GianarosPJ.AbramowitchSM.ManuckSB.HaririAR.Interleukin-6 covaries inversely with hippocampal grey matter volume in middle-aged adults.Biol Psychiatry.20086448449018514163
- CarreroJJ.StenvinkelP.FellströmB.et al.Telomere attrition is associated with inflammation, low fetuin-A levels and high mortality in prevalent haemodialysis patients.J Intern Med.200826330231218070000
- EffrosRB.Kleemeier Award Lecture. 2008--the canary in the coal mine: telomeres and human healthspan.J Gerontol A Biol Sci Med Sci.20096451151519228779
- IrieM.MiyataM.KasaiH.Depression and possible cancer risk due to oxidative DNA damage.J Psychiatr Res.20053955356016005897
- JosephJA.Shukitt-HaleB.CasadesusG.FisherD.Oxidative stress and inflammation in brain aging: nutritional considerations.Neurochem Res.20053092793516187227
- ForlenzaMJ.MillerGE.Increased serum levels of 8-hydroxy-2'-deoxyguanosine in clinical depression.Psychosom Med.2006681716449405
- WolkowitzOM.MellonSH.EpelES.et al.Leukocyte telomere length in major depression: correlations with chronicity, inflammation and oxidative stress - preliminary findings.PLoS One. In press.2011
- TsirpanlisG.ChatzipanagiotouS.BoufidouF.et al.Serum oxidized low-density lipoprotein is inversely correlated to telomerase activity in peripheral blood mononuclear cells of haemodialysis patients.Nephrology (Carlton).20061150650917199788
- GrantMM.BarberVS.GriffithsHR.The presence of ascorbate induces expression of brain derived neurotrophic factor in SH-SY5Y neuroblastoma cells after peroxide insult, which is associated with increased survival.Proteomics.2005553454015627972
- CumurcuBE.OzyurtH.EtikanI.DemirS.KarlidagR.Total antioxidant capacity and total oxidant status in patients with major depression: impact of antidepressant treatment.Psychiatry Clin Neurosci.20096363964519674383
- GrovesJO.Is it time to reassess the BDNF hypothesis of depression?Mol Psychiatry.2007121079108817700574
- ChenB.DowlatshahiD.MacQueenGM.WangJ.-F.YoungLT.Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication.Biol Psychiatry.20015026026511522260
- Bocchio-ChiavettoL.BagnardiV.ZanardiniR.et al.Serum and plasma BDNF levels in major depression: a replication study and meta-analyses.World J Biol Psychiatry20101176377320334574
- SchmidtHD.DumanRS.Peripheral BDNF produces antidepressant-like effects in cellular and behavioral models.Neuropsychopharmacology.2010352378239120686454
- SantarelliL.SaxeM.GrossC.et al.Requirement of hippocampal neurogenisis for the behavioral effects of antidepressants.Science.200330180580912907793
- Ibarguen-VargasY.SurgetA.ToumaC.et al.Deficit in BDNF does not increase vulnerability to stress but dampens antidepressant-like effects in the unpredictable chronic mild stress.Behav Brain Res.200920224525119463708
- JoostenEA.HouwelingDA.Local acute application of BDNF in the lesioned spinal cord anti-inflammatory and anti-oxidant effects.Neuroreport.2004151163116615129166
- FuW.LuC.MattsonMP.Telomerase mediates the cell survival-promoting actions of brain-derived neurotrophic factor and secreted amyloid precursor protein in developing hippocampal neurons.J Neurosci.200222107101071912486164
- ChaldakovGN.TonchevAB.ManniL.et al.Comment on: Krabbe KS, Nielsen AR, Krogh-Madsen R et al. Brain-derived neurotrophic factor (BDNF) and type 2 diabetes.Diabetologia.2007504314381781178217151862
- KrabbeKS.NielsenAR.Krogh-MadsenR.et al.Brain-derived neurotrophic factor (BDNF) and type 2 diabetes. Diabetologia. 200750431438
- BlackburnEH.GreiderCW.SzostakJW.Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging.Nat Med.2006121133113817024208
- AvivA.Telomeres and human aging: facts and fibs.Sci Aging Knowledge Environ.200451pe4315618136
- SimonNM.SmollerJW.McNamaraKL.et al.Telomere shortening and mood disorders: preliminary support for a chronic stress model of accelerated aging.Biol Psychiatry.20066043243516581033
- LungFW.ChenNC.ShuBC.Genetic pathway of major depressive disorder in shortening telomeric length.Psychiatr Genet.20071719519917417064
- HartmannN.BoehnerM.GroenenF.KalbR.Telomere length of patients with major depression is shortened but independent from therapy and severity of the disease.Depress Anxiety.2010271111111621053332
- EpelES.MerkinSS.CawthonR.et al.The rate of leukocyte telomere shortening predicts mortality from cardiovascular disease in elderly men.Aging.20091818820195384
- CawthonRM.SmithKR.O'BrienE.SivatchenkoA.KerberRA.Association between telomere length in blood and mortality in people aged 60 years or older.Lancet.200336139339512573379
- DamjanovicAK.YangY.GlaserR.et al.Accelerated telomere erosion is associated with a declining immune function of caregivers of Alzheimer's disease patients.J Immunol.20071794249425417785865
- WolkowitzOM.MellonSH.EpelES.et al.Resting leukocyte telomerase activity is elevated in major depression and predicts treatment response.Mol Psychiatry. In press.2011
- ChoiJ.FauceSR.EffrosRB.Reduced telomerase activity in human T lymphocytes exposed to cortisol.Brain Behav Immun.20082260060518222063
- LiuJP.ChenSM.CongYS.et al.Regulation of telomerase activity by apparently opposing elements.Ageing Res Rev.2010924525620362078
- EpelE.DaubenmierJ.MoskowitzJT.FolkmanS.BlackburnE.Can meditation slow rate of cellular aging? Cognitive stress, mindfulness, and telomeres.Ann N Y Acad Sci.20091172345319735238
- MattsonMP.DuanW.WanR.GuoZ.Prophylactic activation of neuroprotective stress response pathways by dietary and behavioral manipulations.NeuroRx.2004111111615717011
- SpiresTL.HannanAJ.Nature, nurture and neurology: gene-environment interactions in neurodegenerative disease. FEBS Anniversary Prize Lecture, delivered on 27 June, 2004 at the 29th FEBS Congress in Warsaw.Febs J.20052722347236115885086
- MaesM.YirmyiaR.NorabergJ.et al.The inflammatory neurodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression.Metab Brain Dis.200924275319085093
- McIntyreRS.SoczynskaJK.WoldeyohannesHO.et al.Thiazolidinediones: novel treatments for cognitive deficits in mood disorders?Expert Opin Pharmacother.200781615162817685880
- SanacoraG.ZarateCA.KrystalJH.ManjiHK.Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders.Nat Rev Drug Discov.2008742643718425072
- PaulIA.Antidepressant activity and calcium signaling cascades.Hum Psychopharmacol.200116718012404601
- Berthold-LoslebenM.HeitmannS.HimmerichH.Anti-inflammatory drugs in psychiatry.Inflamm Allergy Drug Targets.2009826627619754410
- BerkM.CopolovDL.DeanO.et al.N-acetyl cysteine for depressive symptoms in bipolar disorder-a double-blind randomized placebo-controlled trial.Biol Psychiatry.20086446847518534556
- ZhangY.PardridgeWM.Conjugation of brain-derived neurotrophic factor to a blood-brain barrier drug targeting system enables neuroprotection in regional brain ischemia following intravenous injection of the neurotrophin.Brain Res.2001889495611166685
- EffrosRB.Telomerase induction in T cells: A cure for aging and disease?Exp Gerontol.20074241642017182206
- HarleyCB.LiuW.BlascoM.et al.A natural product telomerase activator as part of a health maintenance program.Rejuvenation Res. In press.
- OrnishD.ScherwitzLW.DoodyRS.et al.Effects of stress managment training and dietary changes in treating ischemic heart disease.JAMA.198324954596336794
- HajkovaP.Epigenetic reprogramming-taking a lesson from the embryo.Curr Opin Cell Biol.20102234235020537882
- WeaverIC.ChampagneFA.BrownSE.et al.Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life.J Neurosci.200525110451105416306417
- HolsboerF.IsingM.Central CRH system in depression and anxietyevidence from clinical studies with CRH1 receptor antagonists.Eur J Pharmacol.200858335035718272149
- CocchiP.SilenziM.CalabriG.SalviG.Antidepressant effect of vitamin C.Pediatrics.1980658628637367105