
Abstract
The development of the cerebral cortex requires complex sequential processes that have to be precisely orchestrated. The localization and timing of neuronal progenitor proliferation and of neuronal migration define the identity, laminar positioning, and specific connectivity of each single cortical neuron. Alterations at any step of this organized series of events—due to genetic mutations or environmental factors—lead to defined brain pathologies collectively known as malformations of cortical development (MCDs), which are now recognized as a leading cause of drug-resistant epilepsy and intellectual disability. In this heterogeneous group of disorders, macroscopic alterations of brain structure (eg, heterotopic nodules, small or absent gyri, double cortex) can be recognized and probably subtend a general reorganization of neuronal circuits. In this review, we provide an overview of the molecular mechanisms that are implicated in the generation of genetic MCDs associated with aberrations at various steps of neurogenesis and cortical development.
El desarrollo de la corteza cerebral requiere de una secuencia de complejos procesos que tienen que estar coordinados con precisión. La localización y la cronología de la proliferación de las neuronas precursoras y de la migración neuronal definen la identidad, el posicionamiento laminar y la conectividad específica de cada una de las neuronas corticales. Las alteraciones en cualquier etapa de esta serie organizada de acontecimientos- debidas a mutaciones genéticas o a factores ambientales- llevan a patologías cerebrales definidas que en conjunto se denominan malformaciones del desarrollo cortical (MDC), las cuales son reconocidas actualmente como causa de epilepsia resistente a fármacos e incapacidad intelectual. En este grupo heterogéneo de trastornos, las alteraciones macroscópicas de la estructura cerebral (por ej. nódulos heterotópicos, giros pequeños o ausentes, doble corteza) pueden ser reconocidas y es probable que subtiendan a una reorganización general de los circuitos neuronales. En esta revisión se entrega una panorámica de los mecanismos moleculares que se han involucrado en la generación de las MDC asociadas con aberraciones en varias etapas de la neurogénesis y del desarrollo cortical.
Le développement du cortex cérébral fait appel à des processus séquentiels complexes qui doivent être orchestrés précisément. La localisation et la chronologie de la prolifération de neurones précurseurs et celles de la migration neuronale définissent l'identité, le positionnement laminaire et la connectivité spécifique de chaque neurone cortical unique. Toute modification, quel que soit le stade de ces séries organisées d'événements (en raison de mutations génétiques ou de facteurs environnementaux), entraîne des pathologies cérébrales définies, globalement connues sous le terme de malformations du développement cortical (MDC). Ces malformations sont maintenant reconnues comme principalement responsables de la résistance aux médicaments contre l'épilepsie et du déficit intellectuel. Dans ce groupe hétérogène de maladies, les modifications macroscopiques de la structure cérébrale (par exemple, nodules hétérotopiques, gyrus petit ou absent, double cortex) peuvent être identifiées et probablement sous-tendre une réorganisation générale des circuits neuronaux. Cet article présente une vue d'ensemble des mécanismes moléculaires impliqués dans l'apparition de MDC génétiques associées à des aberrations à des stades différents de la neurogenèse et du développement cortical.
Introduction
The cerebral cortex is a laminar structure in which neurons are spatially organized in horizontal layers and vertical columns that reflect a highly precise map of connectivity. The formation of such a defined structure is achieved through a coordinated and tightly regulated orchestration of neurogenesis, neuronal migration, and neuronal wiring. In particular, the neocortex—the phylogenetically newest portion of the cortex, evolved for executing higher brain functions—is composed of six layers of defined excitatory and inhibitory neuronal subtypes.Citation1 Excitatory pyramidal neurons are generated from radial glial cells ( ), cortical progenitor cells that constitute the wall of the lateral ventricles (ie, the ventricular zone, VZ). At the onset of neurogenesis, these cells start to divide asymmetrically and generate either one postmitotic neuron or one intermediate progenitor cell, which further divide in the subventricular zone to generate two postmitotic neurons.Citation2 Newly born neurons radially migrate along the radial glia scaffold towards the pial surface, thus populating the cortical plate and constituting the different layers in an “inside-out” progression from the deeper to more superficial ones.Citation3 While migrating, neurons undergo specific morphological changes: newborn bipolar neurons rapidly move from the ventricular to the subventricular zone (SVZ), acquire a multipolar morphology and pause, then retrogradely migrate towards the ventricle before becoming bipolar again and moving to their final destination in cortical plate.Citation2 Once in the correct position, pyramidal neurons start to differentiate, extending axons and dendrites whose growth, branching, and pathfinding are strictly directed by a complex set of chemoattractant and chemorepellant molecules.Citation4 In contrast to excitatory neurons, inhibitory neurons populating the cortex follow a different development route, as they originate in subcortical regions (medial and caudal ganglionic eminences, preoptic area) and subsequently migrate tangentially to reach the neocortex. A tangential-to-radial migration switch then allows interneurons to position in the appropriate cortical layers in a subtype-specific way.Citation5
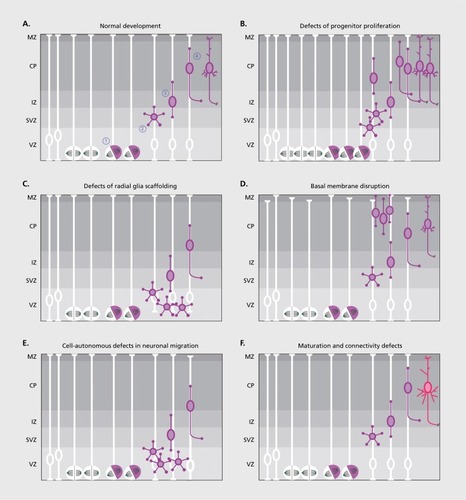
Aberrations at each of these steps—due to genetic alterations and/or environmental insults—are at the basis of diverse pathological conditions. Macroscopic alterations in brain structure due to defective or excessive neurogenesis, neuronal migration, or differentiation are recognized in the so-called malformations of cortical development (MCDs), a heterogeneous group of disorders with variable severity usually associated with cognitive impairment and drug-resistant epilepsy.Citation6 Still, subtler alterations in neuronal layering, particularly of inhibitory interneurons, and connectivity of specific circuits may be at the basis of epilepsy and neuropsychiatric disorders such as autism and schizophrenia.Citation7
In this review we will provide a summary of the current knowledge about the cellular and molecular mechanisms leading to MCDs, with some cardinal examples of genes and pathways whose disruption causes specific phenotypes. A detailed description of the clinical classification and of the genetics underlying the disorders that will be mentioned has been provided elsewhere.Citation8 Our purpose is to provide a step-by-step description of what can go wrong during brain development, and how this translates into defined pathologies (Figure 1).
Defects of progenitor proliferation
Overproliferation
Intuitively, an increase in neuronal progenitor proliferation is expected to modify brain size as it directly impacts on the number of postmitotic neurons that will be generated. Classical examples of this kind of defect are megalencephaly and hemimegalencephaly, in which brain size is macroscopically larger than normal in a symmetric or asymmetric way. This is usually accompanied by important developmental delay, intellectual disability, and drug-resistant childhood epilepsy.Citation8 More localized aberrations of progenitor proliferation have been postulated to be also at the basis of focal cortical dysplasia type II, characterized by localized malformations of the cortex, lamination defects, and dysmorphic neurons, again accompanied by drug-resistant childhood epilepsy.Citation8 For all these diseases, genetic studies have led to the identification of several germline and brain somatic mutations that converge on the mTOR (mammalian target of rapamycin) pathway. Indeed, mutations have been found in MTOR itself and in its upstream regulators, including PIK3CA, PIK3R2, AKT1, AKT3, TSC1, TSC2, NPRL2, DEPDC5, and STRADA, always leading to a hyperactivation of the mTOR kinase.Citation6,Citation9-Citation12 mTOR is a crucial regulator of cell growth and proliferation, as it controls protein synthesis, protein degradation, lipid metabolism, and cytoskeletal dynamics according to extracellular stimuli.Citation13 High levels of activation of the AKT-mTOR pathway have been detected in proliferating radial glial cells in the VZ, suggesting a prominent role in these cells.Citation14,Citation15 In the mouse, deletion of mTOR in neuronal progenitors results in decreased brain size and thinning of the cerebral cortex due to a reduced proliferation of radial glial cells and reduced production of intermediate progenitor cells.Citation14 Functional studies taking advantage of rodent models are starting to characterize the specific effects of single human mutations in the mTOR pathway on cortical development. The use of in utero electroporation approaches is particularly suitable to mimic the low-percentage mosaicism observed for brain somatic mutations. Increased progenitor proliferation, defective neuronal migration with heterotopic neurons, aberrant cortical layering and thickening, and the increased size of neuronal somata have been variably reported upon expression of mutant mTOR-related proteins.Citation11,Citation12,Citation16-Citation18 Interestingly, the hyperactivation of the mTOR pathway in animal models—even in a small number of neurons—is usually sufficient to cause the occurrence of spontaneous generalized seizures that can be significantly ameliorated by treatment with mTOR or PI3K inhibitors.Citation16,Citation17,Citation19 This suggests that this mechanism-based therapeutic intervention may be a valuable strategy to be transferred to clinical practice for the treatment of epilepsy in MCDs. Along this line, the adjunctive treatment with the mTOR inhibitor everolimus has proved effective in reducing seizure frequency in tuberous sclerosis complex patients.Citation20 One fascinating question that arises from these genetic and functional studies is how different mutations in the same pathway can lead to more or less extensive malformations. Growing evidence points to the fact that somatic mutations can differentially affect brain development depending on the timing and cell type in which they occur, meaning that earlier mutations will affect a higher number of progenitor cells as compared with later-occurring ones, thus causing more severe phenotypes. Indeed, it was recently suggested that the extent of somatic mosaicism of mTOR-related mutations (ie, the number of cells positive for a somatic mutation in a brain sample, as assessed through single cell sequencing) directly correlates with the severity of the associated disease.Citation12 In addition, it is possible that, depending on how upstream in the pathway is the studied mutation, effectors in pathways other than mTOR are also misactivated leading to additional molecular events that participate to the pathogenic outcomes. This was exemplified in a recent study reporting that the expression of a mutant AKT3 associated with hemimegalencephaly in a small number of cortical cells through in utero electroporation in the mouse brain causes an heterotopic secretion of reelin—an extracellular glycoprotein that instructs neuronal migration and positioning during brain development—through the aberrant activation of the transcription factor FOXG1. This in turn drives a non-cell autonomous migration defect in wild-type neighboring neurons, thus extending the affected area to a larger population of neurons than that actually carrying the mutation.Citation18 Another possible target of misactivation after mutations in PI3K-AKT is cyclin D2 (CCDN2), a regulator of cell cycle progression whose degradation is promoted by GSK3β-dependent phosphorylation. De novo germline mutations in CCND2 have been identified in patients affected by the megalencephaly-polymicrogyria-polydactyly-hydrocephalus (MPPH) syndrome. These mutations lead to an accumulation of degradation-resistant cyclin D2—probably because the mutation induces a lack of GSK3β-dependent phosphorylation—and to an expansion of proliferating radial glia and intermediate progenitor pools.Citation21 The activity of the GSK3β kinase is negatively regulated by the PI3K-AKT pathway. Indeed, the accumulation of cyclin D2 has been described in cell lines derived from megalencephalic patients with gain-of-function mutations in PI3K or AKT3, thus suggesting that this may represent a shared molecular mechanism underlying megalencephaly of various genetic origins.Citation21
Reduced proliferation
On the opposite side of the coin, reduced proliferation of neuronal progenitor cells is at the basis of primary microcephaly, in which patients display a substantial decrease in brain size—even though brain structure and cortical layering is generally preserved—and intellectual disability, with all other organs growing normally. Primary microcephaly is considered to be a disorder of neurogenic mitosis.
Several germline mutations have been identified in genes whose protein product is directly involved in centrosome maturation and mitotic spindle formation, such as CDK5RAP2, ASPM, CENPJ, STIL, CEP135, CEP152, CEP63, NDE1, CDK6, WDR62. Citation22-Citation27 All of these proteins are highly expressed in the proliferative neuronal progenitors of the VZ/SVZ during brain development. Their loss of function leads to various cellular defects depending on the exact function that they absolve. In some cases, extranumerary centrosomes are produced, leading to multipolar mitotic spindles, aberrant chromosome segregation, random aneuploidy, mitotic arrest, and neuronal progenitor cell death.Citation24,Citation28,Citation29 In others, aberration of the mitotic spindle orientation with respect to the neuroepithelial plane leads to premature symmetric-to-asymmetric switch in cell division, thus causing a reduced expansion of the progenitor pool and consequent reduced production of postmitotic neurons.Citation30
Mutations in genes coding for nuclear proteins have also been described in microcephalic patients. These include loss-of-function mutations in MCPH1 (microcephalin), ZNF335, and PHC1.Citation31,Citation32 MCPH1 has been described as a protein implicated in DNA repair and chromosome condensation during mitosis, as well as in cell-cycle checkpoints. Its ablation leads, through unclear mechanisms, to misregulation of Checkpoint kinase 1 (Chk1) expression and recruitment to centrosomes during mitosis, thus leading to premature mitotic entry and uncoordinated centrosome maturation, defective mitotic spindle alignment, and consequently increased cell death and preferential asymmetric division of progenitor cells.Citation33 ZNF335 interacts with a histone methyltransferase complex and directly regulates the expression of a variety of genes implicated in cell proliferation and postmitotic differentiation. One of these target genes is the transcription factor REST/NRSF, a master repressor of neuronal gene expression in non-neuronal cells. The ablation of ZNF335 causes a reduction in the levels of REST, which probably contributes to premature neuronal fate specification.Citation31 PHC1 is part of Polycomb chromatin remodeling complexes and participates in histone ubiquitination and transcriptional repression. The mechanisms through which PHC1 mutations may lead to microcephaly remain to be elucidated, but dysregulation of a number of genes involved in cell cycle regulation has been reported in patient-derived lymphoblastoid cell lines. This was accompanied by polyploidy, defective cell cycle progression and defective DNA repair response.Citation32
It is worth noting that in this case all the mentioned genes are extensively expressed outside the central nervous system and the mutations reported are all germline or zygotic, not somatic. The reasons why patients display a prevalent effect on brain and not on other tissues' growth are still not understood.
Heterotopic localization of progenitor cells
A third kind of defect that can affect the neurogenic phase of cortical development has been outlined by the identification of mutations in the EML1 gene in patients presenting subcortical heterotopia, ie, heterotopic groups of neurons lying outside the cortical plate, usually in the subcortical white matter.Citation34 Interestingly, the authors firstly identified EML1 as the mutant gene responsible for cortical heterotopia in the spontaneous mutant mouse line named HeCo (for heterotopic cortex), for which a defined genetic cause was missing, and then successfully identified mutations in the human ortholog gene in two families with severe bilateral ribbon-like heterotopia. EML1 is a microtubule-associated protein expressed in progenitor cells of the VZ during cortical development, as well as in postmitotic neurons in the cortical plate. Its loss of function leads to the misplacement of progenitor cells outside the VZ/SVZ in the intermediate zone and cortical plate, where proliferation is normally absent. This causes the production of heterotopic neurons and the creation of a physical barrier for otherwise normally migrating neurons produced from non-heterotopic progenitors in VZ/SVZ, leading to the formation of extended heterotopia.Citation34 This scenario substantially differs from classical band heterotopia and periventricular heterotopia that arise from defects in neuronal migration due to cell-autonomous dysfunction or to aberrations in the radial glia scaffold, as will be described in the following section. The exact mechanism through which heterotopic progenitors are produced upon EML1 loss-of-function is unclear, but may be related to altered microtubule-based regulation of apical-basal attachment of radial glial cells.
Defects of neuronal migration
Alterations in the radial glia scaffold
In addition to their role as neurogenic progenitors, radial glial cells also support and guide the migration of postmitotic neurons from the VZ/SVZ to the cortical plate. As such, disruption of the radial glia scaffold easily leads to non-cell autonomous neuronal migration defects. This has been described for X-linked periventricular nodular heterotopia (PNH) caused by loss-of-function mutations in the filaminA FLNA gene. PNH patients display bilateral nodules of heterotopic neurons protruding in the lateral ventricles, below an apparently normal cerebral cortex. Hemizygous males have high prenatal lethality, while heterozygous females present seizures with variable onset and cardiovascular abnormalities.Citation6,Citation35 FLNA is a pleiotropic protein involved in actin cytoskeleton regulation: it binds and crosslinks actin filaments in orthogonal networks, it scaffolds and regulates signaling molecules involved in cell adhesion and migration (eg, integrins), and it modulates the activation of small GTPases involved in actin remodeling (eg, Rac1).Citation36 The protein is highly expressed in the developing cortex, particularly in radial glia cells, but persists also in the adult brain in postmitotic neurons. In utero electroporation of short interfering RNAs against FLNA in rat embryos showed that FLNA depletion leads to a significant delay in radial neuronal migration and to the formation of heterotopic nodules lining the ventricular surface, as observed in PNH patients. This is associated with a disruption of the ventricular neuroepithelium—normally composed of juxtaposed radial glial cell apical endfeet—with radial glial cells detaching from the ventricular surface, probably because of a misregulation of β1-integrin and cadherin/catenin-mediated anchoring. A similar phenotype was observed in brain biopsies from FLNA-mutant PNH patients, which display clear discontinuity in the neuroepithelial lining. Even though additional cell-autonomous mechanisms probably occur, it is worth noting that the ventricular nodules formed upon FLNA silencing include both electroporated and non-electroporated neurons, supporting the idea of a non cell-autonomous mechanism contributing to heterotopia caused by FLNA deficiency.Citation37
Cell-autonomous alteration of neuronal migration
Classical examples of disorders caused by intrinsic defects in neuronal migration are lissencephaly (smooth cortex, characterized by increased thickness and the production of only 2-4 cortical layers) and subcortical band heterotopia (or double cortex, with the presence of a band of gray matter between a pachygyral 6-layered cortex and the ventricular wall). These two conditions represent the two extremes of a spectrum of disorders, ranging from complete agyria to pachygyria with subcortical band heterotopia, to double cortex only. They are generally associated with drug-resistant epilepsy arising in childhood, delayed motor milestones, and cognitive deficits. The most common genetic cause of these diseases is represented by germline and somatic heterozygous mutations in LIS1 and DCX genes.Citation38-Citation40 Both LIS1 and DCX are implicated in the modulation of the microtubule cytoskeleton in migrating neurons. LIS1 interacts with the microtubule molecular motor dynein and with accessory proteins such as dynactin, thus regulating in a complex way the cargo transport activity of the motor complex.Citation41 DCX (doublecortin) binds to microtubules and regulates their stability and dynamics integrating different signaling pathways in migrating cells.Citation42,Citation43 The two proteins have also been found to physically interact, suggesting that they crosstalk in the regulation of microtubule function.Citation44 In utero downregulation of either protein in rat embryos causes a defective migration of neurons from the VZ to the cortical plate, with an abnormal accumulation of multipolar neurons in the SVZ or intermediate zone, suggesting than neurons depleted of LIS1 or DCX cannot undergo proper multipolar-bipolar transition and proceed to radial migration.Citation45,Citation46 In the case of DCX downregulation, the formation of subcortical heterotopic bands has also been observed in postnatal rats, likely due to the stalled neuronal migration within the intermediate zone.Citation45 Interestingly, the inhibition or knockdown of calpain—a protease responsible for LIS1 degradation—in Lis1 +/- mouse embryos leads to an accumulation of LIS1 and rescues the neuronal migration and layering defects, thus providing evidence for a possible therapeutic strategy for human lissencephaly associated with LIS1 mutations.Citation47 Unfortunately, the use of lissencephalic rodent models is poorly informative when looking for pathogenic mechanisms underlying defective gyrification. Indeed, in gyrencephalic species like humans the formation of gyri seems to rely on a significant expansion of a specific type of progenitors called basal radial glial cells, which are instead scarce in rodents. Similarly to apical radial glia, basal radial glial cells extend a basal process that contacts the pial surface, but lack an apical process connecting to the ventricular lining. In gyrencephalic species, the abundance of basal radial glia leads to a thickening of the SVZ, which can thus be further subdivided in outer and inner SVZ. Basal radial glial soma usually lie in the outer SVZ, and their basal projections multiply the radial structural scaffolding necessary for extending the pial surface of the brain while keeping the ventricular surface limited.Citation48 It is possible that lissencephaly in humans arises because of a defect in the basal radial glia scaffold upon LIS1 or DCX mutation, an hypothesis that needs to be tested in gyrencephalic models such as the ferret or the monkey.
Additional mutations in the lissencephalic spectrum have been identified in genes coding for tubulin subunits (TUBA1A, TUBB2B, TUBG1) and molecular motor proteins (KIF2A, KIF5C, DYNC1H1), further highlighting the importance of microtubule-dependent regulation of neuronal migration in these diseases.Citation6 Similarly, mutations in TUBB2B have been identified in patients affected by polymicrogyria, a condition in which an unlayered cerebral cortex folds into very small gyri, and neuronal migration defects have been demonstrated upon in utero silencing of this gene.Citation49,Citation50
Compound heterozygous and homozygous recessive mutations in TBC1D24 have been identified in several epileptic syndromes, such as familial infantile myoclonic epilepsy, epileptic encephalopathy and DOORS syndrome (deafness, onychodystrophy, osteodystrophy, mental retardation, seizures). Patients usually display severe drug-resistant epilepsy and intellectual disability. An association with sudden unexpected death in epilepsy has been suggested. Structural abnormalities identified by brain imaging are highly variable, ranging from normality to cerebral and cerebellar atrophy, agenesis of corpus callosum, delayed demyelination.Citation51,Citation52 In utero downregulation of TBC1D24 expression in rat embryos causes a delay in radial neuronal migration that is eventually caught up, with no obvious layering defects in postnatal animals. This is due to a delay in the completion of the multipolar-bipolar transition in the intermediate zone, required for timely progression of neuronal migration from the VZ to the cortical plate.Citation53 TBC1D24 interacts with the small GTPase ARF6, and several mutations identified in patients lie in the ARF6 interacting domain of TBC1D24. Indeed, the expression of a constitutively inactive form—but not of a constitutively active form—of ARF6 is able to rescue the migration delay induced by TBC1D24 downregulation, suggesting that TBC1D24 inhibits ARF6 in developing neurons and that this is required for proper radial migration.Citation53 Even though the migration defect is not long-lasting in this case, probably explaining the absence of defined common cortical malformations in patients, the precise timing of neuronal integration in the developing cortical plate is required for establishing appropriate synaptic connections and aberrations in this process likely lead to abnormal wiring and excitation/inhibition imbalance.
Defective formation of the pial basement membrane leading to over-migration
Dystroglycan was originally identified in skeletal muscle as a core component of the dystrophin-glycoprotein complex, a transmembrane protein complex connecting the extracellular matrix and the actin cytoskeleton. It is anyway highly expressed in many other tissues, including the brain, where it is required for the correct deposition and organization of basement membranes. Dystroglycan is composed of two subunits, the extracellular protein α-dystroglycan tightly associated with the transmembrane β-dystroglycan. The α subunit is implicated in the interaction of extracellular ligands, such as laminin, in a glycosylation-dependent manner. The β subunit interacts with cytoskeletal and signaling molecules. Several loss-of-function recessive mutations have been identified in genes coding for proteins implicated in the glycosylation of α-dystroglycan (ISPD, POMT1, POMT2, POMGNT1, LARGE1, FKTN, FKRB) in severe syndromes with muscular dystrophy, eye involvement and brain malformations, such as the Walker-Warburg syndrome.Citation54-Citation56 The cause of muscular, ocular, and brain abnormalities reside in the defective formation of the basement membrane in these tissues, due to a reduced interaction of dystroglycan with the extracellular matrix proteins. In particular, in the brain the dystroglycan expressed in the endfeet of radial glial cells contacts the pial basement membrane, and this interaction is required to maintain the integrity of the basement membrane and to form the glia limitans that defines the brain surface.Citation57 A perturbation of this interaction (ie, upon α-dystroglycan deletion or because of its defective glycosylation) leads to an impairment of radial glial basal anchorage and to the formation of breaches in the pial surface, thus causing aberrant neuronal layering and lissencephaly, as well as overmigration of neurons that accumulate in the so-called cobblestone malformations, neuronal ectopias beyond the glia limitans.Citation57-Citation59
A similar overmigration defect has been described in cobblestone malformations caused by loss-of-function mutations in GPR56, coding for an adhesion G-protein coupled receptor that binds to collagen III produced by meningeal fibroblasts.Citation60,Citation61 GPR56 is expressed by radial glial cells as well as by migrating neurons. Binding to collagen III leads to the activation of Gα12-13 and subsequently of the small GTPase RhoA, which has a role in the modulation of actin cytoskeleton. Interestingly, the activation of GPR56 inhibits neuronal migration, suggesting that during cortical development it may mediate a stop signal for migrating neurons reaching the pial surface to impede basement membrane transmigration. Indeed, mice deleted for either Gpr56 or collagen III display abnormal cortical lamination and frontal subpial ectopias that include neurons with both deep and superficial layer identity.Citation61
Impact of developmental defects on neuronal maturation and connectivity
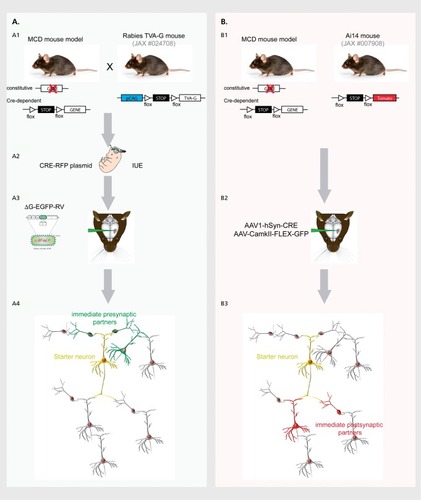
As evidenced in the previous sections, modifications of the correct program of neurogenesis and neuronal development lead to a spectrum of disorders that ranges from macroscopic malformations and more or less severe alterations of cortical layering, to very subtle delays in the timing of neuronal migration resulting in an apparently normal cortex at the macroscopic level. Even in the last scenario, the final outcome is probably an alteration of neuronal wiring, given that the establishment of proper neuronal circuits requires a precise regulation of neuronal positioning, dendritogenesis, axogenesis and input/output synaptic connectivity in space and time. This is emerging from recent functional studies, described hereafter, that demonstrate the presence of defects in neurite outgrowth, synapse formation, synaptic transmission and long-range connectivity in models of diseases with variable degrees of cortical malformation. In addition, more and more information will probably arise on these aspects thanks to the continuous development of new technical approaches to monitor synaptogenesis and connectivity at the single cell level ( ).
In utero silencing of DCX in rat embryos revealed that heterotopic neurons stacked in the ventricular white matter are functionally connected to other brain regions, but display features of immature neurons at postnatal stages, impaired dendritic development and impaired formation of glutamatergic and γ-aminobutyric (GABA) ergic synapses.Citation62,Citation63 Interestingly, non-electroporated neurons in the normal-appearing cortex overlying the heterotopia have an increase in spontaneous and synchronized activity, as well as an imbalance of glutamatergic and GABAergic inputs towards increased excitation that renders them abnormally prone to the generation of epileptiform discharges.Citation62 Accordingly, adult electroporated rats show spontaneous focal seizures even when they present very small subcortical heterotopia, indicating that this is sufficient to drive a larger maladaptive synaptic reorganization of the cortex.Citation64 In addition, electrophysiological characterization of DCX knocked-down rat slices aimed at localizing the spatial origin of epileptiform activity show that the heterotopia is dispensable for seizure generation and that the interictal-like activity originates in the overlying normal-appearing cortex and only subsequently propagates to heterotopic neurons.Citation65
In the case of TBC1D24, no evident cortical malformation is present upon silencing, and knocked-down neurons eventually reach their final position in the cortical plate, even though with some delay. Still, knocked-down neurons in the postnatal brain display a significant impairment in the maturation of their dendritic arbor that likely impact on the distribution of their synaptic contacts and on their activity.Citation53
We recently observed that the conditional ablation of FLNA in a cohort of cortical projection neurons in the mouse leads to excessive dendritic growth, with a consequent disorganization of synaptic contacts and alterations in synaptic activity in the postnatal cortex. Interestingly, in this case we found an increase of inhibitory transmission in knock-out neurons (Falace et al, manuscript in preparation). This may seem counterintuitive considering that loss-of-function mutations of FLNA cause epilepsy in humans, but deserves further exploration as it may give important hints on the specific mechanisms leading to epilepsy in FLNA mutant patients and on the antiseizure medications that may be more suitable in these cases.
Concerning long-range connectivity, a missense mutation in TUBB2B identified in a family affected by congenital fibrosis of extraocular muscles (CFEOM) and polymicrogyria led to a reduction of homotopic callosal projections, as assessed with diffusion tensor imaging performed in patients. In utero electroporation of the mutant TUBB2B in mouse embryos reproduces the human phenotype, with decreased homotopic connectivity in the corpus callosum and perturbed innervation of both local and long-range targets in postnatal animals.Citation66 Interestingly, this phenotype was not detected in patients affected by polymicrogyria without CFEOM, thus suggesting that the presence of cortical dysplasia is not sufficient to drive callosal aberrations and underlying the importance of matching genetic diagnosis, brain imaging and molecular studies for a complete understanding of the MCD spectrum.Citation66
Conclusions and future perspectives
We provided a general description of the main possible defects that may arise during neurogenesis and cortical development in MCDs, giving some examples that could be explicative, although not exhaustive. We would like to emphasize a few concepts that clearly emerge when surveying the literature in the field of cortical malformations and that we think will need special attention in the near future: (i) the expanding evidence that many cortical malformations are caused by somatic brain mutations points to a clear diagnostic limit when analyzing only blood samples and opens an unexpected level of complexity in the genetic spectrum of these disorders, as we can now envisage cases with germline mutations summing to somatic brain mutations, as well as cases in which different somatic brain mutations occur in the same patient but maybe in different cell types. This complexity will surely be more and more addressed with advances in single-cell sequencing approaches, and will probably allow us to eventually identify and/or better specify the genetic causes and complex molecular mechanisms of a much larger number of cases; (ii) a straightforward 1:1 relation between a specific clinical phenotype and a specific cellular mechanism is clearly an oversimplification. Indeed, even though a prevalent defect may be recognized, the coexistence and the consequentiality of different cellular defects are more often described in many of the diseases we mentioned; (iii) research efforts in the last 20 years have led to an explosion of information about the genetics and macroscopic cellular defects, often proliferative or migratory, that ultimately alter neuronal positioning and wiring in MCDs. The next step will be to precisely characterize the final outcomes of these primary defects in terms of neuronal connectivity and network activity in the mature brain in order to understand the exact origin of drug-resistant epilepsy and cognitive impairment in MCDs. This will be useful for guiding the choice of the best suitable therapeutic interventions for each type of malformation, including surgery and pharmacotherapy, as well as for a rationale development of new approaches; (iv) related to the previous point, the most important question that remains is: can we correct neurodevelopmental defects after birth? Recent elegant studies demonstrated that the delivery of the transcription factor Fezf2—a master regulator of identity specification for layer 5 corticofugal neurons—in upper layer postmitotic neurons in mouse pups was sufficient to convert them to layer 5 neurons, with a complete reprogramming of their molecular identity, electrophysiological properties and, more strikingly, of their input and output connectivity.Citation67,Citation68 This indicates that, at least at early postnatal stages, there is a high degree of plasticity that can be exploited to redirect specific circuits in vivo and opens the fascinating possibility of applying this concept to correct pathological alterations of cortical organization.
AF, AdeC, and CC were supported by funding from the European Community 7th Framework programs (Development and Epilepsy—Strategies for Innovative Research to improve diagnosis, prevention and treatment in children with difficult to treat Epilepsy [DESIRE]), Health-F2-602531-2013 (CC and AF), Fritz Thyssen Stiftung - 10.15.2.022MN (CC and AF), Fondation Jérôme Lejeune - 1159-CC2013A (CC and AF) and 1666-DA2017A (AdeC), Fondation Recherche sur le Cerveau (CC and AdeC). FCG. was supported by a postdoctoral fellowship from Fondazione Umberto Veronesi. The authors would like to thank Marco Gervasoni for the help with graphics. The authors declare no financial conflicts of interest.
REFERENCES
- LodatoS.ArlottaP.Generating neuronal diversity in the mammalian cerebral cortex.Annu Rev Cell Dev Biol.20153169972026359774
- NoctorSC.Martinez-CerdenoV.IvicL.KriegsteinAR.Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases.Nat Neurosci.20047213614414703572
- HeS.LiZ.GeS.YuYC.ShiSH.Inside-out radial migration facilitates lineage-dependent neocortical microcircuit assembly.Neuron.20158651159116626050035
- McAllisterAK.Conserved cues for axon and dendrite growth in the developing cortex.Neuron.20023312411779471
- MarinO.Cellular and molecular mechanisms controlling the migration of neocortical interneurons.Eur J Neurosci.20133812019202923651101
- ParriniE.ContiV.DobynsWB.GuerriniR.Genetic basis of brain malformations.Mol Syndromol.20167422023327781032
- MarinO.Interneuron dysfunction in psychiatric disorders.Nat Rev Neurosci.201213210712022251963
- GuerriniR.DobynsWB.Malformations of cortical development: clinical features and genetic causes.Lancet Neurol.201413771072624932993
- OrlovaKA.ParkerWE.HeuerGG.et al.STRADalpha deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice.J Clin Invest.201012051591160220424326
- MirzaaGM.CampbellCD.SolovieffN.et al.Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism.JAMA Neurol.201673783684527159400
- RibierreT.DeleuzeC.BacqA.et al.Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy.J Clin Invest.201812862452245829708508
- D'GamaAM.WoodworthMB.HossainAA.et al.Somatic mutations activating the mtor pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias.Cell Rep.201721133754376629281825
- RyskalinL.LazzeriG.FlaibaniM.et al.mTOR-dependent cell proliferation in the brain.Biomed Res Int.20172017708269629259984
- KaM.CondorelliG.WoodgettJR.KimWY.mTOR regulates brain morphogenesis by mediating GSK3 signaling.Development.2014141214076408625273085
- PoduriA.EvronyGD.CaiX.et al.Somatic activation of AKT3 causes hemispheric developmental brain malformations.Neuron.2012741414822500628
- RoyA.SkiboJ.KalumeF.et al.Mouse models of human PIK3CA-related brain overgrowth have acutely treatable epilepsy.Elife.20154
- LimJS.KimWl.KangHC.et al.Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy.Nat Med.201521439540025799227
- BaekST.CopelandB.YunEJ.et al.An AKT3-FOXG1-reelin network underlies defective migration in human focal malformations of cortical development.Nat Med.201521121445145426523971
- HsiehLS.WenJH.ClaycombK.et al.Convulsive seizures from experimental focal cortical dysplasia occur independently of cell misplacement.Nat Commun.201671175327249187
- FrenchJA.LawsonJA.YapiciZ.et al.Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study.Lancet.2016388100562153216327613521
- MirzaaG.ParryDA.FryAE.et al.De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome.Nat Genet.201446551051524705253
- AlkurayaFS.CaiX.EmeryC.et al.Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected].Am J Hum Genet.201188553654721529751
- BondJ.RobertsE.SpringellK.et al.A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size.Nat Genet.200537435335515793586
- SirJH.BarrAR.NicholasAK.et al.A primary microcephaly protein complex forms a ring around parental centrioles.Nat Genet.201143111147115321983783
- HussainMS.BaigSM.NeumannS.et al.A truncating mutation of CEP135 causes primary microcephaly and disturbed centrosomal function.Am J Hum Genet.201290587187822521416
- HussainMS.BaigSM.NeumannS.et al.CDK6 associates with the centrosome during mitosis and is mutated in a large Pakistani family with primary microcephaly.Hum Mol Genet.201322255199521423918663
- FaragHG.FroehlerS.OexleK.et al.Abnormal centrosome and spindle morphology in a patient with autosomal recessive primary microcephaly type 2 due to compound heterozygous WDR62 gene mutation.Orphanet J Rare Dis.2013817824228726
- MarthiensV.RujanoMA.PennetierC.TessierS.Paul-GilloteauxP.BastoR.Centrosome amplification causes microcephaly.Nat Cell Biol.201315773174023666084
- MclntyreRE.Lakshminarasimhan ChavaliP.IsmailO.et al.Disruption of mouse Cenpj, a regulator of centriole biogenesis, phenocopies Seckel syndrome.PLoS Genet.2012811e100302223166506
- FishJL.KosodoY.EnardW.PaaboS.HuttnerWB.Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells.Proc Natl Acad Sci U S A.200610327104381044316798874
- YangYJ.BaltusAE.MathewRS.et al.Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation.Cell.201215151097111223178126
- AwadS.Al-DosariMS.Al-YacoubN.et al.Mutation in PHC1 implicates chromatin remodeling in primary microcephaly pathogenesis.Hum Mol Genet.201322112200221323418308
- GruberR.ZhouZ.SukchevM.JoerssT.FrappartPO.WangZQ.MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1-Cdc25 pathway.Nat Cell Biol.201113111325133421947081
- KielarM.TuyFP.BizzottoS.et al.Mutations in Eml1 lead to ectopic progenitors and neuronal heterotopia in mouse and human.Nat Neurosci.201417792393324859200
- SheenVL.DixonPH.FoxJW.et al.Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females.Hum Mol Genet.200110171775178311532987
- NakamuraF.StosselTP.HartwigJH.The filamins: organizers of cell structure and function.Cell Adh Migr.20115216016921169733
- CarabalonaA.BeguinS.Pallesi-PocachardE.et al.A glial origin for periventricular nodular heterotopia caused by impaired expression of Filamin-A.Hum Mol Genet.20122151004101722076441
- PilzDT.MatsumotoN.MinnerathS.et al.LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation.Hum Mol Genet.1998713202920379817918
- HaverfieldEV.WhitedAJ.PetrasKS.DobynsWB.DasS.Intragenic deletions and duplications of the LIS1 and DCX genes: a major disease-causing mechanism in lissencephaly and subcortical band heterotopia.Eur J Hum Genet.200917791191819050731
- MatsumotoN.LeventerRJ.KucJA.et al.Mutation analysis of the DCX gene and genotype/phenotype correlation in subcortical band heterotopia.Eur J Hum Genet.20019151211175293
- BaumbachJ.MurthyA.McClintockMA.et al.Lissencephaly-1 is a context-dependent regulator of the human dynein complex.Elife.20176.
- HoreshD.SapirT.FrancisF.et al.Doublecortin, a stabilizer of microtubules.Hum Mol Genet.1999891599161010441322
- SchaarBT.KinoshitaK.McConnellSK.Doublecortin microtubule affinity is regulated by a balance of kinase and phosphatase activity at the leading edge of migrating neurons.Neuron.200441220321314741102
- CaspiM.AtlasR.KantorA.SapirT.ReinerO.Interaction between LIS1 and doublecortin, two lissencephaly gene products.Hum Mol Genet.20009152205221311001923
- BaiJ.RamosRL.AckmanJB.ThomasAM.LeeRV.LoTurcoJJ.RNAi reveals doublecortin is required for radial migration in rat neocortex.Nat Neurosci.20036121277128314625554
- TsaiJW.ChenY.KriegsteinAR.ValleeRB.LIS1 RNA interference blocks neural stem cell division, morphogenesis, and motility at multiple stages.J Cell Biol.2005170693594516144905
- YamadaM.YoshidaY.MoriD.et al.Inhibition of calpain increases LIS1 expression and partially rescues in vivo phenotypes in a mouse model of lissencephaly.Nat Med.200915101202120719734909
- FernandezV.Llinares-BenaderoC.BorrellV.Cerebral cortex expansion and folding: what have we learned?EMBO J.201635101021104427056680
- JaglinXH.PoirierK.SaillourY.et al.Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria.Nat Genet.200941674675219465910
- GuerriniR.MeiD.CordelliDM.PucattiD.FranzoniE.ParriniE.Symmetric polymicrogyria and pachygyria associated with TUBB2B gene mutations.Eur J Hum Genet.201220999599822333901
- CampeauPM.KasperaviciuteD.LuJT.et al.The genetic basis of DOORS syndrome: an exome-sequencing study.Lancet Neurol.2014131445824291220
- BalestriniS.MilhM.CastiglioniC.et al.TBC1D24 genotype-phenotype correlation: Epilepsies and other neurologic features.Neurology.2016871778527281533
- FalaceA.BuhlerE.FaddaM.et al.TBC1D24 regulates neuronal migration and maturation through modulation of the ARF6-dependent pathway.Proc Natl Acad Sci U S A.201411162337234224469796
- WillerT.LeeH.LommelM.et al.ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome.Nat Genet.201244557558022522420
- Beltran-Valero de BernabeD.CurrierS.SteinbrecherA.et al.Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome.Am J Hum Genet.20027151033104312369018
- van ReeuwijkJ.GrewalPK.SalihMA.et al.Intragenic deletion in the LARGE gene causes Walker-Warburg syndrome.Hum Genet.2007121668569017436019
- MyshrallTD.MooreSA.OstendorfAP.et al.Dystroglycan on radial glia end feet is required for pial basement membrane integrity and columnar organization of the developing cerebral cortex.J Neuropathol Exp Neurol.201271121047106323147502
- HuH.YangY.EadeA.XiongY.QiY.Breaches of the pial basement membrane and disappearance of the glia limitans during development underlie the cortical lamination defect in the mouse model of muscle-eye-brain disease.J Comp Neurol.2007501116818317206611
- AckroydMR.WhitmoreC.PriorS.et al.Fukutin-related protein alters the deposition of laminin in the eye and brain.J Neurosci.20113136129271293521900571
- Bahi-BuissonN.PoirierK.BoddaertN.et al.GPR56-related bilateral frontoparietal polymicrogyria: further evidence for an overlap with the cobblestone complex.Brain.2010133113194320920929962
- LuoR.JeongSJ.JinZ.StrokesN.LiS.PiaoX.G protein-coupled receptor 56 and collagen III, a receptor-ligand pair, regulates cortical development and lamination.Proc Natl Acad Sci U S A.201110831129251293021768377
- AckmanJB.AniksztejnL.CrepelV.et al.Abnormal network activity in a targeted genetic model of human double cortex.J Neurosci.200929231332719144832
- MartineauFS.SahuS.PlantierV.et al.Correct laminar positioning in the neocortex influences proper dendritic and synaptic development.Cereb Cortex.20182882976299029788228
- LaprayD.PopovaIY.KindlerJ.et al.Spontaneous epileptic manifestations in a DCX knockdown model of human double cortex.Cereb Cortex.201020112694270120164125
- PetitLF.JalabertM.BuhlerE.et al.Normotopic cortex is the major contributor to epilepsy in experimental double cortex.Ann Neurol.201476342844225074818
- CederquistGY.LuchniakA.TischfieldMA.et al.An inherited TUBB2B mutation alters a kinesin-binding site and causes polymicrogyria, CFEOM and axon dysinnervation.Hum Mol Genet.201221265484549923001566
- De la RossaA.BelloneC.GoldingB.et al.In vivo reprogramming of circuit connectivity in postmitotic neocortical neurons.Nat Neurosci.201316219320023292682
- RouauxC.ArlottaP.Direct lineage reprogramming of post-mitotic callosal neurons into corticofugal neurons in vivo.Nat Cell Biol.201315221422123334497
- WallNR.WickershamIR.CetinA.De La ParraM.CallawayEM.Monosynaptic circuit tracing in vivo through Cre-dependent targeting and complementation of modified rabies virus.Proc Natl Acad Sci U S A.201010750218482185321115815
- ZinggB.ChouXL.ZhangZG.et al.AAV-mediated anterograde trans-synaptic tagging: mapping corticocollicular input-defined neural pathways for defense behaviors.Neuron.2017931334727989459