
Abstract
Prospective, community-based studies allow evaluation of associations between cognitive functioning and synaptic measures, controlled for age-related pathologies. Findings from >400 community-based participants are reviewed. Levels of two presynaptic proteins, complexin-I (inhibitory terminals), and complexin-II (excitatory terminals) contributed to cognitive variation from normal to dementia. Adding the amount of protein-protein interaction between two others, synaptosome-associated protein-25 and syntaxin, explained 6% of overall variance. The presynaptic protein Munc18-1 long variant was localized to inhibitory terminals, and like complexin-I, was positively associated with cognition. Associations depended on Braak stage, with the level of complexin-I contributing nearly 15% to cognitive variation in stages 0-II, while complexin-II contributed 7% in stages V-VI. Non-denaturing gels identified multiple soluble N-ethylmaleimide-sensitive factor attachment protein receptor protein-protein (SNARE) complexes in frontal and in temporal lobes, making specific contributions to cognitive functions. Multiple mechanisms of presynaptic plasticity contribute to cognitive function during aging.
Los estudios prospectivos realizados en la comunidad permiten evaluar las asociaciones entre el funcionamiento cognitivo y las mediciones sinápticas, controladas por patologías relacionadas con la edad. Se revisan los hallazgos de más de 400 participantes de la comunidad. Los niveles de dos proteínas presinápticas, complexina-I (terminales inhibitorios) y complexina-II (terminales excitatorios) contribuyeron a la variación cognitiva entre la situación normal y la demencia. Al agregar la interacción proteína-proteína entre la proteína 25 asociada al sinaptosoma y la sintaxina, se explicó el 6% de la varianza general. La variante larga de la proteína presináptica Munc 18-1 se localizó en terminales inhibitorios y, al igual que la complexina I, se asoció positivamente con la cognición. Las asociaciones dependían de la etapa de Braak, siendo el nivel de complexina-I responsable de casi el 15% de la variación cognitiva para las etapas 0-II, mientras que la complexina-II contribuyó con el 7% en las etapas V-VI. Los geles no desnaturalizantes identificaron múltiples complejos de proteína-proteína del receptor de proteína de unión al factor sensible a N-etilmaleimida soluble (SNARE) en los lóbulos frontales y temporales, los cuales contribuyen de manera específica a las funciones cognitivas. Durante el envejecimiento participan múltiples mecanismos de plasticidad presináptica en la función cognitiva.
Des études prospectives communautaires permettent d’évaluer des associations entre le fonctionnement cognitif et des mesures synaptiques, contrôlées pour des pathologies liées à l’âge. Des résultats issus de plus de 400 participants communautaires sont analysés. Les taux de complexine-I (terminaisons inhibitrices) et de complexine-II (terminaisons excitatrices), deux protéines présynaptiques, contribuent aux variations cognitives de la normalité à la démence. S’y ajoute l’interaction protéine-protéine entre la protéine SNAP25 (protéine 25 associée au synaptosome) et la syntaxine, expliquant 6 % de la variance totale. Le variant long de la protéine pré-synaptique Munc18-1 est localisé sur les terminaisons inhibitrices et associé positivement à la cognition, comme la complexine-I. Les associations dépendent du stade Braak, le taux de complexine-I étant responsable de presque 15 % de la variation cognitive pour les stades 0-II alors que la complexine-II contribue pour 7 % aux stades V-VI. Des gels non dénaturants identifient des complexes multiples protéine-protéine SNARE (Soluble N-ethylmaleimide-sensitive factor Attachment protein REceptor) dans les lobes frontaux et temporaux, contribuant spécifiquement aux fonctions cognitives. Au cours du vieillissement, de nombreux mécanismes de plasticité présynaptique participent à la fonction cognitive.
Background
Memories, combined with a current state of being, create thoughts, emotions, and actions through high-fidelity transmission of chemical information at synapses—the subcellular elements that link neurons into cognitive networks. Cataloguing the molecular constituents of the pre- and postsynaptic components of synapses followed earlier anatomical and electrophysiological descriptions. Citation1 As tools to investigate the proteins enriched in synaptic terminals became available, research into disturbances of synapses in neuropsychiatric disease expanded in parallel with studies of animal models. Human studies of synapses based on case-control autopsy series contrast pathologically defined disease states with samples free from pathological insult, and have been reviewed in detail, including a meta-analysis. Citation2 , Citation3 Of note, the average number of cases of Alzheimer disease (AD) versus healthy controls in the studies included in the meta-analysis was 10 per group. Investigations with prospective assessment of cognitive function allowing clinical-pathological correlation are less common, and rarer still are community- rather than clinic-based studies.

A previous review of presynaptic proteins in AD made five broad conclusions. Citation4 First, not all presynaptic proteins were comparably affected. Second, the levels of presynaptic proteins in different brain regions were differentially affected. Third, associations between neurofibrillary pathology and presynaptic proteins were stronger than with amyloid pathology. Fourth, more severe cognitive impairment was generally associated with more severe presynaptic pathology. Fifth, the relationship of stage of illness and presynaptic pathology was complex, with a suggestion of early increases in protein levels, followed by decline in the more severe stages of illness.
Since the previous review, the conceptual distinction between AD pathology and the Alzheimer dementia syndrome has gained importance. Citation5 Multiple, and only partially overlapping, risk factors contribute to the multiple pathologies (including AD pathology) that are associated with the Alzheimer dementia syndrome. The present review focuses on reports from the Rush Memory and Aging Project (MAP), a relatively large, community-based study with prospective enrolment of participants free from dementia, annual assessment of cognitive function using a comprehensive battery of 21 tests, with an overall follow-up rate of more than 90%, and an autopsy rate in excess of 80%. Citation6 , Citation7 The opportunity to study a wide range of cognitive function, and to prospectively assess cognitive decline in a large number of cases from MAP allows re-evaluation of the earlier conclusions, with a focus on the relationship between cognitive function and presynaptic proteins.
Are all presynaptic proteins comparably affected in aging and Alzheimer dementia?
The antibody tools used to assay presynaptic proteins in MAP are well characterized, with most produced in-house (Honer), and supplemented with commercial reagents. Citation8 - Citation12 The availability of large volumes of monoclonal antibodies allowed development of a high-throughput enzyme-linked immunoadsorbent assay (ELISA) configured to use 384-well plates. Citation9 , Citation13 Samples of frozen brain tissue from MAP autopsies are dissected from frozen blocks biopsied from frozen slabs, with white matter trimmed away. All brains undergo an extensive neuropathological assessment generating quantitative measures of age-related pathologies. Citation14 - Citation16 An extensive series of publications describes the occurrence of multiple pathologies rather than discrete degenerative diseases in the vast majority of brains. Citation6 , Citation17 These pathologies contribute additively to explaining about 40% of the variance in rate of cognitive decline following joining the study, and about two-thirds of Alzheimer dementia cases. Citation18 However, across the spectrum of aging, substantial variation in cognitive function and rate of decline remains unexplained.
The initial report on the potential role of presynaptic proteins in contributing to cognitive function in the MAP study assayed multiple proteins, in multiple brain regions. Citation19 The proteins studied included synaptophysin, a synaptic vesicle-associated protein commonly assayed in dementia research, and used in clinical pathology. Synaptophysin is present in all synapses, but may be enriched in glutamate-containing terminals. Citation20 The three soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins (syntaxin-1A/1B, synaptosome-associated protein-25 [SNAP-25], and vesicle-associated membrane protein [VAMP]-1/2) were included. These three proteins are vital for fusion of synaptic cvesicles with the presynaptic membrane. Citation21 The SNARE modulating proteins complexin-I and complexin-II, localized to inhibitory (γ-aminobutyric acid - GABA) and excitatory (glutamate) presynaptic terminals respectively, were also assayed. Citation10 , Citation22 For correlative studies with cognitive function, the MAP neuropathology assessments aggregate across brain regions affected by age-related pathologies. Citation15 A similar approach was used for the presynaptic protein studies. The regions currently studied are the hippocampus, dorsolateral prefrontal cortex (Brodmann area [BA] 46/9), inferior temporal cortex (BA20), calcarine cortex (BA17), posterior putamen, and ventromedial caudate. The analytic approach aggregated findings across brain regions; proteins remained distinct.
Reporting on an initial sample of more than 250 brains, we first compared a subset of definite AD cases with controls with no or minimal pathology, an approach similar to most studies in the literature. Citation19 Global cognitive function prior to death was correlated with the level of synaptophysin; the protein level was 30% lower in the definite AD cases. Dementia was modeled as a function of Alzheimer-related pathology, cerebral infarcts, and the level of each of the three presynaptic proteins. Alzheimer-related pathology was associated with higher odds of about 5. By contrast, the odds ratio for infarcts was about 2, similar in magnitude to the protective effect of having intact VAMP or complexin presynaptic proteins (odds of about a half). The level of the presynaptic proteins contributed anywhere from 1% to 9% additional variance in predicting global cognitive function, after the effects of age, sex, education, and pathology were accounted for. Across multiple levels of age-related pathology, participants at higher percentiles for presynaptic markers had better global cognitive function. A subsequent analysis using an extended series of samples modeled the rate of cognitive decline rather than cognitive function at the final visit prior to death. Citation18 The age-related pathologies contributed to rate of decline: Alzheimer pathology just over 30%, cerebrovascular disease only about 2%, and Lewy bodies about 5%. Presynaptic markers were again found to be protective, contributing 6% to the overall model, representing a similar magnitude of effect as cerebrovascular or Lewy body diseases. Figure 1 illustrates updated analyses (n=420) from the ongoing study. Findings remain consistent with the earlier report.
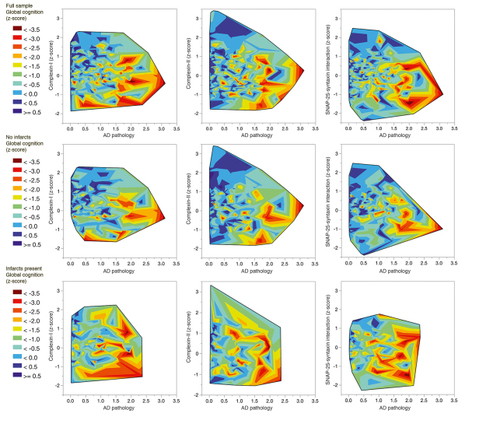
In these studies, effects of the presynaptic marker of inhibitory terminals, complexin-I were greater than those for the marker of excitatory terminals, complexin-II. We probed inhibitory terminals further with assays of the presynaptic protein Munc18-1, a syntaxin-binding protein with two splice variants. Citation23 The long splice variant (M18L) localizes to GABA-containing terminals, while the short variant is broadly distributed in glutamate- and in GABA-containing terminals. The long variant was essentially found in the synaptosomal fraction matching lipid raft domains, while M18S was widely distributed across cytosolic and synaptosomal fractions in water- and lipid-soluble compartments. Studying more than 300 samples of MAP prefrontal cortex, we created a model of global cognitive function with predictors age, sex, education, and multiple pathologies: a standardized composite of neuritic and diffuse plaques and neurofibrillary tangles, Citation15 macro- and microinfarcts, Lewy bodies, hippocampal sclerosis, arteriolosclerosis, and atherosclerosis. After accounting for the effects of these measures on cognitive function, we reported about 50% lower M18L in dementia cases ( P <0.001), which contributed almost 2% of variation across the entire spectrum of cognitive function.
To summarize so far, in the MAP study participants, dementia was associated with lower levels of some, but not all, presynaptic proteins. Using the same panel of antibodies reported here, similar findings were described in a study of samples from participants 70 to 100+ years of age, where lower levels of complexin-I were most prominently associated with dementia. Citation24 Samples from the Oxford study of dementia (some of which were prospectively assessed) studied by us also showed lower levels of SNARE proteins and synaptophysin in frontal lobe, but only with the most severe cognitive impairment. Citation25 An early report from a prospective investigation, the Bronx Aging study, showed weak correlations between the presynaptic protein synaptophysin and cognition in the temporal and parietal cortices, and no correlation with the frontal cortex. Citation26 More recently, select synaptic proteins assayed from frontal cortex with mass spectroscopy showed differences in levels related to neurological diagnosis of degenerative disease, and associations with decline in a screening test, the Mini-Mental State Examination. Citation27 A similar result was reported for synaptophysin in the frontal lobe in a series of 32 prospectively studied cases over 90 years of age, and in more than 100 cases with Lewy body disease or AD. Citation28 , Citation29 A marker of glutamate containing terminals was lower in frontal cortex in a study of nearly 175 cases, with the strongest association with the most severe rating of dementia. Citation30 With a continuous rather than a categorical variable to assess cognition, and a larger sample, we observed associations across the range of cognitive function with levels of specific presynaptic proteins, particularly complexin-I and the long variant of Munc18-1. These findings also applied to the prospective assessments of cognitive decline. This may support models of early decline in inhibitory neuronal function, or a shift in the balance of inhibitory and excitatory terminal function related to cognitive impairment in aging. Citation31 - Citation33 Additionally, the relationship between early loss in other neurotransmitter systems (ie, cholinergic) and effects on presynaptic terminals requires investigation. Citation34
Are presynaptic proteins comparably affected in all brain regions?
The present findings also support the conclusion that not all brain regions are equally affected. This was noted in early studies comparing AD cases with controls, and meta-analysis continues to support the prominence of the hippocampal presynaptic protein loss in AD. Citation3 , Citation35 A limitation of most of the synaptic protein work on MAP samples is reliance on biochemical assays of homogenates. Immunocytochemical studies of discrete neuronal connections such as the perforant pathway projection to the molecular layer of the dentate gyrus may be highly informative. Citation36 , Citation37 A more recent study of more than 150 brains (with a categorical description of three levels of cognitive impairment) showed differences in the molecular layer, again, separating those with the most severe level of impairment from the other groups. Citation38 Ongoing studies with MAP hippocampal sections using immunocytochemistry may reveal more subtle findings at earlier stages of cognitive impairment, and more definitively relate preliminary findings relating impaired neurogenesis to presynaptic protein loss and dysfunction. Citation39
What are the associations between illness stage, age-related pathology, and presynaptic proteins?
Neurofibrillary tangle pathology exhibits a progressive pattern of distribution across brain regions, captured by scoring the Braak stage of illness. Citation40 With more than 400 brains scored for Braak stage, we studied the associations of two presynaptic proteins localized to inhibitory (complexin-I) and excitatory (complexin-II) terminals with this measure. Citation41 In the earliest Braak stages (0-II), cognition was related to one form of pathology only (hippocampal sclerosis), with the overall model explaining a third of the variation in cognitive function. The level of complexin-I contributed nearly 15% additional variation in cognitive function; there was no association with the level of complexin-II or with an overall measure of presynaptic terminals created by calculating the mean level of the three SNARE proteins. In Braak stages III to IV, cognition was related to multiple pathologies (macroinfarcts, hippocampal sclerosis, and tau accumulation), accounting for nearly a quarter of the variation over the range of cognitive function at these stages. There was no association with complexin-I or complexin-II, or with the overall measure of presynaptic terminals. Once the final Braak stages (V-VI) were reached, cognition continued to be associated with pathologies (hippocampal sclerosis, tau accumulation), again accounting for nearly a quarter of the variation of cognition. In contrast to the findings at the least severe stages, at the most severe stages the level of complexin-II contributed 7% to the variation of cognition, while complexin-I and the overall measure of presynaptic terminals did not make a significant contribution. Of note, the above associations between cognitive performance and the inhibitory (complexin-I, M18L), or the excitatory (complexin-II) synaptic deficits were not influenced by the overall synapse loss, as the inclusion of variables accounting for global synaptopathy (eg, synaptophysin levels, or the average of the three SNARE proteins) did not modify the findings. This study also included a preliminary investigation of the relationship between presynaptic proteins and neuritic plaques, using confocal microscopy to assess the neuropil within plaques. The complexins showed minimal colocalization with either amyloid or tau within a plaque, in contrast to VAMP and SNAP-25 that showed focal areas of overlap with both pathological markers.
To summarize, staging of AD, or of age-associated neuropathologies, continues to be of great interest and possible relevance to hypotheses of spread of pathology across synapses. Citation36 , Citation42 - Citation44 A previous prospective study of 48 participants using several of the present panel of antibodies reported higher levels of presynaptic proteins synaptophysin, syntaxin, and SNAP-25 at Braak stage III, with lower levels at stages V-VI. Citation45 In MAP samples, we observed region- and protein-specific associations with cognitive function, depending on early or late Braak stage.
Studies using a range of techniques from electron microscopy to immunocytochemistry demonstrate the presence of synaptic elements in neuritic pathology, and in Lewy bodies. Citation46 - Citation48 Model systems indicate that the neuropil environment adjacent to plaques may differ from plaque-free neuropil. Citation49 , Citation50 As reported previously, not all presynaptic markers from our panel colocalize with neuritic plaques. Citation37 VAMP and other SNARE markers appear most commonly, while complexin proteins are absent. A clearer description of the sequence of loss or accumulation of synaptic markers within neuritic plaques requires further study, as does the possibility to include assessment of synapses in evolving models of the spread of tau pathology, and of relationships between proteins such as Fas-associated death domain, which may link complex neurobiological functions including apoptosis, synaptic elimination, and neural plasticity. Citation51 - Citation53
Presynaptic protein-protein interactions
Studies in other neuropsychiatric disorders indicate the interactions between SNARE proteins may differ more between illness and health than the levels of the individual proteins themselves. Citation8 , Citation13 , Citation54 - Citation56 Early experiments demonstrated that in unboiled samples, SNARE protein-protein interactions have the uncommon feature of being stable in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels (Figure 2A) . Citation57 - Citation59 This property allows quantification of the protein-protein interactions, and spurred the application of additional technical strategies. A high-throughput ELISA, using an immobilized antibody reactive with one SNARE protein to capture or pull-down the target or “bait” antigen, allows “capture” of binding partner proteins that can be detected with a second antibody. Citation13 Non-denaturing (blue-native) gels allow visualization of more protein complexes than seen in SDS-PAGE gels. Citation60 Immunoprecipitation directed at one SNARE protein, followed by mass spectrometry of the total protein complex products, allows description of the SNARE “interactome” in health and diseased human brain tissue. Citation61 , Citation62
We used the ELISA capture assay to measure binary SNARE protein-protein interactions in MAP brain tissues. Citation19 Higher levels of SNAP-25-syntaxin interaction (controlled for the level of SNAP-25 as well as for pathology) were associated with lower likelihood of dementia (odds ratio about a half). The SNAP-25-syntaxin interaction contributed an additional 2% to the variance in cognitive function across the full range of outcomes, controlled for age, sex, education, and pathology. This SNARE protein-protein interaction was part of the 6% protective contribution of presynaptic protein measures to the model of overall rate of cognitive decline in MAP participants. Citation18 An updated illustration of these findings appears in Figure 1.
To further explore the association of cognition with SNARE protein-protein interactions, and to assess a potential role for stage of illness, we studied nearly 200 MAP cases with tissue from the middle frontal gyrus (affected by Alzheimer-type pathology later in the course of illness) and the inferior temporal gyrus (affected earlier). Citation51 Blue-native gels were used to allow identification of 10 low-to-high molecular weight protein complexes (30 to 500 kDa). An interesting double dissociation was observed. In the temporal lobe, the trimeric SNARE protein interaction (SNAP-25, syntaxin, VAMP) was associated with the rate of cognitive decline, and with global cognitive function nearest to death. For the latter model, the SNARE complex contributed just shy of 10% to the variance over the range of cognitive function. The trimeric SNARE complex measured in the frontal lobe was not associated with cognitive function or with decline. In the frontal lobe, the ratio between a high -molecular- weight complex from inhibitory terminals (identified with a complexin-I antibody), and the equivalent complex in excitatory terminals (identified with a complexin-II antibody) showed an association with cognitive decline, and with cognitive function. Similar to the observation for the SNARE complex in temporal cortex, this ratio contributed nearly 10% to the variance over the range of cognitive function. The same inhibitory/excitatory complex ratio measured in the temporal lobe was not associated with cognitive function or with decline. Additional support for differences in molecular pathology or reserve between the two lobes was the observation of divergent associations with domains of cognitive function. The trimeric SNARE complex in the inferior temporal lobe was associated with semantic memory and visuospatial ability, while the inhibitory:excitatory ratio, high-molecular-weight complex in the frontal lobe was associated with episodic memory and perceptual speed.
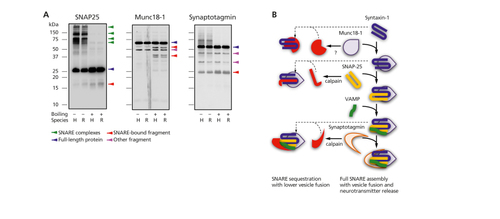
The multimeric SNARE protein complexes comprised of full-length proteins are illustrated in Figure 2B. On -going work identified lower molecular weight fragments of the SNARE and SNARE-associated proteins SNAP-25, Munc18-1 and synaptotagmin (Figure 2A). In other neuropsychiatric illnesses such as schizophrenia, lower expression of these fragments appears to be associated with higher SNARE protein-protein interactions. Citation63 As seen on the left side of Figure 2B, the fragments may sequester full-length SNARE proteins to modulate protein-protein interactions. The potential role of these protein fragments in contributing to the lower level of SNARE protein-protein interaction in cognitive disorders is a focus of current studies.
To summarize, moving beyond assessment of presynaptic protein levels to investigate protein-protein interactions is increasingly common as a research strategy. In schizophrenia, evidence from animal models and postmortem studies supports the importance of increased SNARE protein-protein interactions to pathophysiology. Citation8 , Citation13 , Citation54 - Citation56 In degenerative disorders, regulation of SNARE complex formation received attention from animal models of neurodegeneration related to cysteine string protein alpha (CSP-alpha), and to synuclein. Citation64 - Citation66 In the CSP-alpha report, a small series of 15 samples (5 each control, AD and Parkinson disease) demonstrated lower SNARE complex formation in the diseased samples. Citation65 Our findings in the MAP series are similar, and demonstrate an association between SNARE complex formation and cognitive function that is in part regionally specific, and may be more prominent for specific cognitive domains. The SNARE protein interactome is also of importance as part of the development of biomarkers for AD. Immunoprecipitating proteins using a SNAP-25 antibody from our panel, and a mass spectroscopy assay, demonstrated that fragments of SNAP-25 were elevated in cerebrospinal fluid of patients with cognitive impairment, and in an animal model. Citation61 , Citation67 Of further interest is the possibility that activity of the proteolytic enzyme calpain could be involved in these processes. Citation68 - Citation70
Conclusion
Review of findings from the Memory and Aging Project demonstrates the value of community-based investigation of the range of cognitive function in older persons, along with the multiple age-related pathologies, in order to identify the contribution of presynaptic protein mechanisms to cognition. Emerging findings support complementary roles for inhibitory and excitatory terminals at different stages of illness. Beyond the levels of individual proteins, protein-protein interactions that are critical to the process of vesicular neurotransmission also contribute, and may differ regionally as well as show different strength of association with different cognitive domains. Ongoing research suggests processing of presynaptic proteins by specific enzymatic mechanisms may alter protein-protein interactions, and could provide a target for developing therapeutics.
WGH has received consulting fees and/or sat on Advisory Boards for the Canadian Agency for Drugs and Technology in Health, AlphaSights, In Silico, Otsuka, Lundbeck, Translational Life Sciences and Newron. AMB sat on the Advisory Board for Roche Canada. Other authors have no conflicts of interest to declare. The authors acknowledge grant support from the Canadian Institutes of Health Research (MT-14037, MOP-81112), the National Institute of Aging (RO1AG17917, RO1AG42210), the BC Mental Health and Addictions Research Institute, and the Jack Bell Chair in Schizophrenia.
REFERENCES
- WilhelmBGMandadSTruckenbrodtSet alComposition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteinsScience201434461871023102824876496
- ScheffSWNeltnerJH,NelsonPTIs synaptic loss a unique hallmark of Alzheimer’s disease?Biochem Pharmacol201488451752824412275
- de WildeMCOverkCRSijbenJWMasliah EMeta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerabilityAlzheimers Dement201612663364426776762
- HonerWGPathology of presynaptic proteins in Alzheimer’s disease: more than simple loss of terminalsNeurobiol Aging 2003 24810471062
- JamesBDBennettDACauses and patterns of dementia: an update in the era of redefining Alzheimer’s diseaseAnnu Rev Public Health201940658430642228
- BennettDABuchmanASBoylePABarnes LLWilsonRSSchneiderJAReligious orders study and rush memory and aging projectJ Alzheimers Dis201864suppl 1S161S18929865057
- BennettDASchneiderJABuchmanASMendes de LeonCBieniasJLWilsonRSThe Rush Memory and Aging Project: study design and baseline characteristics of the study cohortNeuroepidemiol2005254163175
- HonerWGFalkaiPBayerTAet alAbnormalities of SNARE mechanism proteins in anterior frontal cortex in severe mental illnessCereb Cortex200212434935611884350
- HonerWGFalkaiPYoungCet alCingulate cortex synaptic terminal proteins and neural cell adhesion molecule in schizophreniaNeuroscience1997781991109135092
- TakahashiSYamamotoHMatsudaZet alIdentification of two highly homologous presynaptic proteins distinctly localized at the dendritic and somatic synapsesFEBS Lett199536834554607635198
- MatthewWDTsavalerLReichardtLFIdentification of a synaptic vesicle-specific membrane protein with a wide distribution in neuronal and neurosecretory tissueJ Cell Biol19819112572697298720
- Gil-PisaIMunarriz-CuezvaERamos-MiguelAUrigüenLMeanaJJGarcía-Sevilla JARegulation of munc18-1 and syntaxin-1A interactive partners in schizophrenia prefrontal cortex: down-regulation of munc18-1a isoform and 75 kDa SNARE complex after antipsychotic treatmentInt J Neuropsychopharmacol201215557358821669024
- BarakauskasVEBeasleyCLBarrAMet alA novel mechanism and treatment target for presynaptic abnormalities in specific striatal regions in schizophreniaNeuropsychopharmacol201035512261238
- SchneiderJABoylePAArvanitakisZBieniasJLBennettDASubcortical infarcts, Alzheimer’s disease pathology, and memory function in older personsAnn Neurol200762596617503514
- BennettDAWilsonRSSchneiderJAet alApolipoprotein E epsilon4 allele, AD pathology, and the clinical expression of Alzheimer’s diseaseNeurology200360224625212552039
- BennettDASchneiderJAArvanitakisZet alNeuropathology of older persons without cognitive impairment from two community-based studiesNeurology200666121837184416801647
- KapasiADecarliCSchneiderJAImpact of multiple pathologies on the threshold for clinically overt dementiaActa Neuropathol2017134217118628488154
- BoylePAYuLHonerWGSchneiderJAMuch of late life cognitive decline is not due to common neurodegenerative pathologiesAnn Neurol201374347848923798485
- HonerWGBarrAMSawadaKet alCognitive reserve, presynaptic proteins and dementia in the elderlyTransl Psychiatry201225e11422832958
- GrønborgMPavlosNJBrunkIChuaJJEMünster-WandowskiAJahnRQuantitative comparison of glutamatergic and GABAergic synaptic vesicles unveils selectivity for few proteins including MAL2, a novel synaptic vesicle proteinJ Neurosci201030121220053882
- SöllnerTBennettMKWhiteheartSWSchellerRHRothmanJEA protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusionCell19937534094188221884
- SawadaKBarrAMNakamuraMet alHippocampal complexin proteins and cognitive dysfunction in schizophreniaArch Gen Psychiatry200562326327215753239
- Ramos-MiguelAHercherCBeasleyCLet alLoss of Munc18-1 long splice variant in GABAergic terminals is associated with cognitive decline and increased risk of dementia in a community sampleMol Neurodegener20151016526628003
- BeeriMSHaroutunianVSchmeidlerJet alSynaptic protein deficits are associated with dementia irrespective of extreme old ageNeurobiol Aging2012331125
- MingerSLHonerWGEsiriMMet alSynaptic pathology in prefrontal cortex is present only with severe dementia in Alzheimer diseaseJ Neuropathol Exp Neurol2001601092993611589423
- DicksonDWCrystalHABevonaCHonerWVincentIDaviesPCorrelations of synaptic and pathological markers with cognition of the elderlyNeurobiol Aging19951632852987566338
- BereczkiEBrancaRMFrancisPTet alSynaptic markers of cognitive decline in neurodegenerative diseases: a proteomic approachBrain2018141258259529324989
- UbhiKPengKLessigSet alNeuropathology of dementia with Lewy bodies in advanced age: a comparison with Alzheimer diseaseNeurosci Lett2010485322222720849919
- HeadECorradaMMKahle-WrobleskiKet alSynaptic proteins, neuropathology and cognitive status in the oldest-oldNeurobiol Aging20093071125113418006193
- PoirelOMellaSVideauCet alModerate decline in select synaptic markers in the prefrontal cortex (BA9) of patients with Alzheimer’s disease at various cognitive stagesSci Rep20188193829343737
- SolodkinAVeldhuizenSDVan HoesenGWContingent vulnerability of entorhinal parvalbumin-containing neurons in Alzheimer’s diseaseJ Neurosci19961610331133218627368
- KoliatsosVEKecojevicATroncosoJCGastardMCBennettDASchneiderJAEarly involvement of small inhibitory cortical interneurons in Alzheimer’s diseaseActa Neuropathol2006112214716216758165
- BellKFSBennettDACuelloACParadoxical upregulation of glutamatergic presynaptic boutons during mild cognitive impairmentJ Neurosci20072740108101081717913914
- BeachTGHonerWGHughesLHCholinergic fibre loss associated with diffuse plaques in the non-demented elderly: the preclinical stage of Alzheimer’s disease?Acta Neuropathol19979321461539039461
- HonerWGDicksonDWGleesonJDaviesPRegional synaptic pathology in Alzheimer’s diseaseNeurobiol Aging19921333753821625766
- SuJHDengGCotmanCWTransneuronal degeneration in the spread of Alzheimer’s disease pathology: immunohistochemical evidence for the transmission of tau hyperphosphorylationNeurobiol Dis1997453653759440125
- WakabayashiKHonerWGMasliahESynapse alterations in the hippocampal-entorhinal formation in Alzheimer’s disease with and without Lewy body diseaseBrain Res1994667124327895080
- RobinsonJLMolina-PorcelLCorradaMMet alPerforant path synaptic loss correlates with cognitive impairment and Alzheimer’s disease in the oldest-oldBrain2014137Pt 92578258725012223
- TobinMKMusaracaKDisoukyAet alHuman hippocampal neurogenesis persists in aged adults and Alzheimer’s disease patientsCell Stem Cell20192497498231130513
- BraakHBraakENeuropathological stageing of Alzheimer-related changesActa Neuropathol19918242392591759558
- Ramos-MiguelASawadaKJonesAAet alPresynaptic proteins complexin-I and complexin-II differentially influence cognitive function in early and late stages of Alzheimer’s diseaseActa Neuropathol2017133339540727866231
- TaiH-CWangBYSerrano-PozoAFroschMPSpires-JonesTLHymanBTFrequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer’s diseaseActa Neuropathol Commun20142114625330988
- de CalignonAPolydoroMSuárez-CalvetMet alPropagation of tau pathology in a model of early Alzheimer’s diseaseNeuron201273468569722365544
- LewisJDicksonDWPropagation of tau pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studiesActa Neuropathol20161311274826576562
- Mukaetova-LadinskaEBGarcia-SieraFHurtJet alStaging of cytoskeletal and beta-amyloid changes in human isocortex reveals biphasic synaptic protein response during progression of Alzheimer’s diseaseAm J Pathol2000157262363610934165
- MasliahEHansenLAlbrightTMalloryMTerryRDImmunoelectron microscopic study of synaptic pathology in Alzheimer’s diseaseActa Neuropathol19918144284331903014
- MasliahEHonerWGMalloryMet alTopographical distribution of synaptic-associated proteins in the neuritic plaques of Alzheimer’s disease hippocampusActa Neuropathol19948721351428171963
- TakedaAMalloryMSundsmoMHonerWHansenLMasliahEAbnormal accumulation of NACP/alpha-synuclein in neurodegenerative disordersAm J Pathol199815223673729466562
- Spires-JonesTLHymanBTThe intersection of amyloid beta and tau at synapses in Alzheimer’s diseaseNeuron201482475677124853936
- KoffieRMMeyer-LuehmannMHashimotoTet alOligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaquesProc Natl Acad Sci U S A2009106104012401719228947
- Ramos-MiguelAJonesAASawadaKet alFrontotemporal dysregulation of the SNARE protein interactome is associated with faster cognitive decline in old ageNeurobiol Dis2018114314429496544
- BraakHDel TrediciKSpreading of tau pathology in sporadic Alzheimer’s disease along cortico-cortical top-down connectionsCereb Cortex20182893372338429982389
- KaufmanSKDel TrediciKThomasTLBraakHDiamondMITau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PARTActa Neuropathol20181361576729752551
- KatranchaSMKoleskeAJSNARE complex dysfunction: a unifying hypothesis for schizophreniaBiol Psychiatry201578635635826296424
- Ramos-MiguelABarakauskasVAlamriJet alThe SNAP25 interactome in ventromedial caudate in schizophrenia includes the mitochondrial protein ARF1Neuroscience2019
- Ramos-MiguelABeasleyCLDworkAJet alIncreased SNARE protein-protein interactions in orbitofrontal and anterior cingulate cortices in schizophreniaBiol Psychiatry201578636137325662103
- HayashiTMcMahonHYamasakiSet alSynaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assemblyEMBO J19941321505150617957071
- PevsnerJHsuSCBraunJEet alSpecificity and regulation of a synaptic vesicle docking complexNeuron19941303533618060616
- HayashiTYamasakiSNauenburgSBinzTNiemannHDisassembly of the reconstituted synaptic vesicle membrane fusion complex in vitroEMBO J19951410231723257774590
- WittigIBraunH-PSchäggerHBlue native PAGENat Protoc20061141842817406264
- BrinkmalmABrinkmalmGHonerWGet alSNAP-25 is a promising novel cerebrospinal fluid biomarker for synapse degeneration in Alzheimer’s diseaseMol Neurodegener2014915325418885
- GoriniGPonomarevaOShoresKSPersonMDHarrisRAMayfieldRDDynamin-1 co-associates with native mouse brain BKCa channels: proteomics analysis of synaptic protein complexesFEBS Lett2010584584585120114047
- Ramos-MiguelAGicasKAlamriJet alReduced SNAP25 Protein Fragmentation contributes to SNARE complex dysregulation in schizophrenia postmortem brainNeuroscience2019
- BurréJSharmaMTsetsenisTBuchmanVEthertonMRSüdhofTCAlpha-synuclein promotes SNARE-complex assembly in vivo and in vitroScience201032959991663166720798282
- SharmaMBurréJSüdhofTCProteasome inhibition alleviates SNARE-dependent neurodegenerationSci Transl Med20124147147ra113
- SharmaMBurréJBronkPZhangYXuWSüdhofTCCSPα knockout causes neurodegeneration by impairing SNAP-25 functionEMBO J201231482984122187053
- BrinkmalmABrinkmalmGHonerWGet alTargeting synaptic pathology with a novel affinity mass spectrometry approachMol Cell Proteomics201413102584259224973420
- AndoKKudoYTakahashiMNegative regulation of neurotransmitter release by calpain: a possible involvement of specific SNAP-25 cleavageJ Neurochem200594365165815992386
- GrumelliCBerghuisPPozziDet alCalpain activity contributes to the control of SNAP-25 levels in neuronsMol Cell Neurosci200839331432318721885
- HassenGWKesnerLStracherAet alEffects of novel calpain inhibitors in transgenic animal model of Parkinson’s disease/dementia with Lewy bodiesSci Rep2018811808330591714