
Abstract
This article describes the history of the diagnostic class of neurodevelopmental disorders (NDDs) up to DSM-5. We further analyze how the development of genetics will transform the classification and diagnosis of NDDs. In DSM-5, NDDs include intellectual disability (ID), autism spectrum disorder (ASD), and attention-deficit/hyperactivity disorder (ADHD). Physicians in German-, French- and English-speaking countries (eg, Weikard, Georget, Esquirol, Down, Asperger, and Kanner) contributed to the phenomenological definitions of these disorders throughout the 18th and 20th centuries. These diagnostic categories show considerable comorbidity and phenotypic overlap. NDDs are one of the chapters of psychiatric nosology most likely to benefit from the approach advocated by the National Institute of Mental Health’s Research Domain Criteria project. Genetic research supports the hypothesis that ID, ASD, ADHD, schizophrenia, and bipolar disorder lie on a neurodevelopmental continuum. The identification of recurrently observed copy number variants and disruptive gene variants in ASD (eg, CDH8, 16p11.2, SCN2A) led to the adoption of the genotype-first approach to characterize individuals at the etiological level.
Este artículo describe la historia de la clase diagnóstica de los trastornos del neurodesarrollo (TND) hasta el DSM-5. Además se analiza cómo el desarrollo de la genética transformará la clasificación y el diagnóstico de los TND. En el DSM-5, los TND incluyen la discapacidad intelectual (DI), los trastornos del espectro autista (TEA) y el trastorno por déficit de atención / hiperactividad (TDAH). Los médicos en países de habla alemana, francesa e inglesa (por ejemplo, Weikard, Georget, Esquirol, Down, Asperger y Kanner) contribuyeron a las definiciones fenomenológicas de estos trastornos a lo largo de los siglos XVIII y XX. Estas categorías diagnósticas muestran una importante comorbilidad y superposición fenotípica. Los TND constituyen uno de los capítulos de la nosología psiquiátrica que tienen más probabilidades de beneficiarse con el enfoque recomendado por el proyecto Research Domain Criteria (Criterios de Dominio de Investigación) del Instituto Nacional de Salud Mental. La investigación genética respalda la hipótesis de que la DI, el TEA, el TDAH, la esquizofrenia y el trastorno bipolar se encuentran en un continuo del neurodesarrollo. La identificación y observación repetida de variaciones en el número de copias y variaciones de genes disruptivos en el TEA (p. Ej., CDH8, 16p11.2, SCN2A) condujeron a la adopción de un enfoque primariamente genético para la caracterización etiológica de los sujetos.
Dans cet article qui décrit l’histoire de la classe diagnostique des troubles neurodéveloppementaux (TND) jusqu’au DSM-5, nous analyserons comment les progrès de la génétique vont transformer leur classification et leur diagnostic. Dans le DSM-5, les TND comprennent les handicaps intellectuels (HI), les troubles du spectre de l’autisme (TSA) et le déficit de l’attention/hyperactivité (TDAHA). Ce sont des médecins issus de pays germanophones, francophones et anglophones (comme Weikard, Georget, Esquirol, Down, Asperger et Kanner) qui ont participé à la définition phénoménologique de ces troubles, du XVIIIe au XXe siècle. On observe dans ces catégories diagnostiques une comorbidité et un chevauchement des phénotypes très importants. En termes de nosologie psychiatrique, les TND seraient les plus susceptibles de bénéficier de l’approche défendue par le NIMH (National Institute of Mental Health) : le projet Research Domain Criteria (Critères des domaines de recherche). Selon la recherche génétique, le HI, les TSA, le TDAHA, la schizophrénie et les troubles bipolaires appartiendraient à un continuum neurodéveloppemental. L’idée d’une caractérisation étiologique des individus en passant d’abord par la génétique est venue de l’observation répétée au sein des TSA de variations du nombre de copies et de variations génétiques perturbatrices (par exemple CDH8, 16p11.2, SCN2A).
The origin of the concept of neurodevelopmental disorder
“Developmental disorders” were included for the first time in DSM-III , Citation1 for the category that comprised autistic disorder. “Neurodevelopmental disorders” (NDDs) were introduced as an overarching disorder category in DSM‑5 . Citation2 This new section replaced a previous chapter that was termed “Disorders usually first diagnosed in infancy, childhood, or adolescence.” In ICD‑11 , the latest revision of the International Classification of Diseases published by the WHO, NDDs gained even more prominence by becoming an integral part of the title of the chapter on psychiatry: “Mental, behavioral or neurodevelopmental disorders.” Figure 1 outlines the main categories comprising neurodevelopmental disorders in DSM-5 , their historical background, and the hypothesis of the genetic spectrum of neurodevelopmental disorders that will be discussed later in this article.
In DSM‑5 , NDDs are defined as a group of conditions with onset in the developmental period, inducing deficits that produce impairments of functioning. NDDs comprise intellectual disability (ID); Communication Disorders; Autism Spectrum Disorder (ASD); Attention-Deficit/Hyperactivity Disorder (ADHD); Neurodevelopmental Motor Disorders, including Tic Disorders; and Specific Learning Disorders. The classification of NDDs in ICD‑11 does not diverge significantly from that in DSM‑5 . Importantly, all NDDs in DSM‑5 may include the specifier “associated with a known medical or genetic condition or environmental factor.” This specifier offers the clinician the possibility to document etiological factors, such as fragile X syndrome, and is a harbinger of the fact that this disorder category will probably be transformed in the coming decades by new data emerging from genetics research.
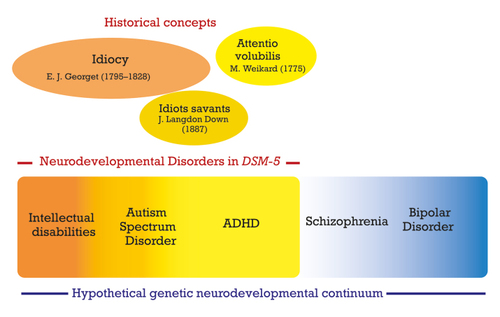
The validity of NDDs as a construct is supported by the high rates of comorbidity between various disorders within this diagnostic grouping. For instance, various studies showed that 22% to 83% of children with ASD have symptoms that satisfy the DSM-IV criteria for ADHD, and vice versa, 30% to 65% of children with ADHD have clinically significant symptoms of ASD. Citation3 Also, although not part of the criteria of ASD, accompanying intellectual or language impairments are frequent in ASD and their presence must be specified. NDDs share the characteristic of being diagnosed more often in males than females; DSM-5 mentions male-to-female ratios of 4:1 for the diagnosis of ASD, 2:1 for the diagnosis of ADHD in children, and 1.6:1 and 1.2:1 for mild and severe ID, respectively. The idea of gathering these various disorders into one single diagnostic group came with DSM-5 . As late as in the 10 th edition of Kaplan & Sadock’s comprehensive textbook of psychiatry (2017), Attention-Deficit Disorders (ADD), ASD, and ID were distinct disorders whose only commonality was being part of child psychiatry. According to German E. Berrios, Citation4 the historian of psychiatry, the concept of developmental disorder in psychiatry appeared for the first time in 1820 in a textbook by Étienne Jean Georget (1795-1828), a student of Philippe Pinel (1745-1826) and Jean-Étienne Esquirol (1772-1840), the pioneers of a modern psychiatric nosology based on mental symptoms instead of humoral concepts. Pinel translated William Cullen’s nosology into French in 1785, and published his own classification of mental illnesses in 1801; in his nosography, idiocy was a psychiatric disorder, along with mania, melancholia, and dementia. Correcting Pinel’s nosography to further delineate the field of mental illness, Georget Citation5 wrote (p 102) that Idiocy is “ a lack of development of intellectual faculties, ” (emphasis added) and he added in a footnote that “a developmental defect is not, strictly speaking, a disease ,” and that “idiots should be classified among monsters .” At a distance of two centuries, the word “monster” should not be read anachronistically as an insult, but as a reference to a genetic abnormality in the human body. Within the larger group of idiots, Georget included a subtype of “imbeciles,” with higher cognitive abilities. Georget died at the early age of 33, and Berrios suggests that Esquirol later adopted Georget’s developmental view, without quoting him. Esquirol Citation6 criticized Pinel for having differentiated dementia and idiocy on the sole basis of the degree of impairment of intelligence—Pinel considered idiocy as the most severe degree of dementia—and for admitting the existence of both acquired and innate idiocy. Instead, Esquirol stated that idiocy was not an illness, but a condition in which intellectual faculties could not achieve sufficient development . This statement by Esquirol has often been quoted in various textbooks over the following two centuries. Esquirol stated that there were innumerable degrees of severity and, like Georget, he distinguished two main categories: idiocy per se, a severe intellectual impairment, and imbecility, a mild or moderate impairment. In Georget’s suggestive words, imbeciles “have memory, can judge simple acts of life … are clean, know how to appreciate the difference between the sexes,” whereas “the idiots urinate and defecate wherever they are and are often very prone to masturbation” (p 104).
Early descriptions of ADHD, intellectual disabilities, and ASD
The symptoms of various NDDs were described long before the diagnostic concepts were delineated from each other in the mid-20 th century.
ADHD
The first known description of attention deficit was published in 1775 by the German physician Melchior Adam Weikard, under the name Mangel der Aufmerksamkeit/Attentio volubilis, in a book entitled Der philosophische Arzt . Citation7 This work was the subject of several editions. Weikard (1742-1803) began his medical career in 1764 in Bavaria, near his birthplace, as a doctor at the Bad Brückenau spa. In 1784 he was appointed doctor at the court of Empress Catherine II of Russia. He was known as a scholar and was a prolific author. In an edition of his book published in 1799, Citation8 Weikard devoted six pages to the description of attention deficit. He describes how sensory stimuli capture the patient’s attention and divert him from his thoughts: “... it is easier to perceive impressions through the sense organs than to form or retain ideas, to recover past memories or to do other reflective operations. Each sense can disturb us in our thoughts or thinking, distract us from our object and draw our attention to something else. Of all the senses, this occurs most often with hearing and sight. The result is distraction, lack of attention, inattention.” The Latin term “volubilis” comes from the verb “volvere” (to turn). The literal meaning of Volubilis is “easily rotating,” and by extension “fickle” or “changing.” This image of a permanent rotation of ideas is sometimes used by some patients who relate the subjective feeling that their attention does not hold in place but twirls.
Separating ASD from mental retardation
The terms “idiocy” and “imbecility” are attested to by the Oxford English Dictionary as early as the 16th century. Also, behaviors consistent with autism were described long before that diagnostic category was named and defined by Leo Kanner Citation9 in 1943 and Hans Asperger Citation10 in 1944. Russian specialists Citation11 suppose that many of the “holy fools,” or Orthodox church ascetics (yuródivïe) who roamed ancient Russia displayed autistic behaviors, being notoriously nonverbal, impervious to social conventions, and indifferent to cold weather or pain. Famous cases showed that some “idiots” had specific problems in social communication—consistent with autism—rather than a mere global mental deficiency. One such case is Victor, the Wild Boy of Aveyron (c 1788-1828), a feral child who was discovered when he was about 12. Victor was taken to Paris, and his case was taken up by a young physician, Jean Itard, who tried to instruct him and repeatedly assessed his sensory, intellectual, and affective progress over 5 years. Edouard Seguin Citation12 (1812-1880), a pioneer of the education of children with intellectual disability, first in France and afterward in the United States, recounted the various diagnostic hypotheses discussed at the time. According to Seguin, Pinel had declared Victor “idiotic” and therefore uneducable; Itard, on the other hand, asserted that the child was wild and entirely untaught. Itard’s observations over the following years suggest that both diagnoses might have been erroneous and that Victor might have been a child with autism, abandoned by his parents. As Itard reports, Victor demonstrated aberrant requesting strategies like the use of hand-over-hand (eg, Victor once seized Itard’s hand and directed it to a locked door to ask him to open it), he was using others as tools to satisfy his needs, a hallmark of the clinical picture was his difficulty initiating and maintaining social relationships, and even though he learned letters and some spelling, he could not use this knowledge in a regular interaction with others.
Dr J. Langdon Down (1828-1896) is best known for having described Down’s syndrome (trisomy 21). In a series of lectures delivered before the Medical Society of London in 1887, Citation13 he also described 10 cases of “idiots savants,” patients with exceptional abilities in an extremely narrow field, such as calendar calculating. While these cases were not all autistic, most of them would now satisfy Criterion B for ASD in DSM‑5 (ie, restricted and repetitive patterns of behavior, interest, or activities). In papers published after these lectures, Citation14 Down based himself on three decades of clinical experience to propose a classification of mental retardation into (i) “congenital;” (ii) “accidental;” and (iii) a third kind which he termed “developmental.” The latter type occurred in children who did not have the usual “physical aspects” of retardation. Some of these children had developed normally “until the period of second dentition,” and then suddenly regressed, lost their “wonted brightness” and speech, lived “in a world of their own,” spoke “in the third person,” had “rhythmical and automatic movements” and “lessened responsiveness to all endearments of friends.” Treffert Citation15 comments on Down’s interesting choice of the term “developmental retardation” for this category that probably corresponds to cases of both early-onset and late-onset regressive autism.
We have so far described the history of the diagnostic categories, based on phenomenological definitions, that came to comprise the NDDs in DSM-5 . We will now discuss how these psychopathological concepts can fare in the era of neurosciences.
How far along are we to a new molecular or genetic classification?
With the increasing appreciation of the considerable phenotypic overlap between NDDs, there has been a general trend to move away from the classification of disorders as discrete entities, to placing them within a spectrum. This is illustrated by the single diagnostic label for ASD in DSM-5 , which subsumes all previously designated subtypes. The classification is still mostly based on behavioral phenotypes and the Research Domain Criteria (RDoC) project by the National Institute of Mental Health has been calling for analysis at the genetic levels. Citation16,Citation17 With rapid advances in molecular biology, genetics, and genomics, neuropsychiatric disorders are increasingly grouped by their biology and in particular, the genes and variants that have been found to cause them. The clinical heterogeneity in NDDs is, however, reflected in extreme genetic heterogeneity, with a genetic diagnosis not possible for most cases. Unlike Mendelian disorders, such complex disorders are defined by a phenotype that is not caused by one or two pathogenic variants in a single gene, but rather by many genetic events with significant contribution from environmental factors. Disorders which have a simple genetic cause comprise a small fraction of the spectrum of NDDs, so the challenge is to develop even better approaches to link phenotype and genotype for NDDs.
Genetic studies of complex diseases traditionally followed a pathway from phenotype to genotype to gene, a so-called “forward genetics” approach. Most disease genes have, however, been identified by an approach known as “reverse genetics,” ie, the identification of a gene via its chromosomal location, without prior knowledge of its protein product or the molecular pathway in which it functions. The development of techniques to generate large amounts of genetic data by genome-wide genotyping has allowed genetic research to increasingly focus on the use of unbiased genome-wide approaches to gene identification. This led to the concept of “reverse phenotyping.” Citation18 In this approach, genetic markers are used to drive, or form the basis of new phenotypic definitions, so that phenotypes are refined based on genetic marker data. The goal is to define phenotypic groupings that are distinguished by higher rates of allele-sharing (linkage data) or more deviant allele frequencies (association data) than are seen in the traditional diagnostic categories. This “genotype-first” approach was elegantly elaborated by Stessman et al Citation19 as a way of defining the subtypes of a complex disease. They defined a “genetic subtype” as a gene in which recurrent mutations show an excess of burden in patients versus controls. A “molecular subtype,” on the other hand, constitutes a group of genetic subtypes that are linked together in a common pathway (eg coexpression, protein interaction network, etc). Citation20
The hypothesis of a genetic neurodevelopmental continuum
Over the past decade, evidence has been accumulating that childhood neurodevelopmental disorders such as ID, ASD, and ADHD share specific genetic risk alleles with each other, as well as with psychiatric disorders, particularly schizophrenia. Citation21 Copy number variants (CNVs) associated with ID were significantly enriched in patients with schizophrenia, supporting the view that many additional ID-related variants also confer risk to schizophrenia, but at reduced penetrance. Citation22 This has led authors to propose the model of a neurodevelopmental continuum, in which neurodevelopmental disorders, including schizophrenia, are seen as representing the diverse range of outcomes that follow from disrupted or deviant brain development. Citation23 Thus, childhood neurodevelopmental disorders (ID, ASD, ADHD) and adult psychiatric disorders (including both bipolar disorder [BPD] and schizophrenia) could better be conceptualized as lying on an etiological and neurodevelopmental continuum, rather than being defined as discrete entities. The model is based on emerging evidence for shared genetic and environmental risk factors and predicts that there are likely overlapping pathogenic mechanisms. The authors have taken this model one step further and have proposed the neurodevelopmental gradient hypothesis, in which disorders are graded according to the severity of neurodevelopmental impairment. Contributing features to this grading are age of onset relative to the typical age of onset for each of the disorders, the severity of associated cognitive impairment and the persistence of functional impairment. Although this model may appear to be a gross oversimplification of the diagnostic conundrum, it posits that the degree of neurodevelopmental impairment is currently the most recognizable of these features and makes clear predictions about the relative importance of the most damaging classes of rare genetic variants. In support of this hypothesis, Girirajan et al Citation24 have shown that the burden of DNA CNVs is positively correlated with the severity of childhood neurodevelopmental disorders, being greater in ID than in ASD, and greater in ASD with ID than in those children without ID. Kirov et al Citation25 have shown that the burden of large, rare CNVs implicated in neurodevelopmental disorders is greater in cases with developmental delay, autism, or congenital malformations, than in schizophrenia. The enrichment of rare mutations appears to be correlated with the degree of cognitive impairment both across and within diagnostic groups, but pathogenic CNVs and rare coding variants are found in ASD and schizophrenia, without gross cognitive impairment. Pathogenic CNVs are also found in individuals with subtle impairments of cognition but who do not have psychiatric diagnosis.
Neurodevelopmental disorders are associated with reduced fecundity. Citation26 One can postulate then, that genetic variants that confer a high risk for those disorders should be rare in the population due to negative selection. The frequency of such variants in the population should be a function of that selection pressure and the rate of replacement due to new, or de novo mutation. The increased rate of de novo variants in most neurodevelopmental disorders, supports this postulation. Individuals with severe, undiagnosed developmental disorders (DDs) are enriched for damaging de novo mutations (DNMs) in developmentally important genes (the Deciphering Developmental Disorders Study [DDDS], 2017). Citation27 In a large whole-exome sequencing study of 4293 families with individuals with developmental disorders and meta-analysis of data with another 3287 individuals with similar disorders, the DDDS identified 94 genes enriched for damaging de novo mutations (DNMs). The authors estimated that 42% of the cohort carried pathogenic DNMs in coding sequences, and approximately half disrupt gene function, with the remainder resulting in altered function. They concluded that de novo mutations account for approximately half of the genetic architecture of severe developmental disorders and more than 40% in intellectual disability. A large study of autism found the de novo mutation rate to be 5.2% in patients compared with 1.6% in unaffected siblings. Citation28 When the relative enrichment for de novo variants is compared across disorders, the rates are once again in decreasing frequency when placed against a gradient, from ID to ASD to schizophrenia, in line with predictions from the gradient hypothesis.
One can also explore whether the same genes or sets of functionally related genes tend to be implicated across neurodevelopmental disorders, and this appears to be the case. The same set of genes affected by loss of function de novo mutations are enriched in data from patients with ID, ASD, and schizophrenia. Citation29 Functionally, rare de novo and rare damaging coding variants tend to cluster broadly in similar biological processes, such as synaptic plasticity, chromatin modification, and targets of fragile X mental retardation protein, indicating that the same pathogenic mechanism may be affected across disorders. Citation30,Citation31
How can these findings be used in diagnosis and classification? ASD as a model for the genotype-first approach
The ASD population is etiologically and phenotypically heterogeneous. The genetic etiology of ASD is no less varied. Heritability of autism is high, with estimates ranging from 50% to 90% Citation32,Citation33 and rates of recurrence among non-twin siblings approaching 20%. Citation34 ASD not only has shared phenotypic overlap with many syndromic forms, such as Down syndrome, Prader-Willi/Angelman syndrome, and Fragile X-linked ID (about 4% to 5% of ASD), Citation35 but as described above, is also one of the disorders for which rare variants have been demonstrated to have a strong effect. Over 100 genes and genomic regions have been associated with ASD, and over 800 genes have been suggested to play a role in ASD.
Additive polygenic factors explain many ASD cases, especially those at the milder ends of the spectrum, and genome-wide association studies (GWAS) have provided support for the contribution of common DNA variants to the broader ASD phenotype. Citation36 Sebat et al Citation37 used a trio approach of an affected proband and their unaffected parents, to perform high-resolution DNA arrays to detect de novo CNVs in affected children. Their studies highlighted the power of identifying de novo events as likely causal variants across a broad range of genetic loci. De novo CNVs were up to ten times more prevalent in patients affected with ASD than in unaffected individuals. Citation38 De novo pathogenic variants and CNVs are now estimated to account for approximately 30% of simplex cases of ASD.
The identification of recurrently observed CNVs and disruptive gene variants in ASD led to the adoption of the genotype-first approach to characterize individuals at the etiological level. The genotype-first approach has led to the identification of ASD-specific genetic subtypes, variant-specific phenotypic spectra, and promising pharmacological treatment targets and psychosocial benefits to affected families. Citation39 One of the first subtypes to be described was that for heterogeneous, disruptive variants in CHD8 . CDH8 is involved in chromatin remodeling and its targets include many other genes that have also been associated with ASD. Citation40 The CDH8 genetic subtype includes physical characteristics such as macrocephaly, dysmorphic facial features, and gastrointestinal complaints and is more specific to ASD (≥87%) than to ID (~60%).
In contrast to the fairly narrow phenotype defined in carriers of pathogenic CDH8 variants, Steinman et al Citation41 were able to delineate the differences between carriers of the 16p11.2 CNV. The 16p11.2 deletion neurologic phenotype was characterized by highly prevalent speech articulation abnormalities, limb and trunk hypotonia with hyporeflexia, abnormalities of agility, sacral dimples, seizures/epilepsy, large head size/macrocephaly, and Chiari I/cerebellar tonsillar ectopia. Duplication carriers demonstrated more prominent hyperreflexia; less, though still prevalent, hyporeflexia; highly prevalent action tremor; small head size/microcephaly; and cerebral white matter/corpus callosum abnormalities and ventricular enlargement. The neurologic phenotypes of these reciprocal 16p11.2 CNVs included both shared and distinct features. The 16p11.2 phenotype eludes simple classification, spanning more than 20 different disorders in older DSM classifications. Citation42 Although the majority of patients would not qualify as autistic by this definition, some aspects of the 16p11.2 deletion phenotype are remarkably consistent and reminiscent of a “type of autism” not yet recognized by the DSM . These conclusions highlight the power of the genotype-first-based approach.
A similar dichotomy was observed in individuals with pathogenic SCN2A variants. Disruption of the gene SCN2A has been identified as one of the main causes of a wide variety of neurodevelopmental disorders, including benign familial neonatal-infantile seizures, infantile epileptic encephalopathy and ASD/ID.Citation43-Citation47 Gain of function variants resulting in increased Nav1.2 channel function have been associated with infant-onset seizures and encephalopathy, whereas loss-of-function variants conveying diminished channel function led to ASD and/or intellectual disability.Citation48 The consequences of disease-associated variants on protein function can have predictive value regarding the nature and severity of the resulting phenotype. SCN2A encodes a subunit of the neuronal voltage-gated sodium channel NaV1.2, which is involved in the initiation and the propagation of action potentials. Knowledge of the physiology of sodium channels and the known phenotype-genotype correlations relating to SCN2A, make it an important target for a precision medicine approach to therapy for ASD and other neurodevelopmental disorders.Citation49
Conclusion
The historic use of categorical diagnoses and classifications has failed NDDs in that the boundaries between disorders are not clear and comorbidity is common. The neurodevelopmental continuum underscores the need for new and flexible approaches to diagnosis and patient stratification, and the high degree of pleiotropy suggests that therapeutic approaches may be fruitful across diagnostic boundaries.
The rate of disease gene identification has accelerated dramatically over the past decade and with whole-exome and genome sequencing becoming increasingly routine practice, the genotype-first approach will likely soon spread beyond autism and developmental delay to include genes and CNVs associated with other psychiatric disorders. This surge in technological development to generate large datasets has been accompanied by increasing sophistication in the statistical methods used for data analysis. While genetically informed targeted therapies are the ultimate goal of precision medicine, there are substantial clinical and psychosocial benefits to the genotype-first approach. For families, this will translate into better diagnosis and counseling, and the formation of patient-driven support groups. Family groups associated with specific genetic subtypes can be mobilized to support one another, share experiences, and work closely with clinicians and researchers to enrol participants for research projects, to provide valuable phenotypic data and to place them in the front line for potential clinical trials.
The authors have no conflict of interest in relation to the content of this article
REFERENCES
- American Psychiatric AssociationDiagnostic and Statistical Manual of Mental Disorders1980Washington, DCAmerican Psychiatric Association Publishing
- American Psychiatric AssociationDiagnostic and Statistical Manual of Mental Disorders2013Arlington, VAAmerican Psychiatric Association Publishing
- SokolovaEOerlemansAMRommelseNRet alA causal and mediation analysis of the comorbidity between attention deficit hyperactivity disorder (ADHD) and ASDJ Autism Dev Disord20174761595160428255761
- BerriosGPorterRA History of Clinical Psychiatry. The Origin and History of Psychiatric Disorders1995 London, UK The Athlone Press 228
- GeorgetEJDe la folie. Considérations sur cette maladie1820 Paris, France Crévot 102
- EsquirolEDes maladies mentales1838 Paris, France Baillière 284
- BarkleyRAPetersHThe earliest reference to ADHD in the medical literature? Melchior Adam Weikard’s description in 1775 of “Attention Deficit” (Mangel der Aufmerksamkeit, Attentio Volubilis)J Attention Disord2012168623630
- WeikardMADer philosophische Arzt17993641 Available at:https://hdl.handle.net/2027/ucm.532772908x
- KannerLAutistic disturbances of affective contactNervous Child19432217250
- AspergerHDie „Autistischen Psychopathen“ im KindesalterArchiv für Psychiatrie und Nervenkrankheiten19441171132135
- Mal’tinskayaNAThe history of the concept of autismKoncept2017 Accessed August 10, 2019 S11 Available at:http://e-koncept.ru/2017/470137.htm (in Russian)
- SeguinEIdiocy and Its Treatment by the Physiological Method1866 New York, NY William Wood
- TreffertDAThe savant syndrome: an extraordinary condition. A synopsis: past, present, futurePhil Trans R Soc B20093641351135719528017
- DownJLOn some of the mental affections of childhood and youth, being the Lettsomian Lectures delivered before the Medical Society of London in 18871887 London, UK J & A Churchill
- TreffertDADr Down and “developmental disorders.”J Autism Dev Disord20063696596610.1007/s10803-006-0183-1 16960762
- InselTCuthbertBCommentary. Research Domain Criteria (RDoC): Toward a new classification framework for research on mental disordersAm J Psychiatry2010167774875120595427
- CrocqMACan psychopathology and neuroscience coexist in psychiatric classifications?Dialogues Clin Neurosci20182015516030581284
- SchulzeTGMcMahonFJDefining the phenotype in human genetic studies: Forward genetics and reverse phenotypingHum Hered201458131138
- StessmanHABernierREichlerEEA genotype-first approach to defining the subtypes of a complex diseaseCell2014156587287710.1016/j.cell.2014.02.002 24581488
- O’RoakBJVivesLGirirajanSet alSporadic autism exomes reveal a highly interconnected protein network of de novo mutationsNature201248524625010.1038/nature10989 22495309
- OwenMJO‘DonovanMCThaparACraddockNNeurodevelopmental hypothesis of schizophreniaBr J Psychiatry2011198317317510.1192/bjp.bp.110.084384 21357874
- SinghTWaltersJTRJohnstoneMet alThe contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disabilityNat Genet20174981167117328650482
- OwenMJO‘DonovanMCSchizophrenia and the neurodevelopmental continuum: evidence from genomicsWorld Psychiatry201716322723510.1002/wps.20440 28941101
- GirirajanSBrkanacZCoeBPet alRelative burden of large CNVs on a range of neurodevelopmental phenotypesPLoS Genet20117e100233422102821
- KirovGReesEWaltersJTRet alThe penetrance of copy number variations for schizophrenia and developmental delayBiol Psychiatry20147537838523992924
- PowerRAKyagaSUherRet alFecundity of patients with schizophrenia, autism, bipolar disorder, depression, anorexia nervosa, or substance abuse vs their unaffected siblingsJAMA Psychiatry201370223023147713
- McRaeJFClaytonSFitzgeraldTWet alDeciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disordersNature2017542764243343810.1038/nature21062 28135719
- SandersSJHeXWillseyAJet alInsights into autism spectrum disorder genomic architecture and biology from 71 risk lociNeuron20158761215123326402605
- FromerMPocklingtonAJKavanaghDHet alDe novo mutations in schizophrenia implicate synaptic networksNature20145061798424463507
- IossifovIO’RoakBJSandersSJet alThe contribution of de novo coding mutations to autism spectrum disorderNature2014515216 22125363768
- SugathanABiagioliMGolzioCet alCHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitorsProc Natl Acad Sci U S A2014111E4468E447725294932
- HallmayerJClevelandSTorresAet alGenetic heritability and shared environmental factors among twin pairs with autismArch Gen Psychiatry201168111095110221727249
- TickBBoltonPHappéFRutterMRijsdijkFHeritability of autism spectrum disorders: a meta-analysis of twin studiesJ Child Psychol Psychiatry201657558559526709141
- OzonoffSYoungGSCarterAet alRecurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium studyPediatrics20111283e488e49521844053
- TammimiesKMarshallCRWalkerSet alMolecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorderJAMA2015314989590326325558
- WeissLAArkingDEDalyMJChakravartiAGene Discovery Project of Johns Hopkins & the Autism Consortium. A genome-wide linkage and association scan reveals novel loci for autismNature2009461726580280819812673
- SebatJLakshmiBMalhotraDet alStrong association of de novo copy number mutations with autismScience2007316582344544917363630
- SandersSJErcan-SencicekAGHusVet alMultiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autismNeuron201170586388521658581
- ArnettABTrinhSBernierRAThe state of research on the genetics of autism spectrum disorder: methodological, clinical and conceptual progressCurr Opin Psychol2019271530059871
- CotneyJMuhleRASandersSJet alThe autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopmentNat Commun201566404643425752243
- SteinmanKJSpenceSJRamockiMBet al16p.2 deletion and duplication: characterizing neurologic phenotypes in a large clinically ascertained cohortAm J Med Genet A20161702943295527410714
- DuyzendMHEichlerEEGenotype-first analysis of the 16p.2 deletion defines a new type of “autism”Biol Psychiatry20157776977125843334
- HeronSECrosslandKMAndermannEet alSodium-channel defects in benign familial neonatal-infantile seizuresLancet200236085185212243921
- BerkovicSFHeronSEGiordanoLet alBenign familial neonatal-infantile seizures: characterization of a new sodium channelopathyAnn. Neurol20045555055715048894
- OgiwaraIItoKSawaishiYet alDe novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsiesNeurology2009731046105319786696
- LiaoYAnttonenA-KLiukkonenEet alSCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and painNeurology2010751454145820956790
- SandersSJMurthaMTGuptaARet alDe novo mutations revealed by whole-exome sequencing are strongly associated with autismNature201248523724122495306
- Ben-ShalomRKeeshenCMBerriosKNAnJYSandersSJBenderKJOpposing effects on Na(V)1.2 function underlie differences between SCN2A variants observed in individuals with autism spectrum disorder or infantile seizuresBiol Psychiatry20178222423228256214
- SandersSJCampbellAJCottrellJRet alProgress in understanding and treating SCN2A-mediated disordersTrends Neurosci201841744245629691040