
Abstract
Oropharyngeal candidiasis is caused by Candida sp., opportunistic yeasts that infect immunocompromised patients. Chemotherapies are based on antifungal drugs against which yeast overexpress and address to the plasma membrane ATP-binding cassette (ABC) pumps for expelling these drugs out of the cell. More critical—because these pumps translocate structurally unrelated drugs—they confer to the yeast a broad resistance to antifungals when expressed, hampering the efficacy of these treatments whatever the drug used. We review here the disease, its treatment, and the role played by multidrug resistance ABC, and strategies to overcome this problem.
Oropharyngeal candidiasis (OPC) is a fungal infection that affects oral and pharyngeal mucosa. In most cases, OPC is caused by an overgrowth of yeast from Candida sp., albicans glabrata, tropicalis, and krusei (Citation1). These yeast are opportunistic and most of the time nonpathogenic; up to 60% stays in the buccal space of healthy people (Citation1). They become aggressive in favorable conditions, typically those of immunocompromised patients after surgery or HIV-infected immunodeficient people, leading to superficial to life-threatening systemic infections with an elevated mortality level (Citation2). OPC displays a large variety of clinical forms and classifications. The lesions can be acute or chronic and can involve other microorganisms such as bacteria (candida-associated injury). A brief description is summarized in .
Table 1 Main clinical manifestation forms of OPC
Antifungal agents
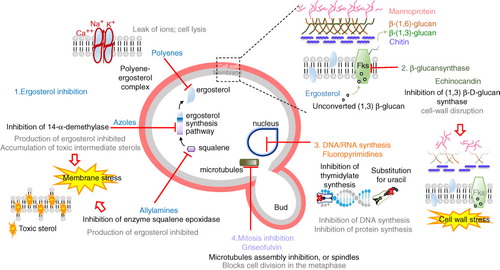
Due to the similarities between fungal and mammalian cells, therapeutic options for fungal infections are limited compared to antibacterial treatments. Only four distinct fungal metabolic pathways are targeted (): (i) inhibition of ergosterol biosynthesis (azole derivatives and allylamines) and alteration of membrane function through ergosterol complex (polyenes); (ii) inhibition of glucan synthesis (echinocandins); (iii) inhibition of macromolecule synthesis (fluorinated pyrimidine analogs); and (iv) interaction with microtubules (griseofulvin).
Azoles such as miconazole, fluconazole, and abafungi inhibit the sterol 14 α-demethylase, a protein encoded by ERG11, causing an ergosterol depletion and accumulation of 14-α-methyl-3,6-diol, a toxic sterol produced by the Δ-5,6-desaturase encoded by ERG3 (Citation5).
Allylamines such as terbinafin and naftifin inhibit the squalene epoxidase, encoded by ERG1, responsible for the first step of the biosynthesis of ergosterol. However, these inhibitors have a poor efficacy, being fungistatic for most of the Candida sp. (Citation6). They are thus used mostly as topical agents (Citation7).
Polyenes such as nystatin and amphotericin B are cyclic amphiphilic molecules binding to the lipid bilayer and to ergosterol. They generate pores in the membrane promoting the leak of small cations such as K+, Na+, and Ca2 + (Citation7).
Echinocandins, such as caspofungin, micafungin, and anidulafungin, are noncompetitive inhibitors of (Citation1, Citation3) β-D-glucan synthase, resulting in a cell wall unable to withstand osmotic stress.
Fluoropyrimidines such as 5-fluorocytosin (5-FC) are first transported into the cell by cytosine permeases and pyrimidine transporters. Once in the cytoplasm, they are converted into 5-fluorouracil (5-FU) by cytosine deaminase, then phosphorylated to give the 5- fluorouracil monophosphate (5-FUMP), and converted into either the 5-fluorouracil triphosphate (5-FUTP) or the 5-fluorodeoxyuridine monophosphate (5-FdUMP); 5-FUTP can be incorporated into RNA while the 5-FdUMP interferes with DNA replication (Citation8).
Griseofulvin binds to tubulin, disrupting the mitotic spindle formation, and thus prevents the yeast division (Citation7).
Fig. 1. Antifungal drugs and their mechanisms of action. Adapted from ref. (Citation4) and http://www.doctorfungus.org/thedrugs/antif_pharm.php.
Oral candidiasis treatment
The first line of treatment for mild and localized candidiasis usually consists of topical antifungal drugs. A systemic therapy is recommended for patients with compromised immunology defenses. summarizes the formulation of these antifungals.
Table 2 Antifungal medications of OPC
Resistance mechanisms to antifungal chemotherapies
Despite the appropriate administration of antifungal drugs, the number of cases of persistence or infection progression is increasing. Factors that drive fungi resistance either take place prior to the drug treatment (natural resistance) or occur after the exposure to a drug (acquired resistance) (reviewed in (Citation6–(Citation8, Citation11). Four to ten percent of Candida species are resistant to fluconazole and voriconazole (Citation12). Each species can display a specific molecular mechanism of resistance to antifungal drugs (Citation2, Citation5); however, four mechanisms are common and can function synergistically: (Citation1) altered drug metabolism, (Citation2) mutations in gene encoding target proteins, (Citation3) prevented entry of the drug, and (Citation4) removal of the drug from the cell through the upregulation of the expression of multidrug efflux pumps. Mutations in cytosine permease (uptake) or deficiency in enzymes implicated in the metabolism of fluoropyrimidines are a frequent cause of antifungal drug resistance. Mutations in ERG11, or in FKS1 (which encodes the β-1,3-glucan synthase), result in resistance to azoles and echinocandins, respectively. A recent study showed that upon fluconazole treatment, Candida species overexpress Candida drug resistance (CDR) membrane pumps to eliminate the drug (Citation13). Overexpressed in Saccharomyces cerevisiae, they increase the resistance to fluconazole 600 times (Citation14).
Multidrug efflux pumps
The most ubiquitous mechanism resistant to xenobiotic toxicity is the elimination of drugs out of the cell mediated by ATP-binding cassette (ABC) membrane transporters. These act as molecular pumps that actively translocate drugs through the plasma membrane by using the energy gained from ATP hydrolysis. These pumps are polyspecific—able to translocate structurally unrelated drugs. This property has a deep impact on chemotherapies because once the resistance is acquired for one drug, it is also acquired for most of them, from common to distinct chemical classes. These proteins thus cover a critical field in drug disposition and drug resistance to chemotherapeutic treatments, for which solutions remain to be found. The pleiotropic drug resistance (PDR) in fungi displays several similarities to the multidrug resistance (MDR) phenotype in humans (Citation15, Citation16) and bacteria (Citation17–(Citation19). Thirty-one ABC transporters have been identified in S. cerevisiae (Citation20), compared with the 48 ABC transporters found in humans (Citation21). They are classified in five subfamilies according to their phylogenetic relationships: PDR, MDR, multidrug-resistance protein (MRP)/cystic fibrosis transmembrane (TM) conductance regulator (CFTR), adrenoleukodystrophy protein (ALDP), and yeast elongation factor 3 (YEF3)/RNase L inhibitor 1 (RLI) (Citation22). Among them, Pdr5p is the most studied, with Snq2p (Citation20) and Yor1p (Citation23). These pumps constitute the main shield against xenobiotics, including antifungal drugs. Most ABC transporters in Candida species (http://www.candidagenome.org) are orthologs of S. cerevisiae Pdr5 and are equally implicated in MDR (Citation22). However, a few of them have been studied in detail so far; they are summarized in .
Table 3 ABC proteins in pathogenic candida species
ABC transporter topology
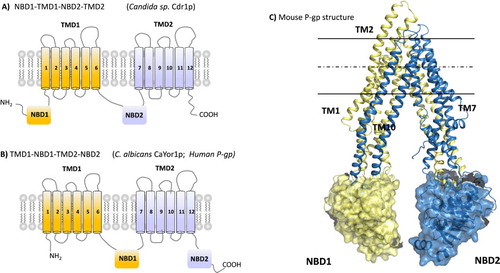
The basic structure that defines the members of the ABC transport family is a combination of a cytosolic nucleotide-binding domain (NBD) and a transmembrane domain (TMD) organized either in 1 to 4 polypeptides (). These domains are arranged in any possible combination in a protein that includes two NBDs and two TMDs. Some members have additional TM segments or cytosolic domains. The human P-glycoprotein (P-gp) has a TMD1-NBD1-TMD2-NBD2 topology in a single polypeptide while PDR or CDR pumps, which are functionally close in terms of polyspecificity, display a reverse topology: NBD1-TMD1-NBD2-TMD2. The reason for such specificities is not known. The TMD has six hydrophobic α-helices, poorly conserved in length or amino acid sequence. Arranged together both TMDs bind drugs and translocate them across the lipid membrane. Note that in that frame, strictly speaking, drugs are not substrates because they do not undergo a molecular transformation by the pump; but because the term is widely used, we will keep it here. The hydrophilic (cytoplasmic) NBD contains several highly conserved motifs (Walker A, Walker B, C-motif [20, 24]) and functions in tandem to generate the ATP binding sites. ATP is thought to be hydrolyzed to reset the protein to its initial state after drug translocation.
Fig. 2. A–B. Topology of ATP-binding cassette (ABC) transporters belonging to pleiotropic drug resistance (PDR) and multidrug resistance (MDR) subfamily. C. 3D-structure of mouse P-gp in cartoon and colored in yellow and blue illustrating each moiety of the protein.
Transport cycle
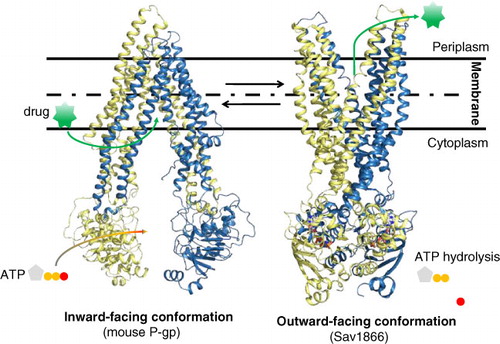
In a typical drug efflux cycle, the drug binds first to the TMDs. This binding triggers a conformational change by which each NBD comes closer to the other, generating the ATP binding sites. The binding of two ATPs blocks the protein in a close state that leads it to undergo a second large conformational change from the inward-facing to the outward-facing conformation (). In this new conformation, both NBDs remain bound together with their ATP while the outer leaflet part of each TMD becomes distant, open to the extracellular face of the drug-binding sites. Drugs are released by changes in the affinity of the sites. Finally, the protein comes back to the initial inward-facing conformation by hydrolyzing ATP, which gives the energy to the protein to allow separation of each moiety (Citation25). Several x-ray structures of bacterial and eukaryotic ABC transporters have now been resolved at high to medium resolution, 2.2–5.5 Å, BtuCD (Citation26), HI1470/1 (Citation27), HmuUV (Citation28), ModBC (Citation29), MalFGK2 (Citation30), MetNI (Citation31), Sav1866 (Citation32), MsbA (Citation33), ABCB1 (Citation34, Citation35), ABCB10 (Citation36), and TM287/288 heterodimer (Citation37). They give some hints about the putative mechanism; however, the structure of intermediate conformational states is required to elucidate on a molecular level the mechanism by which drugs are translocated. In this way, three new conformations in the inward-facing conformation of P-gp could be solved (Citation38).
Fig. 3. Conformational changes of ATP-binding cassette (ABC) exporters. The 3D-structure of mouse P-gp in the inward-facing conformation (Citation34, Citation35) is shown on the left and the homodimer Sav1866 in the outward-facing conformation (Citation32) is displayed on the right. Nonhydrolysable ATP-analog AMPPNP (adenosine-5′(βγ-imido)triphosphate) bound to Sav1866 is shown in CPK-colored stick molecules (red, O atom; gray, C atom). The drug is symbolized in green.
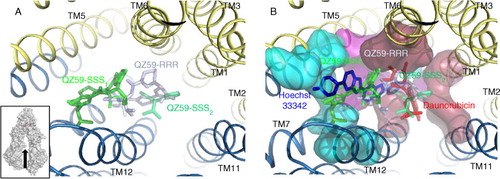
The human P-gp contains at least two well-identified drug-binding sites, one binding Hoechst 33342 (the H site) and the other rhodamine 123 (the R site) (Citation39, Citation40). Both dyes are also transported by PDR/CDR transporters because the structural organization of their drug-binding sites is probably close to that of the P-gp. In that frame, 3D models could be proposed (Citation41) that may be helpful in studying such proteins for which no structural information has yet been released. More importantly, no structural information is available for CDR pumps, while the present 3D structures cannot be transposed to them due to their reversed topology. Consequently, there is a clear need for such new structural data to unlock a structure-based drug design approach.
Regardless, these structures open the way to molecular enzymology to elucidate how the drug-binding sites are formed, organized, and how they bind drugs. The cocrystallization of two cyclic hexapeptides with the mouse P-gp (Citation34, Citation35, Citation38) allowed us to precisely locate the H and R drug-binding sites by characterizing the mode of inhibition of these compounds on the transport of drugs binding to each site () (Citation42).
Fig. 4. Drug binding site of the mouse P-gp. A. The 3D-structure of mouse P-gp in the inward-facing conformation with the hexapeptides inhibitors QZ59SSS and QZ59RRR bound in separate P-gp (Citation34, Citation35). The inset shows the direction of observation. B. This time, the same view is displayed showing the location of Hoechst 33,342 (H site) and daunorubicin (R site) as determined in (Citation42). Blue, red, and magenta areas correspond to residues belonging to the H site, the R site, and to both sites.
Modulation of MDR ABC transporters for restoring antifungal drug sensitivity
Altering the capacity of pathogenic yeast that overexpress MDR ABC pumps to expel antifungal drugs can be achieved with inhibitors. Ideally such compounds display the following characteristics: effective at low concentrations, specific to the targets, no pharmacokinetic interactions with the coadministrated antifungal drug, and nontoxic (Citation43). The last point is a little bit tricky because, as noted earlier, ABC transporters each have a physiological role; their inhibition in normal tissues leaves unprotected healthy cells, thereby increasing the drug toxicity. Using specific inhibitors for a single transporter can overcome this obstacle. Some substrates and inhibitors of these efflux pumps are displayed in .
Table 4 Substrates and inhibitors of ABC transporters from Candida sp. and human P-glycoprotein
Conflict of interest and funding
The authors declare no conflict of interest and funding.
Acknowledgements
LM was supported by the Rhône-Alpes region (Cluster 10-ARC1 santé). PF and LM are supported by the Ligue Contre le Cancer; PF was supported by ANR-SVE5-CLAMP and ARC1 santé.
References
- Pankhurst CL. Candidiasis (oropharyngeal). Clin Evid. 2013;2013:1304
- Tscherner M Schwarzmüller T Kuchler K. Pathogenesis and antifungal drug resistance of the human fungal pathogen Candida glabrata. Pharmaceuticals. 2011;4:169-86.
- Farah CS Lynch N McCullough MJ. Oral fungal infections: An update for the general practitioner. Aust Dent J. 2010;55(Suppl 1):48-54.
- Cowen LE. The evolution of fungal drug resistance: Modulating the trajectory from genotype to phenotype. Nat Rev Microbiol. 2008;6:187-98.
- Shapiro RS Robbins N Cowen LE. Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiol Mol Biol Rev. 2011;75:213-67.
- Cannon RD Lamping E Holmes AR Niimi K Baret PV Keniya MV et al Efflux-mediated antifungal drug resistance. Clin Microbiol Rev. 2009;22:291-321.
- Vandeputte P Ferrari S Coste AT. Antifungal resistance and new strategies to control fungal infections. Int J Microbiol. 2012;2012:713687
- Morschhauser J. Regulation of multidrug resistance in pathogenic fungi. Fungal Genet Biol. 2010;47:94-106.
- Williams D Lewis M. Pathogenesis and treatment of oral candidosis. J Oral Microbiol. 2011 3
- Dangi YS Soni M Namdeo KP. Oral candidiasis: A review. Int J Pharm Pharm Sci. 2010;2:36-41.
- Nasira S Vilas J Sarita K Basavraj N. Antifungal drug resistance in Candida species. Eur J Gen Med. 2013;10:254-8.
- Pfaller MA Diekema DJ Gibbs DL Newell VA Ellis D Tullio V et al Results from the ARTEMIS DISK Global Antifungal Surveillance Study, 1997 to 2007: A 10.5-year analysis of susceptibilities of Candida species to fluconazole and voriconazole as determined by CLSI standardized disk diffusion. J Clin Microbiol. 2010;48:1366-77.
- Chen LM Xu YH Zhou CL Zhao J Li CY Wang R. Overexpression of CDR1 and CDR2 genes plays an important role in fluconazole resistance in Candida albicans with G487T and T916C mutations. J Int Med Res. 2010;38:536-45.
- Lamping E Monk BC Niimi K Holmes AR Tsao S Tanabe K et al Characterization of three classes of membrane proteins involved in fungal azole resistance by functional hyperexpression in Saccharomyces cerevisiae . Eukaryot Cell. 2007;6:1150-65.
- Wu CP Hsieh CH Wu YS. The emergence of drug transporter-mediated multidrug resistance to cancer chemotherapy. Mol Pharm. 2011;8:1996-2011.
- Szakacs G Paterson JK Ludwig JA Booth-Genthe C Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219-34.
- Nikaido H. Multidrug resistance in bacteria. Ann Rev Biochem. 2009;78:119-46.
- Piddock LJ. Multidrug-resistance efflux pumps – not just for resistance. Nat Rev Microbiol. 2006;4:629-36.
- Lubelski J Konings WN Driessen AJ. Distribution and physiology of ABC-type transporters contributing to multidrug resistance in bacteria. Microbiol Mol Biol Rev. 2007;71:463-76.
- Prasad R Goffeau A. Yeast ATP-binding cassette transporters conferring multidrug resistance. Ann Rev Microbiol. 2012;66:39-63.
- Dean M Hamon Y Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res. 2001;42:1007-17. [PubMed Abstract]
- Klein C Kuchler K Valachovic M. ABC proteins in yeast and fungal pathogens. Essays Biochem. 2011;50:101-19.
- Nagy Z Montigny C Leverrier P Yeh S Goffeau A Garrigos M et al Role of the yeast ABC transporter Yor1p in cadmium detoxification. Biochimie. 2006;88:1665-71.
- Prasad R Sharma M Rawal MK. Functionally relevant residues of Cdr1p: A multidrug ABC transporter of human pathogenic Candida albicans. J Amino Acids. 2011;2011:531412
- Linton KJ Holland IB. The ABC transporters of human physiology and disease: The genetics and biochemistry of ATP binding cassette transporters. 2011 USA: World Scientific
- Locher KP Lee AT Rees DC. The E. coli BtuCD structure: A framework for ABC transporter architecture and mechanism. Science. 2002;296:1091-8.
- Pinkett HW Lee AT Lum P Locher KP Rees DC. An inward-facing conformation of a putative metal-chelate-type ABC transporter. Science. 2007;315:373-7.
- Woo JS Zeltina A Goetz BA Locher KP. X-ray structure of the Yersinia pestis heme transporter HmuUV. Nat Struct Mol Biol. 2012;19:1310-5.
- Hollenstein K Frei DC Locher KP. Structure of an ABC transporter in complex with its binding protein. Nature. 2007;446:213-16.
- Oldham ML Khare D Quiocho FA Davidson AL Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515-21.
- Kadaba NS Kaiser JT Johnson E Lee A Rees DC. The high-affinity E. coli methionine ABC transporter: Structure and allosteric regulation. Science. 2008;321:250-3.
- Dawson RJ Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180-5.
- Ward A Reyes CL Yu J Roth CB Chang G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc Natl Acad Sci U S A. 2007;104:19005-10.
- Aller SG Yu J Ward A Weng Y Chittaboina S Zhuo R et al Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718-22.
- Li J Jaimes KF Aller SG. Refined structures of mouse P-glycoprotein. Protein Sci. 2014;23:34-46.
- Shintre CA Pike AC Li Q Kim JI Barr AJ Goubin S et al Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc Natl Acad Sci U S A. 2013;110:9710-5.
- Hohl M Briand C Grutter MG Seeger MA. Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat Struct Mol Biol. 2012;19:395-402.
- Ward AB Szewczyk P Grimard V Lee C-W Martinez L Doshi R et al Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc Natl Acad Sci U S A. 2013;110:13386-91.
- Shapiro AB Ling V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur J Biochem. 1997;250:130-7.
- Shapiro AB Fox K Lam P Ling V. Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone. Evidence for a third drug-binding site. Eur J Biochem. 1999;259:841-50.
- Rutledge RM Esser L Ma J Xia D. Toward understanding the mechanism of action of the yeast multidrug resistance transporter Pdr5p: A molecular modeling study. J Struct Biol. 2011;173:333-44.
- Martinez L Arnaud O Henin E Tao H Chaptal V Doshi R et al Understanding polyspecificity within the substrate-binding cavity of the human multidrug resistance P-glycoprotein. FEBS J. 2014;281:673-82.
- Zhang L Ma S. Efflux pump inhibitors: A strategy to combat P-glycoprotein and the NorA multidrug resistance pump. ChemMedChem. 2010;5:811-22.
- Eckford PD Sharom FJ. ABC efflux pump-based resistance to chemotherapy drugs. Chem Rev. 2009;109:2989-3011.