Abstract
Background : Species in the Culex pipiens complex are common almost all over the world and represent important vectors for many serious zoonotic diseases. Even if, at the moment, many of the pathogens potentially transmitted by Cx. pipiens are not a problem in northern Europe, they may, with increasing temperatures and changing ecosystems caused by climate change, move northward in the future. Therefore, the question whether or not the Cx. pipiens populations in northern Europe will be competent vectors for them is of high importance. One way to estimate the similarity and the rate of contact between European Cx. pipiens populations is to look at the gene exchange between these populations.
Methods : To test the genetic diversity and degree of differentiation between European Cx. pipiens populations, we used eight microsatellite markers in 10 mosquito populations originating from northern, central, and southern Europe.
Results : We found that three of the analyzed populations were very different from the rest of the populations and they also greatly differed from each other. When these three populations were removed, the variance among the rest of the populations was low, suggesting an extensive historic gene flow between many European Cx. pipiens populations.
Conclusions : This suggests that infectious diseases spread by this species may not be associated with a certain vector genotype but rather with suitable environmental conditions. Consequently, we would expect these pathogens to disperse northward with favorable climatic parameters.
Mosquitoes are found throughout the world except in locations that are permanently frozen Citation1 Citation2. In almost all mosquitoes, the female feed on vertebrate blood to obtain the protein they need for egg development. A complex salivary secretion in mosquitoes helps the feeding process, and it is the injection of this fluid to the host capillaries that enables several kinds of simple organisms – viruses, protozoa and nematodes – to enter the vertebrate blood system Citation1 Citation2. This process makes it possible for pathogens to exploit mosquitoes as a principal vector of infectious diseases in both developing countries and in industrial nations Citation3. In many, mainly tropical and economically challenged parts of the world, mosquito-borne diseases are flourishing and they are responsible for a large proportion of the global morbidity and mortality Citation2.
Species in the Culex pipiens complex are common almost all over the world and act as important vectors for many serious zoonotic diseases, mostly because they often are the most common mosquitoes in urban areas Citation4. The taxonomy and systematics of the Cx. pipiens complex is a challenge. The form molestus that was originally described as a species by the Linnaean disciple Forsskål in 1775 from Egypt has since been reduced to represent a bioform (bf.) of the species Cx. pipiens. This decision was based upon the fact that no morphological characters were found that would allow the discrimination of one species from the other. Fonseca et al. Citation4 showed that there were distinct genetic differences using microsatellites between populations of bf. pipiens and molestus and that they probably had undergone a recent speciation event. Because bf. pipiens is strictly ornithophilic while molestus readily bites humans and other mammals, this is of epidemiological importance when discussing diseases such as West Nile fever (WNF). Supposedly, molestus will act as a bridge vector between the bird reservoirs and susceptible mammal hosts, while pipiens only will maintain the bird-to-bird transmission. Except for some underground populations of Cx. pipiens bf. molestus, in northern European cities, most specimens of this temperate species require a blood meal for egg development Citation4.
Anthropogenic climate change is presently exerting multifaced effects on ecosystems, communities, and populations Citation5. The ecology, behavior, development, and survival of mosquitoes and the disease transmission dynamics are strongly influenced by climatic factors. Temperature, rainfall, and humidity are thought to be the main components influencing the life history of mosquitoes, even if other factors, such as the wind and photoperiod can have significant influence Citation1 Citation6. The same factors also influence the survival and transmission pattern of mosquito-borne pathogens – for example, the temperature affects the rate of pathogen multiplication within the insect, which in turn affects the rate of infective organisms in the salivary secretion and thus the likelihood of successful transmission to another host Citation1 Citation2 Citation7.
Understanding the connection between infectious diseases and climate is difficult because of the multivariate nature of climate change and non-linear thresholds in both disease and climate change processes. It is commonly assumed that the distribution of infectious diseases is determined by climate and that the present climate change will increase their incidence and geographic distribution. Consequently, the relationship between temperature and the transmission of pathogens has gained substantial attention Citation8. However, just because a vector species can transmit a certain pathogen in one geographical area, that does not necessarily mean that the populations of the same species are competent hosts for the same pathogen in another geographic location Citation9 as the vector competence of mosquitoes can be influenced by both temperature and viral genetics Citation8.
A wide variety of pathogens can be transmitted by Cx. pipiens. For example, these mosquitoes are important vectors of West Nile virus that was first characterized in Uganda in 1937 but has spread over large areas of Africa, Europe, the Middle East, Central Asia, and the Americas. Usutu virus is another example of a previously tropical mosquito-borne virus that has now been introduced and established in temperate climate – originally isolated from a bird-biting mosquito, Culex univittatus, in South-Africa, and historically detected in various places in sub-Saharan Africa, the virus, in 2001, suddenly emerged in the Austrian capital of Vienna Citation10 and recently in Italy Citation11 Citation12 and in southern Germany Citation13. Dirofilariases are important vector-borne parasitic zoonoses that mainly affect the canine population. Cx. pipiens has been suggested to be one of the main vectors for the heartworm disease by Dirofilaria immitis that can be rather common in some areas in southern Europe Citation14. Rift Valley fever virus is another virus that can be carried by Cx. pipiens, and can cause severe disease in both animals and humans in various parts of Africa (however, carried by other mosquito species in African cases) Citation15. In September 2000, Rift Valley fever cases were diagnosed in Saudi Arabia and Yemen, raising concerns that it could extend to other parts of Asia and Europe.
The above-mentioned pathogens have not yet been causing problems in northern Europe, however with increasing temperatures and changing ecosystems caused by climate change, it is possible that they continue to move northward. Therefore, the question whether or not the Cx. pipiens populations of northern Europe will be as competent vectors for mentioned pathogens as the more southern populations is of high importance for risk analysis.
One way to estimate the similarity and the rate of contact between European Cx. pipiens populations is to look at the gene exchange between mosquito populations. Since gene flow is always lower than the actual movements of individuals, this will give a rather conservative estimate of the amount of spread of individuals between parts of Europe. By analyzing the extent of contact between populations that have been shown to be able to carry certain pathogens and populations in northern Europe that have yet not been seen to transmit them, we can estimate the possibility of natural pathogen dispersal under changing climatic conditions. To estimate the genetic diversity and degree of differentiation between European Cx. pipiens populations, we used eight microsatellite markers in 10 mosquito populations originating from northern, central, and southern Europe.
Methods
Sampling
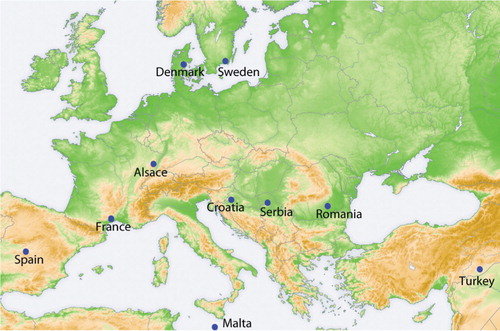
Summer 2009, adult individuals of Culex pipiens were caught by using different models of CO2 and light traps in rural habitats at 10 different European locations (). Mosquitoes were preserved in 90% ethanol and sent to Uppsala by mail by the local mosquito researchers. Each Cx. pipiens population was represented by 30 adult individuals, all from wild, over-ground populations.
Fig. 1. Geographic origin of mosquito populations.
Molecular analysis
DNA from individual mosquitoes was extracted by a Chelex procedure proteinase-K-based extraction Citation16 using the protocol provided by the manufacturer. We used eight primers developed for Cx. pipiens Citation4 Citation17–Citation19 (CQ13, CQ29, CxpGT9, CxpGT20, CxpGT40, CxpGT46, CxpGT51, and CxpGT53) and followed the PCR-protocols given in the original paper. The PCR-products were run on a MegaBase using dyed primers.
Statistics
We used the software Microchecker Citation20 to check for null alleles and other technical problems. We detected a significant number of null alleles in several loci, apparent as an uncommonly large proportion of homozygotes for a few alleles, which created large deviations from Hardy–Weinberg equilibrium. The repeatability of the amplifications was 95%; so, there are no reasons to believe that technical problems caused this pattern. Since several samples did not amplify at all for a given locus, there are reasons to suspect that there are alleles present that did not amplify. Null alleles are commonly found in Lepidoptera Citation21 and seem to be present also in this Dipterian sample. The main effect of null alleles seems to be an overestimate of F ST Citation22, but no major distortions of genetic structure. We used the method by Chapuis and Estoup Citation23 to correct for the presence for null alleles since this method performed best in a comparative study. The method is implemented in the software FreeNA Citation23.
To perform basic statistics, such as the number of alleles, allelic richness, and expected heterozygosity, we used the softwares Arlequin 3.5.1.2 Citation24 and Fstat 2.9.3.2 Citation25. To calculate pairwise F ST, we used FreeNA using both uncorrected data and data corrected for null alleles; 95% confidence intervals were obtained through 1000 bootstrap replicates. Since F ST is dependent on the level of heterozygosity, a low estimate can be due to a high within population variation rather than lack of differentiation. We therefore also calculated G’ST Citation26, which rescales F ST to have bounds between zero and one to get an intuitive picture of the degree of differentiation. To tests for possible isolation by distance patterns, we used the program Mantel 2.0 Citation27. Finally, to make neighbor-joining (NJ) trees based on the distances obtained, we used MEGA5 Citation28. To assess if the sampling and the number of loci used was enough to detect a structure if there is one (i.e. power of the tests), we used the program POWSIM Citation29. We used all loci and simulated F ST-values for two populations being separated for 250 generations, each with an effective population size (N e) of 5000 and with a sampling effort of 30 individuals (as in this study). The values were used to simulate a case where the split of the populations are very recent, and the populations are very large. Hence, this is a very conservative test of power, as if time is longer and N e is smaller, it would be much easier to find significant results. Nevertheless, even with these very conservative start values, we found that the expected F ST is 0.025 (95% range, 0.0139–0.0392) and the power 0.99. Thus, we are confident that, given the number of loci and sampling effort, we can find differences if they exist.
Because of some possible technical errors, we had to adjust our data. Consequently, we were not able to estimate genetic differentiation, as our data adjustments cannot cope with functions such as, for example, the estimation of gene flow. When the data are adjusted for null alleles (alleles that do not amplify), multilocus estimates of gene flow are no longer possible because the different loci are now made independent. Instead we used so-called private alleles (i.e. alleles only found in one population; Citation30) assuming that there were a number of private alleles that did not amplify (null alleles). With help of GenePop and by using private alleles, we estimated the number of migrants per generation across all populations. The aim of estimating the gene flow with this method was to test if the observed amount of divergence was affected by gene flow or if it was what we could expect when no gene flow has occurred since separation. To implement this, we calculated expected F ST by simulations using POWSIM with N e values ranging from 100 to 10,000 and time since separation (t) ranging from 100 to 5000 generations and allele frequencies taken from the observed data. This will give us a range of expected F ST-values for different combinations of N e and t (for results, see ). Populations from Romania, Turkey, and France were excluded from this calculation since they probably belong to another group of Culex. To estimate N e, we used the method of Xu and Fu 2004 Citation31 designed for highly variable markers such as microsatellites, where θ = 4 N eµ, and µ is the mutation rate. If the parameter values needed to get the observed F ST-values are outside what is realistic in this system, we can reject the hypothesis of no gene flow between them.
Results
The number of alleles, allelic richness, and expected heterozygosity did not differ much between the populations (). The global F ST was 0.090 (95% CI: 0.049–0.14) using the uncorrected data set and 0.054 (95% CI: 0.032–0.80). The pairwise F ST-values were low but were not different from zero, except in one case; between Denmark and Serbia (). This result was consistent regardless of which data set was used. However, the comparisons that involved the samples from France, Rumania, and Turkey were highly differentiated from the rest of the populations. This can be illustrated in the NJ-trees made (). As can be seen in , the distance between populations was on average lower using the corrected data set. If we exclude the three deviating populations, global F ST was now only 0.029 (0.018–0.041) and 0.022 (0.015–0.028), respectively, which corresponds to G’ST-values of 0.20 and 0.15, respectively. The two estimates of F ST was strongly correlated (r=0.99, P<0.001; Mantel test). There was a significant correlation between geographic and genetic distances [log (distance) versus F ST/(1 − F ST), r=0.31, P=0.0.032 for the corrected data set, Mantel test].
Fig. 2. Neighbor-joining (NJ) trees illustrating the genetic distance between populations (a) without correction for null alleles, (b) with correction, (c) using the lower 2.5 interval, and (d) using the upper 2.5 interval.
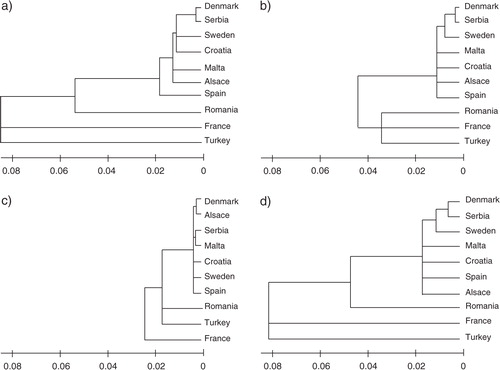
Table 1. Number of individuals sampled, number of alleles (SE), allelic richness, AR (SE), and expected heterozygosity (SE) for the different populations
Table 2. Pairwise distances with (a) uncorrected F ST and (b) corrected values
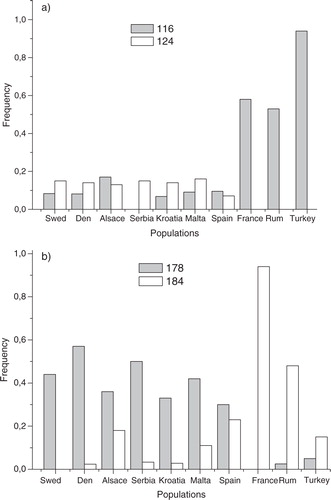
Two of the markers used here were the same that were used in Fonseca et al. (Citation4), where two distinct lineages were found. In one of the loci CxpGT9, the allele frequency spectrum of the three outlying populations (France, Romania, and Turkey) was similar to the underground populations discovered in Ref. (Citation4) and the allele frequency spectrum for the rest was close to the one found in the above-ground populations found in Ref. Citation4. The difference between the two populations can be seen in a, where the allele size 116 [corresponding to allele size 114 in Ref. Citation4] is shown to be very frequent in France, Romania, and Turkey. In Ref. Citation4, the same allele reached a frequency of around 0.5, while in the other populations, the frequencies were much lower. Allele size 124, on the other hand, was not found in Romania, Turkey, or France, but in all other populations, at about the same frequency. It is also worth noting that allele size 116, being the most frequent one in the putative underground populations, was found in almost all of the other populations, albeit at a much lower frequency. This suggests a gene flow between the underground populations and the above-ground populations. The same pattern can be seen in locus CQ29 where allele size 178 reaches a high frequency in the above-ground populations, but not in the below-ground populations (b), while allele size 184 is very common in France, being almost the only allele found, and relatively common in Romania, but having a low frequency in Turkey. Both alleles are found in both groups, but at a much lower frequency, again indicating some gene flow between the groups.
Fig. 3. Frequencies of two alleles in (a) locus CxpGT9 and (b) QT29 in the different populations.
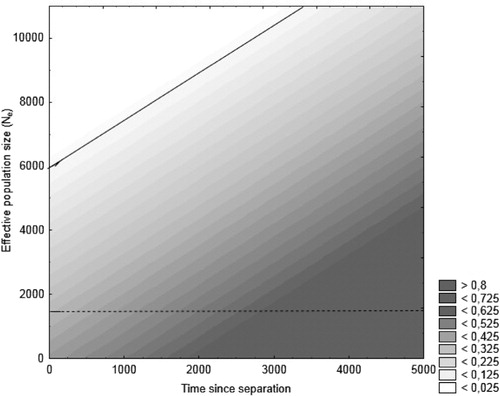
The mean frequency of private alleles was 0.026, which when corrected for sample size yields an estimate of the number of migrants of 5.74. The number of migrants between Malta and Sweden (the most distant populations) was as high as 2.04.We found that the estimate of effective population, θ = 2.85, which assuming a mutation rate for microsatellites (µ) of 5×10−4, gives an estimate of N e≈1500, and with a more conservative estimate of 10−4, gives N e≈7100. To obtain the range of observed F ST-values by drift alone, we would expect N e in the range of at least 6000, which is possible given the conservative estimate of mutation, but only at short times since separation (). Thus, even if the observed F ST-values are not unattainable, the conditions under which this might happen are quite restricted.
Fig. 4. Expected F ST values (shaded areas) in relation to time to separation in generations (x-axis) and effective population size (N e) based on simulations in POWSIM. The line indicates the upper limit of F ST-levels observed in the current study, and thus the observed values are only possible under a model of pure genetic drift for parameter values above the line. The dotted line indicates the average of the estimated N e over all populations.
Discussion
The study showed two major results: first, three of the analyzed populations (France, Romania, and Turkey) were very different from the rest of the populations (Sweden, Denmark, Alsace, Croatia, Serbia, Spain, and Malta) and they also greatly differed from each other (, ). In fact, they were so different that there are reasons to believe that they belong to differentiated lineages. Furthermore, the allele frequency distribution in these populations match the one that has been earlier found in the underground populations (Citation4). Second, most pairwise differences were low and showed a shallow structure among the populations apart from the three deviating ones (). The low F ST-values between populations suggest that there is an extensive gene flow between the populations, and this was confirmed by the simulations where the conditions for differentiation by drift alone was far too stringent given what is known about these populations. A low F ST by itself does not mean that there is extensive gene flow Citation32 since F ST is dependent on the level of heterozygosity. However, even if we rescale the values to account for within-population heterozygosity, the overall level of differentiation is still low in the above-ground populations. Thus, we are confident that basically all European populations of the above-ground Cx. pipiens share genes. The estimate of gene flow was high, which together with the simulations, suggests that the genetic similarity among populations is due to ongoing gene flow rather than a result of a recent shared history and expansion.
An alternative explanation is that the species only very recently spread throughout Europe, and thus due to incomplete lineage sorting and subsequent local stochastic effects, the populations have not diverged more, which is reflected in the unclear relationships between the populations. However, this species were described by Linnaeus in 1758 Citation33, and there are no reasons to believe that it was an uncommon species at that time. Even if the species had very recently arrived to Northern Europe when Linnaeus described it, 250 years is ample time to develop a population structure if gene flow is restricted or absent. The results from the power analysis confirm this; the expected F ST-values are in our simulation higher than observed here, even though we used very conservative input-values. This strengthens the support for a wide-ranging dispersal of mosquito-individuals across large distances in Europe, and hence the possibility of a spread of vector-borne pathogens.
Vector competence of mosquitoes describes their ability to transmit pathogens after taking an infected blood meal. The ability of vectors transmitting a pathogen is known to vary between populations of mosquito species. The exact relationship between vector competence and environmental factors is however not clear and can be influenced by many external factors Citation8. According to our study, an extensive amount of gene exchange, and hence exchange of individuals, occurs between populations of Cx. pipiens in Europe. Therefore, infectious diseases spread by this species may not be associated with a certain vector genotype, but rather with suitable environmental conditions. Consequently, we would expect these pathogens to disperse northward with favorable climatic parameters.
The distribution and intensity of mosquito-borne infections is strongly influenced by the interactions between temperature, vectors, hosts, and pathogen genetics. Temperature often determines both the latitudinal boundary and upper elevation limit of pathogen transmission when the extrinsic incubation period becomes greater than the life span of the vector Citation8. Temperature is also linked to the intensity and transmission of pathogens. In temperate regions, seasonality is one of the key components of the climate. In many temperate regions, however, the summer temperatures can be as high as temperatures in the tropics. The difference with tropics lays in the absence of winters. Therefore, tropical mosquito-borne diseases can be introduced and transmitted during the right season in the temperate regions, but in most cases, they will be eliminated during the winter (Citation1). Some viruses, however, such as the Usutu virus in Austria, have been shown to be able to maintain in a natural cycle with a minimum of at least 10 days/year with a temperature >30°C (Citation10). It is also predicted that Usutu virus will become endemic in large parts of Central Europe before the end of the century and that optimal environmental conditions for outbreaks of this virus will occur in about 10 years from now Citation10 Citation34.
A concern associated with pathogens carried by Cx. pipiens mosquitoes is the distribution of hybrid populations between bf. pipiens and molestus. Because pipiens is strictly ornithophilic while molestus has a more catholic taste and readily bites humans and other mammals, these hybrid populations are thought to be the most high-capacity vectors in southern Europe and North America (Citation35). This gives rise to concern, should those hybrids spread to northern Europe or if another hybridization event should occur. It is possible that presently the populations of Cx. pipiens in Sweden and the rest of northern Europe are less likely to be involved in zoonotic transmission of, for example, WNF since they are not constituted of hybrids. However, in a recent paper, Reusken et al. Citation35 showed the presence of hybrid pipiens×molestus mosquitoes in the Netherlands. Since our results imply an extensive gene flow between populations of Cx. pipiens in Europe, hybrid mosquitoes could spread fast over the whole Europe. This could potentially alter the epidemiology of many arthropod-borne viruses in Northern Europe giving rise to epidemics/epizootics similar to the WNF outbreak in North America from 1999 and onward. Consequently, up-to-date surveillance systems are called for all over the Europe as, without them, detection and response to emerging vector-borne diseases can be severely impaired Citation36. The emergence of WNF in 1999 in New York is a good example of the consequences that a delayed response can have on the outcome of novel pathogen introduction. Studies show that impaired mosquito surveillance can make the management costs of epidemics several times higher than applying a strategy with sustained surveillance and early case detection Citation36.
Conflict of interest and funding
The authors have not received any funding or benefits from industry or elsewhere to conduct this study.
References
- Reiter P. Climate change and mosquito-borne disease. Environ Health Perspect. 2001; 109: 141–61.
- Tolle MA. Mosquito-borne diseases. Curr Probl Pediatr Adoles Health Care. 2009; 39: 97–140. 10.3402/iee.v2i0.12001.
- Kalluri S, Gilruth P, Rogers D, Szczur M. Surveillance of arthropod vector-borne infectious diseases using remote sensing techniques: a review. PLoS Pathog. 2007; 3: e116. 10.3402/iee.v2i0.12001.
- Fonseca DM, Keyghobadi N, Malcolm CA, Mehmet C, Schaffner F, Mogi M, et al.. Emerging vectors in the Culex pipiens complex. Science. 2004; 303: 1535. 10.3402/iee.v2i0.12001.
- Harvell CD, Mitchell CE, Ward JR, Altizer S, Dobson AP, Ostfeld RS, et al.. Climate warming and disease risks for terrestrial and marine biota. Science. 2002; 296: 2158. 10.3402/iee.v2i0.12001.
- Shaman J, Day JF. Reproductive phase locking of mosquito populations in response to rainfall frequency. PLoS One. 2007; 2: e331. 10.3402/iee.v2i0.12001.
- Dohm DJ, O'Guinn ML, Turell MJ. Effect of environmental temperature on the ability of culex pipiens (diptera: Culicidae) to transmit west nile virus. J Med Entomol. 2002; 39: 221–5. 10.3402/iee.v2i0.12001.
- Kilpatrick AM, Meola MA, Moudy RM, Kramer LD. Temperature, viral genetics, and the transmission of west nile virus by culex pipiens mosquitoes. PLoS Pathog. 2008; 4: e1000092. 10.3402/iee.v2i0.12001.
- Bennett KE, Olson KE, Muñoz MdL, Fernandez-Salas I, Farfan-Ale JA, Higgs S, et al.. Variation in vector competence for dengue 2 virus among 24 collections of Aedes aegypti from Mexico and the United States. Am J Trop Med Hyg. 2002; 67: 85–92.
- Pfeffer M, Dobler G. Emergence of zoonotic arboviruses by animal trade and migration. Parasit Vectors. 2010; 3: 1–15. 10.3402/iee.v2i0.12001.
- Pecorari M, Longo G, Gennari W, Grottola A, Sabbatini AM, Tagliazucchi S, et al.. First human case of usutu virus neuroinvasive infection, Italy, August–September 2009. Euro Surveill. 2009; 14: pii. 19446.
- Tamba M, Bonilauri P, Bellini R, Calzolari M, Albieri A, Sambri V, et al.. Detection of Usutu virus within a West Nile virus surveillance program in Northern Italy. Vector Borne Zoonotic Dis. 2011; 11: 551–7. 10.3402/iee.v2i0.12001.
- Jöst H, Bialonski A, Maus D, Sambri V, Eiden M, Groschup MH, et al.. Isolation of Usutu virus in Germany. Am J Trop Med Hyg. 2011; 85: 551–3. 10.3402/iee.v2i0.12001.
- Cancrini G, Scaramozzino P, Gabrielli S, Di Paolo M, Toma L, Romi R. Aedes albopictus and Culex pipiens implicated as natural vectors of Dirofilaria repens in central Italy. J Med Entomol. 2007; 44: 1064–6. 10.3402/iee.v2i0.12001.
- Moutailler S, Krida G, Schaffner F, Vazeille M, Failloux AB. Potential vectors of rift valley fever virus in the mediterranean region. Vector Borne Zoonotic Dis. 2008; 8: 749–54. 10.3402/iee.v2i0.12001.
- Walsh PS, Metzger DA, Higuchi R. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques. 1991; 10: 506.
- Keyghobadi N, Matrone MA, Ebel GD, Kramer LD, Fonseca DM. Microsatellite loci from the northern house mosquito (Culex pipiens), a principal vector of West Nile virus in North America. Mol Ecol Resour. 2004; 4: 20–2.
- Smith JL, Keyghobadi N, Matrone MA, Escher RL, Fonseca DM. Cross species comparison of microsatellite loci in the Culex pipiens complex and beyond. Mol Ecol Notes. 2005; 5: 697–700. 10.3402/iee.v2i0.12001.
- Kent RJ, Harrington LC, Norris DE. Genetic differences between Culex pipiens f. molestus and Culex pipiens pipiens (diptera: Culicidae) in New York. J Med Entomol. 2007; 44: 50–9. 10.3402/iee.v2i0.12001.
- Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P. Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes. 2004; 4: 535–8. 10.3402/iee.v2i0.12001.
- Meglecz E, Petenian F, Danchin E, D'Acier AC, Rasplus JY, Faure E. High similarity between flanking regions of different microsatellites detected within each of two species of lepidoptera: Parnassius apollo and Euphydryas aurinia. Mol Ecol. 2004; 13: 1693–700. 10.3402/iee.v2i0.12001.
- Carlsson J. Effects of microsatellite null alleles on assignment testing. J Hered. 2008; 99: 616. 10.3402/iee.v2i0.12001.
- Chapuis MP, Estoup A. Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol. 2007; 24: 621. 10.3402/iee.v2i0.12001.
- Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online. 2005; 1: 47.
- Goudet J. Fstat (version 1.2): a computer program to calculate f-statistics. J Hered. 1995; 86: 485.
- Meirmans PG, Hedrick PW. Assessing population structure: FST and related measures. Mol Ecol Resources. 2011; 11: 5–18. 10.3402/iee.v2i0.12001.
- Liedloff A. Mantel nonparametric test calculator for windows version 2. Brisbane: School of Natural Resource Sciences. , Queensland University of Technology. 1999.
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. Mega5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011; 28: 2731–9. 10.3402/iee.v2i0.12001.
- Ryman N, Palm S. Powsim: a computer program for assessing statistical power when testing for genetic differentiation. Mol Ecol Notes. 2006; 6: 600–2. 10.3402/iee.v2i0.12001.
- Barton NH, Slatkin M. A quasi-equilibrium theory of the distribution of rare alleles in a subdivided population. Heredity. 1986; 56: 409–15. 10.3402/iee.v2i0.12001.
- Xu H, Fu YX. Estimating effective population size or mutation rate with microsatellites. Genetics. 2004; 166: 555. 10.3402/iee.v2i0.12001.
- Whitlock MC, McCauley DE. Indirect measures of gene flow and migration: Fst[ne]1/(4nm + 1). Heredity. 1999; 82: 117–25. 10.3402/iee.v2i0.12001.
- Linnaeus C. Systema naturae per regna tria naturae, secundum classes, ordines, genera, species, cum characteribus, differentiis, synonymis, locis (System of nature through the three kingdoms of nature, according to classes, orders, genera and species, with characters, differences, synonyms, places). Tomus i. Editio decima, reformata. vol. 1. 10th ed.Uppsala: Holmiæ (Salvius). 1758; 824. p.
- Brugger K, Rubel F. Simulation of climate-change scenarios to explain Usutu-virus dynamics in Austria. Prev Vet Med. 2009; 88: 24–31. 10.3402/iee.v2i0.12001.
- Reusken C, De Vries A, Buijs J, Braks M, Den Hartog W, Scholte EJ. First evidence for presence of Culex pipiens biotype molestus in the Netherlands, and of hybrid biotype pipiens and molestus in Northern Europe. J Vector Ecol. 2010; 35: 210–2. 10.3402/iee.v2i0.12001.
- Vazquez-Prokopec GM, Chaves LF, Ritchie SA, Davis J, Kitron U. Unforeseen costs of cutting mosquito surveillance budgets. PLoS Negl Trop Dis. 2010; 4: e858. 10.3402/iee.v2i0.12001.