
Abstract
Background
Hepatitis C virus (HCV) is a major public health concern and data on its molecular epidemiology in Sweden is scarce. We carried out an 8-year population-based study of newly diagnosed HCV cases in one of Sweden's centrally situated counties, Södermanland (D-county). The aim was to characterize the HCV strains circulating, analyze their genetic relatedness to detect networks, and in combination with demographic data learn more about transmission.
Methods
Molecular analyses of serum samples from 91% (N=557) of all newly notified cases in D-county, 2002–2009, were performed. Phylogenetic analysis (NS5B gene, 300 bp) was linked to demographic data from the national surveillance database, SmiNet, to characterize D-county transmission clusters. The linear-by-linear association test (LBL) was used to analyze trends over time.
Results
The most prevalent subtypes were 1a (38%) and 3a (34%). Subtype 1a was most prevalent among cases transmitted via sexual contact, via contaminated blood, or blood products, while subtype 3a was most prevalent among people who inject drugs (PWIDs). Phylogenetic analysis revealed that the subtype 3a sequences formed more and larger transmission clusters (50% of the sequences clustered), while the 1a sequences formed smaller clusters (19% of the sequences clustered), possibly suggesting different epidemics.
Conclusion
We found different transmission patterns in D-county which may, from a public health perspective, have implications for how to control virus infections by targeted interventions.
Hepatitis C virus (HCV) poses a major challenge for public health with 130–150 million chronic carriers worldwide and an estimated 500,000 related deaths annually (Citation1). In Sweden (population: approximately 9 million people), at least 45,000 individuals are estimated to be chronic carriers of HCV and it is one of the leading causes of liver transplantation (Citation2–Citation4). Before HCV was identified in 1989, and before implementation of blood donor screening in 1991, HCV was allowed to spread through blood transfusions and poorly sterilized medical equipment (Citation5). Today, the Swedish HCV epidemic is driven by people who inject drugs (PWIDs) and is likely related to a substantial increase of this group in the late 1960s and 1970s (Citation3, Citation4) (Citation6).
HCV exhibits a high degree of genetic diversity (Citation7) and is characterized by regional variations in genotype prevalence (Citation8, Citation9). It has been classified into seven genetically different genotypes, 1–7, 67 confirmed subtypes, and an additional 21 unassigned subtypes (Citation10, Citation11). There is no HCV vaccine available and increased knowledge on genotype, or even subtype, is critical for treatment duration and outcome prediction with many of the new direct acting antivirals (DAAs) (Citation12–Citation14). Considering the high costs of the latest pan-genotyping DAAs, the lack of a vaccine, as well as a constant recruitment of young PWIDs, the eradication of HCV is likely out of reach in the near future. Molecular characterization of HCV is important for both disease surveillance to guide implementation of preventive and control measures and for providing information for treatment and vaccine strategies.
In Sweden, it is mandatory for clinicians and laboratories to report and add demographic information on diagnosed cases of HCV to the national surveillance database SmiNet governed by The Public Health Agency. The report rate of genotype results, 2005–2014, was approximately 5%, and in cases of sequence-based genotyping the sequences are rarely, if at all, made public. To date, only fragmented genotyping data and few Swedish HCV sequences have been published (Citation15–Citation19) and further knowledge of the molecular epidemiology and circulating HCV strains in Sweden is needed. Here, we report on a population-based study on all first time notified cases of hepatitis C in the Swedish county, Södermanland (D-county), between 2002 and 2009. The aim was to molecular characterize the HCV strains circulating, analyze their genetic relatedness to detect networks, and in combination with demographic data learn more about transmission.
Materials and methods
Study population
Serum samples from 557 of 613 (91%) newly notified HCV-positive cases in D-county were collected between 2002 and 2009. All HCV confirmatory tests were performed at Capio Diagnostics (Eskilstuna, D-county); detection of antibodies with ARCHITECT Anti-HCV (Abbott, Chicago, USA) and INNO-LIA HCV Score (Fujirebio, Tokyo, Japan). RNA qualitative tests were performed using COBAS AMPLICOR HCV Test (Roche, Pleasanton, California). Demographic information was retrieved from SmiNet. All serum samples were coded with lab nr-ID and could only be associated with an identifiable individual by authorized staff at Public Health of Sweden working with hepatitis C case notifications in SmiNet. This study did not modify the existing diagnosis or the therapeutic strategy.
RNA extraction and sequencing
HCV RNA was extracted from 100 µl serum using an in-house phenol-chloroform extraction method and stored in −70°C. Synthesis of cDNA was performed using SuperScript III reverse transcriptase (Invitrogen, Carlsbad, California) and random hexamers (Roche Diagnostics, Pleasanton, California) according to manufacturer's guidelines. Primarily semi-nested PCR reactions were performed targeting NS5B; PCR1, 467 bp, using primers hep-101 and hep-120 (Citation20) and PCR II, 383 bp, using primers hep-101 and hep-105 (Citation21). If negative in the NS5B region, PCR reactions were amplified targeting the core gene; PCR1, 725 bp, using primers 186 and NCR3 (Citation21) and PCRII, 709 bp, using primers 186 and univ-1 (Citation20). The nested PCR products were purified and sequenced using BigDye Terminator Cycle Sequencing Ready reaction kit version (Applied Biosystems, Foster City, USA) and the primers were used in the PCR as sequencing primers. The ABI PRISM 3100 genetic analyzer (Applied Biosystems, Foster City, USA) was used for capillary electrophoresis sequencing and data collection. Sequences were edited using the CLC Main workbench 6 software (CLCbio, Aarhus, Denmark).
Genotyping and phylogenetic analysis
Determination of the HCV subtype was done by phylogenetic analysis using reference sequences from the Los Alamos HCV data base (www.hcv.lanl.gov/content/index).
To obtain a reference data set, all HCV sequences available at the time of analysis (March 20, 2014) were downloaded from the NCBI nucleotide collection (N=147,534). A BLAST database was built from these sequences using makeblastdb (BLAST+2.2.29). All query (NS5B) sequences were matched against the BLAST database using BLASTN. Identity score results of 83 and 87%, empirically tested, were used to exclude sequences from other subtypes than 3a and 1a, respectively. After phylogenetic analysis of the reference data set, large clusters belonging to one patient were removed to reduce bias in sequence diversity. All remaining reference sequences with overlap ≥280 nt with the Swedish NS5B sequences (300 nt) were collected in a reference data set. The reference sequences and Swedish sequences were aligned using Clustal Omega v1.2.0. Garli 2.01 was used for maximum likelihood phylogenetic analysis using the general time-reversible (GTR) model. The reliability of each tree split was estimated by the approximate likelihood-ratio test Shimodaira–Hasegawa (aLRT-SH) using PhyML 3.0.1 (Citation22, Citation23). Trees were visualized and edited in FigTree 1.4.2.
Definition of a transmission cluster
A transmission cluster was determined by statistically supported nodes in the phylogeny as strongly supported clusters **aLRT-SH >0.9 and as moderately supported clusters *aLRT-SH >0.85 (based on expansive studies on HIV clusters, Joakim Esbjörnsson, unpublished observations). Transmission clusters with ≥80% of the sequences from D-county were defined as Swedish ‘D-county-clusters’. If>80% of sequences belonged to female or male cases, we defined the clusters as a ‘female’ cluster or ‘male’ cluster, respectively, and otherwise as ‘mixed’ clusters. Clusters were determined as ‘dyads’ (two sequences), 3–10 sequences as networks and larger clusters if>10 sequences.
Statistics
The linear-by-linear association test (LBL) was used to analyze trends over time. Other statistical analyses were performed using the two-tailed Fisher's exact test. Statistical analysis was performed using IBM SPSS Statistics for Windows, Version 21.0., IBM Corp, Armonk, NY.
Results
Study population and demographic characteristics
The demographic characteristics of the first time notified HCV-positive population in Södermanland (D-county) in 2002–2009 (age, gender, country of transmission, risk factors, and reason for diagnosis) were in line with data on cases reported from all of Sweden (data extracted from SmiNet). The reason for HCV test analysis, as reported in SmiNet (D-county, 2002–2009; N=613), were screening of high-risk groups (38%), other reasons (29%), no information reported (23%), due to symptoms (7%), and epidemiological contact tracing (2%). HCV was most common among men, 397 of 613 (65%), and the average ages were 38 for men and 40 for women on the date of notification. Due to the often asymptomatic nature of hepatitis C, the age reported on the date of notification may considerably differ from the actual date of transmission. The male/female ratio (2:1) was consistent during the study period. The majority of cases, 354 of 613 (58%), were reported with injecting drug use as risk factor (male/female; 2.8). Transmission via injecting drug use increased among females over the study period (from 12 to 40%, 2002–2009, p=0.044, LBL); however, no relative increase of the entire group of PWIDs was observed (from 16 to 18%, 2002–2009, p=0.25, LBL). Unknown or no reported risk factor was reported with 177 of 613 (29%) frequency (male/female; 1.4), followed by 35 of 613 (6%) infected via contaminated blood/blood products (male/female; 0.67), and 31 of 613 (5%) via sexual contact (male/female; 0.55). A minority, 16 of 613 (2.5%), was reported with other routes of transmission. A majority, 425 of 613 (69%), of individuals stated Sweden as the most likely country of infection, 54 of 613 (9%) were reported with infection acquired in other countries, and 134 of 613 (22%) had no information on the country of infection. Overall, a non-significant decreasing trend of transmission within Sweden was found over the study period (from 92 to 82%, 2002–2009, p=0.090, LBL). However, when the analysis was stratified by gender, this decreasing trend was significant among females (100 to 75%, 2002–2009, p=0.029, LBL).
Genotype distribution
Genotyping was possible for 403 of 557 (72%) serum samples collected between 2002 and 2009. The remaining 154 of 557 (28%) had too low RNA concentration for PCR amplification and sequencing. The genotyping results were based on sequencing of the NS5B gene, 367 of 403 (91%), and the core gene, 36 of 403 (9%). Around 100 samples, 2002–2009, were sequenced in both the NS5B and the core gene and no recombinant was found (data not shown). Based on this and that recombinants are considered rare (Citation24), we have combined the genotype results in our analyses. The subtypes 1a and 3a were most prevalent, 38 and 34%, respectively, followed by 2b (19%) and 1b (6%). A few cases of minority subtypes were found (1g, 2a, 3h, 4a, 6a and 6e), of which all reported that they likely acquired the infection outside Sweden. The overall subtype distribution was relatively stable over the years except for the last year of sampling (2009), when eight 1b samples were collected compared to an average of 1.6 cases/year, 2002–2008. During the study period subtype, 1b increased among females (from 10 to 32%, 2002–2009, p=0.061, LBL). Also, minority subtypes increased both overall (from 0 to 6%, 2002–2009, p=0.007, LBL) and among men (from 0 to 10%, 2002–2009, p=0.010, LBL). The proportion of subtype 3a among females decreased (from 55 to 21%, 2002–2009, p=0.014, LBL), and among PWIDs subtype 1a increased significantly over the study period, both among males and females (from 29 to 57%, 2002–2009, p=0.003, LBL).
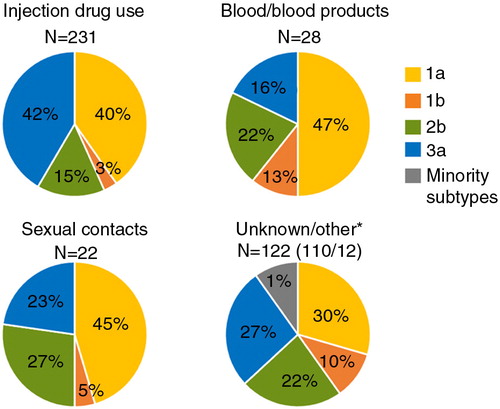
Overall, subtype 1a dominated the prevalence among the cases transmitted via contaminated blood or blood products and among sexual transmissions in D-county during 2002–2009 (). Among the cases transmitted via injecting drug use, subtype 1a and 3a was equally prevalent over 2002–2009. However, 3a occurred with higher prevalence among PWIDs as compared to cases transmitted via other routes (p=0.0007, two-tailed Fisher's exact test) ().
Fig. 1 Genotype distribution found among the risk factors associated with HCV in patients first notified during 2002–2009. *Unknown=unknown or no information reported. Other=care-related transmission as patient/staff, mother to child/pregnancy, tattoo/piercing, other.
Analysis of transmission clusters
Transmission patterns were analyzed focusing on NS5B sequences from the most prevalent subtypes, 1a and 3a. A total of 154 genotype 1a and 138 genotype 3a samples collected from D-county were sequenced in NS5B and included in the phylogenetic analysis (including 23 1a sequences from D-county cases first notified during 1991–2001, 2 1a sequences from 2010, and 25 3a sequences from cases notified during 1991–1999). The reference sequences, 1a N=2493 and 3a N=998, originated from 38 and 32 different countries, respectively.
The overall topologies of the 1a and 3a trees showed distinctly different patterns in the distribution of the Swedish D-county sequences. The 3a sequences from D-county were to a greater extent found in clusters compared with the 1a sequences from D-county (51 and 18%, respectively, and ). The 1a sequences were more highly interspersed as single sequences among the reference sequences compared with the 3a sequences (42 and 12%, respectively). A majority of these single sequences belonged to PWIDs. For subtype 1a, 13 D-county clusters were found; 11 dyads and two networks with three sequences each. For subtype 3a, 17 D-county clusters were found; six dyads and 11 networks with 3–10 sequences (). No large cluster was found; however, many of the 3a dyads and networks were part of a large cluster (just below the threshold of what we defined as moderately supported clusters) with 87 cases (N=74 D-county sequences, aLRT-SH 0.85), b. Many of the international reference sequences (N=13) in this cluster were isolated from PWIDs in the United Kingdom and France. Overall, this large cluster was overrepresented with strains from PWIDs first time notified in D-county in the beginning of the 90s till 2009.
Fig. 2 Maximum likelihood phylogenetic analysis of NS5B sequences of subtype 1a and 3a. (a) Subtype 1a and (b) subtype 3a and a zoom-in of a large non-supported cluster N=74 (aLRT-SH=0.8), including 10 of 17 3a D-county clusters (five dyads and five networks with high statistical support (aLRT-SH >0.85). The tree is displayed as midpoint rooted. Branches in red denote Swedish sequences from D-county– 1a: N=122 (2002–2009), N=23 (1991–2001), and 3a: N=115 (2002–2009), N=25 (1991–1999). Branches in gray denote all overlapping reference sequences from GenBank (1a: N=2493 and 3a: N=998) originating from 38 and 32 countries, respectively, worldwide. The Swedish D-county clusters: dyads (two sequences) and networks (3–10 sequences) are highlighted in blue (SH-aLRT >0.85).
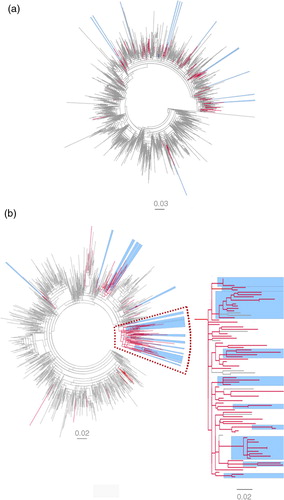
Table 1 1a and 3a Swedish D-county clusters and gender
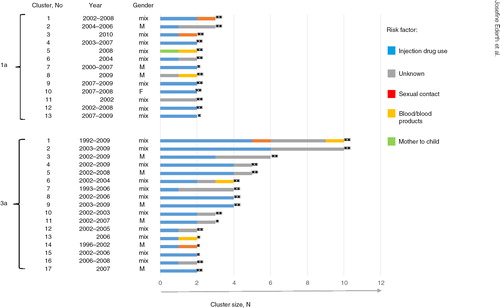
All of the 3a clusters contained sequences isolated from cases with injection drug use as transmission source, while among the 1a clusters, three dyads were found isolated from cases with no reported relation to injection drug use (). Unknown source of infection was common among cases of both subtypes and were often found together with sequences isolated from PWIDs in the clusters (). None of the cases (infected with 1a, N=12, or 3a, N=6) transmitted via contaminated blood/blood products were found to have closely related strains. However, the cases that inferred one of the 1a dyads (No. 5 in ) included a strain likely indirectly received from the same source of contaminated blood. Of the sexual transmissions reported (infected with 1a, N=14, or 3a, N=6), none were directly related in the phylogeny. The sexual transmissions found in dyads or networks were often connected to PWIDs. The majority of 1a and 3a clusters had mixed gender and only one female cluster was found, a 1a dyad consisting of two cases transmitted via injecting drug use ().
Fig. 3 D-county clusters and risk factors for transmission. Subtypes 1a and 3a D-county clusters are displayed as horizontal bars (N cases on x-axis; strongly supported clusters **aLRT-SH >0.9 and moderately supported clusters *aLRT-SH >0.85). The clusters are color coded according to the number of cases belonging to different risk factors for transmission (color-risk factor to the right). Gender and year of notification are shown to the left of the bars.
Discussion
This study was initiated due to the limited amount of molecular data on HCV strains circulating in Sweden. Here, we provide a population-based study from a Swedish county, Södermanland (D-county), with molecular data on >70% of all newly diagnosed cases of HCV, 2002–2009. Subtype 1a and 3a were most prevalently detected over the study period, and an increase in subtype 1b and minority subtypes was observed. The proportions of subtype 3a were higher among PWIDs than the rest of the population, where 1a dominated.
The majority of HCV tests in D-county were performed by testing of individuals of high-risk groups, such as PWIDs. This could explain the relatively high prevalence of 1a and 3a found in D-county since these subtypes have been observed among PWIDs in many European countries as well as North Africa and the Mediterranean basin (Citation25–Citation27).
The increase in minority subtypes was likely caused by a change in migration flows since all cases had reported transmission in endemic countries as probable route of infection. Subtype 1b, which increased at the end of the study period, is so far considered relatively rare in Sweden (Citation19). However, in neighboring countries such as the Baltic countries and Russia, as well as other Mediterranean countries, there is high prevalence of subtype 1b (Citation28, Citation29). Several of the 1b subtypes isolated in D-county were retrieved from PWIDs (>25%) with transmission in Sweden. Emergence of new subtypes into the PWID networks can potentially bring about a change in the relative prevalence (Citation20, Citation30–Citation32). This has been observed, for example, with a 3a-to-1b shift among PWIDs in Estonia and increased prevalence of genotype 4 in Europe and subtype 6a in Vietnam and Hong Kong (Citation19, Citation32) (Citation33). These observations suggest that any subtype has an epidemic potential under the right circumstances. At present, the treatment strategies are dependent on genotype when it comes to duration and outcome prediction using standard of care combination treatments with interferon, ribavirin, and DAAs (Citation34).
Substantial research on new DAAs has been directed toward genotype 1, which is the most prevalent genotype worldwide (Citation8), with a history of being one of the more difficult genotypes to treat. Today, genotype 3, with high prevalence among PWIDs (Citation35, Citation36), is considered to be the new challenge for effective treatment with many of the recent DAAs (Citation12, Citation13) (Citation37). Until the latest pan-genotypic DAAs is affordable for the large mass of HCV-patients that needs to be treated, surveillance of genotype prevalence is of importance for national treatment strategies as well as better understanding of treatment of minority genotypes is needed. Over the past 10 years, the majority (95%) of notified cases in the national surveillance database lacked information on genotype (data extracted from SmiNet). It is unclear whether this is due to a low rate of genotyping tests performed or just low rate of reporting. However, improved reporting of genotyping data would allow tracking fluctuation in genotype prevalence in various epidemiological settings to signal possible changes in transmission. However, phylogenetic analysis of sequence data is required to reveal or verify detailed networks of transmission.
Subtype 1a and 3a were equally prevalent among the dominating risk group, PWIDs, while 1a was found to be the most common subtype among transmission via sexual contact as well as via contaminated blood or blood products. The second most common route of transmission reported was ‘unknown’ and many of the D-county clusters, both 1a and 3a, included sequences from cases with unknown routes of transmission intermixed with strains from PWIDs (). Individuals notified with unknown routes of transmission were often associated with high risk for drug-related care (Citation4). It is possible that ‘unknown’ could indicate injection drug use, but that this was not reported due to reluctance in admitting persistent or sporadic drug use. Sexual transmissions could also be considered even if anticipated as a low-rate transmission route (Citation38). However, the spread of HCV has emerged among men who have sex with men (MSM) in Europe (Citation39) during the past years. A few cases with reported sexual contact were also interspersed among the cases transmitted via injection drug use, both for subtype 1a and 3a, further implying sexual transmission from PWIDs to their partners. Other explanations for unknown transmission may be transmission at clinical settings, which has, for example, occurred in Sweden even after HCV was discovered (Citation34, Citation40) (Citation41).
The subtype 1a and 3a sequences showed distinct patterns of distribution among the reference sequences in the phylogeny, suggesting different epidemics (a and b). Subtype 1a sequences did not cluster as often and generally formed smaller clusters (2–3 sequences) as compared with subtype 3a sequences (19% vs. 51% of the sequences formed D-county clusters, respectively). Subtype 1a sequences were also more intermixed with international reference sequences compared with subtype 3a, suggesting that extant subtype 1a lineages were introduced to D-county from multiple independent sources. The majority of the clustering 1a sequences were dyads with mixed gender and transmission via injection drug use, suggesting transmission between couples or in smaller PWIDs networks with more restricted needle sharing.
We also observed a general increase, irrespective of infecting subtype, among female PWIDs (from 12 to 40%, 2002–2009, p=0,040) as well as a rise in subtype 1a among PWIDs over the study period. This suggests that there may be gender-based behavioral differences which need to be explored further. The increase in 1a among PWIDs indicated an upcoming rise in the number of subtype 1a cases in D-county. With the subclinical nature of HCV, the lag time between infection and actual diagnosis can be years to decades, and the many small subtype 1a clusters may reflect recent introductions that could expand into future larger networks.
Subtype 3a was most prevalent among PWIDs compared to the other risk groups in D-county. Subtype 3a is known to be highly prevalent among PWIDs and show increased incidence in various parts of the world (Citation42–Citation46). Large, non-exclusive transmission clusters of subtype 3a among PWIDs worldwide has led to the suggestion that 3a spread through a unique worldwide epidemic driven by injection drug use (Citation27, Citation35) (Citation36, Citation45) (Citation47, Citation48). The 3a clusters in D-county likely grew over a long time, with cases reported as early as in the beginning of the 1990s. The larger 3a networks observed in D-county suggest that subtype 3a were more common in groups of PWIDs practicing high-risk behavior such as needle sharing and perhaps socializing in larger networks. In a Greek study, subtype 3a was in analogy characterized by faster epidemic growth over time compared with subtype 1a (Citation45).
Finally, single sequences, not forming clusters, also gave valuable information on transmission behavior in D-county. The phylogeny revealed that 42% of the subtype 1a sequences and 12% of the subtype 3a sequences were interspersed among the international reference sequences. A majority of these sequences, both for 1a and 3a, belonged to cases that acquired the infection in Sweden and had stated injection drug use as the likely source of infection. Many PWIDs are HCV positive or will contract the virus within 3 years after active injection drug use (Citation49), and it is possible that unreported cases connected to these individuals exist. Other explanations for single sequences may be the location of treatment facilities for PWIDs in D-county that may have contributed to the introduction HCV strains from other parts of Sweden. Single sequences from PWIDs may further reflect on injection drug use in the past, eliminated risk behaviors, emigration to other regions, or difficulties in contact tracing related individuals.
In conclusion, molecular characterization of HCV strains circulating in D-county over 8 years showed different transmission patterns for the most common subtypes 1a and 3a. Improved resolution of transmission patterns could be achieved by sequencing longer fragments or ultimately complete genomes. In addition, more sequences are needed, both national and international, to further explore and map clusters as regional or part of the nationwide spread.
From a public health perspective, molecular surveillance together with epidemiological data would allow survey of the transmission dynamics to promote targeted interventions.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
Thanks to Romanico Arrighi for a critical reading of the manuscript.
Funding
Joakim Esbjörnsson was supported by a postdoctoral fellowship from the Swedish Research Council (350-2012–6628).
References
- WHO. Hepatitis C: fact sheet no. 164. 2012; Geneva. Available from: http://www.who.int/mediacentre/factsheets/fs164/en/ (cited 30 December 2015]..
- Lavanchy D. The global burden of hepatitis C. Liver Int. 2009; 29(Suppl 1): 74–81.
- Duberg AS, Blach S, Falconer K, Kaberg M, Razavi H, Aleman S. The future disease burden of hepatitis C virus infection in Sweden and the impact of different treatment strategies. Scand J Gastroenterol. 2015; 50: 233–44.
- Duberg AS, Pettersson H, Aleman S, Blaxhult A, Daviethsdottir L, Hultcrantz R, etal. The burden of hepatitis C in Sweden: a national study of inpatient care. J Viral Hepat. 2011; 18: 106–118.
- Choo QL, Kuo G, Weiner A, Wang KS, Overby L, Bradley D, etal. Identification of the major, parenteral non-A, non-B hepatitis agent (hepatitis C virus) using a recombinant cDNA approach. Semin Liver Dis. 1992; 12: 279–88.
- Duberg A, Janzon R, Back E, Ekdahl K, Blaxhult A. The epidemiology of hepatitis C virus infection in Sweden. Euro Surveill . 2008. pii: 18882.
- Jackowiak P, Kuls K, Budzko L, Mania A, Figlerowicz M, Figlerowicz M. Phylogeny and molecular evolution of the hepatitis C virus. Infect Genet Evol. 2014; 21: 67–82.
- Messina JP, Humphreys I, Flaxman A, Brown A, Cooke GS, Pybus OG, etal. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology. 2015; 61: 77–87.
- Trimbitas RD, Serghini FZ, Lazaar F, Baha W, Foullous A, Essalhi M, etal. The “hidden” epidemic: a snapshot of Moroccan intravenous drug users. Virol J. 2014; 11: 43.
- Nakano T, Lau GM, Lau GM, Sugiyama M, Mizokami M. An updated analysis of hepatitis C virus genotypes and subtypes based on the complete coding region. Liver Int. 2012; 32: 339–45.
- Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, etal. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014; 59: 318–27.
- Ampuero J, Romero-Gomez M, Reddy KR. Review article: HCV genotype 3 – the new treatment challenge. Aliment Pharmacol Ther. 2014; 39: 686–98.
- Lawitz E, Gane EJ. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 2013; 369: 678–9.
- Li HC, Lo SY. Hepatitis C virus: virology, diagnosis and treatment. World J Hepatol. 2015; 7: 1377–89.
- Westin J, Lindh M, Lagging LM, Norkrans G, Wejstal R. Chronic hepatitis C in Sweden: genotype distribution over time in different epidemiological settings. Scand J Infect Dis. 1999; 31: 355–8.
- Lindh M, Hannoun C. Genotyping of hepatitis C virus by Taqman real-time PCR. J Clin Virol. 2005; 34: 108–14.
- Lidman C, Norden L, Kaberg M, Kall K, Franck J, Aleman S, etal. Hepatitis C infection among injection drug users in Stockholm Sweden: prevalence and gender. Scand J Infect Dis. 2009; 41: 679–84.
- Danielsson A, Palanisamy N, Golbob S, Yin H, Blomberg J, Hedlund J, etal. Transmission of hepatitis C virus among intravenous drug users in the Uppsala region of Sweden. Infect Ecol Epidemiol. 2014; 4 , 22251, doi: http://dx.doi.org/10.3402/iee.v4.22251 .
- Bruggmann P, Berg T, Ovrehus AL, Moreno C, Brandao Mello CE, Roudot-Thoraval F, etal. Historical epidemiology of hepatitis C virus (HCV) in selected countries. J Viral Hepat. 2014; 21(Suppl 1): 5–33.
- Kalinina O, Norder H, Vetrov T, Zhdanov K, Barzunova M, Plotnikova V, etal. Shift in predominating subtype of HCV from 1b to 3a in St. Petersburg mediated by increase in injecting drug use. J Med Virol. 2001; 65: 517–24.
- Norder H, Bergstrom A, Uhnoo I, Alden J, Weiss L, Czajkowski J, etal. Confirmation of nosocomial transmission of hepatitis C virus by phylogenetic analysis of the NS5-B region. J Clin Microbiol. 1998; 36: 3066–9. [PubMed Abstract] [PubMed CentralFull Text].
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010; 59: 307–21.
- Anisimova M, Gil M, Dufayard JF, Dessimoz C, Gascuel O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst Biol. 2011; 60: 685–99.
- Folch C, Casabona J, Espelt A, Majo X, Merono M, Gonzalez V, etal. Gender differences in HIV risk behaviours among intravenous drug users in Catalonia, Spain. Gac Sanit. 2013; 27: 338–43.
- Mahfoud Z, Kassak K, Kreidieh K, Shamra S, Ramia S. Distribution of hepatitis C virus genotypes among injecting drug users in Lebanon. Virol J. 2010; 7: 96.
- Raptopoulou M, Touloumi G, Tzourmakliotis D, Nikolopoulou G, Dimopoulou M, Giannoulis G, etal. Significant epidemiological changes in chronic hepatitis C infection: results of the nationwide HEPNET-GREECE cohort study. Hippokratia. 2011; 15: 26–31. [PubMed Abstract] [PubMed CentralFull Text].
- Calado RA, Rocha MR, Parreira R, Piedade J, Venenno T, Esteves A. Hepatitis C virus subtypes circulating among intravenous drug users in Lisbon, Portugal. J Med Virol. 2011; 83: 608–15.
- Stamenkovic G, Zerjav S, Velickovic ZM, Krtolica K, Samardzija VL, Jemuovic L, etal. Distribution of HCV genotypes among risk groups in Serbia. Eur J Epidemiol. 2000; 16: 949–54.
- Vince A, Iscic-Bes J, Zidovec Lepej S, Baca-Vrakela I, Bradaric N, Kurelac I, etal. Distribution of hepatitis C virus genotypes in Croatia – a 10 year retrospective study of four geographic regions. Coll Antropol. 2006; 30(Suppl 2): 139–43. [PubMed Abstract].
- Schroter M, Zollner B, Laufs R, Feucht HH. Changes in the prevalence of hepatitis C virus genotype among injection drug users: a highly dynamic process. J Infect Dis. 2004; 190: 1199–200.
- Treso B, Takacs M, Dencs A, Dudas M, Par A, Rusvai E. Molecular epidemiology of hepatitis C virus genotypes and subtypes among injecting drug users in Hungary. Euro Surveill. 2013; 18 pii: 20639.
- Tallo T, Norder H, Tefanova V, Krispin T, Schmidt J, Ilmoja M, etal. Genetic characterization of hepatitis C virus strains in Estonia: fluctuations in the predominating subtype with time. J Med Virol. 2007; 79: 374–82.
- Ngui SL, Brant L, Markov PV, Tung JP, Pybus OG, Teo CG, etal. Hepatitis C virus genotype 4 in England: diversity and demographic associations. J Med Virol. 2015; 87: 417–23.
- Lagging M, Duberg AS, Wejstal R, Weiland O, Lindh M, Aleman S, etal. Treatment of hepatitis C virus infection in adults and children: updated Swedish consensus recommendations. Scand J Infect Dis. 2012; 44: 502–21.
- Morice Y, Cantaloube JF, Beaucourt S, Barbotte L, De Gendt S, Goncales FL, etal. Molecular epidemiology of hepatitis C virus subtype 3a in injecting drug users. J Med Virol. 2006; 78: 1296–303.
- Pybus OG, Cochrane A, Holmes EC, Simmonds P. The hepatitis C virus epidemic among injecting drug users. Infect Genet Evol. 2005; 5: 131–9.
- Kylefjord H, Danielsson A, Sedig S, Belda O, Wiktelius D, Vrang L, etal. Transient replication of a hepatitis C virus genotype 1b replicon chimera encoding NS5A-5B from genotype 3a. J Virol Methods. 2014; 195: 156–63.
- Terrault NA, Dodge JL, Murphy EL, Tavis JE, Kiss A, Levin TR, etal. Sexual transmission of hepatitis C virus among monogamous heterosexual couples: the HCV partners study. Hepatology. 2013; 57: 881–9.
- Boesecke C, Grint D, Soriano V, Lundgren JD, d'Arminio Monforte A, Mitsura VM, etal. Hepatitis C seroconversions in HIV infection across Europe: which regions and patient groups are affected?. Liver Int. 2015; 35: 2384–91.
- Widell A, Christensson B, Wiebe T, Schalen C, Hansson HB, Allander T, etal. Epidemiologic and molecular investigation of outbreaks of hepatitis C virus infection on a pediatric oncology service. Ann Intern Med. 1999; 130: 130–4.
- Waldenstrom J, Konar J, Ekermo B, Norder H, Lagging M. Neonatal transfusion-transmitted hepatitis C virus infection following a pre-seroconversion window-phase donation in Sweden. Scand J Infect Dis. 2013; 45: 796–9.
- Choudhary MC, Natarajan V, Pandey P, Gupta E, Sharma S, Tripathi R, etal. Identification of Indian sub-continent as hotspot for HCV genotype 3a origin by Bayesian evolutionary reconstruction. Infect Genet Evol. 2014; 28: 87–94.
- Paintsil E, Verevochkin SV, Dukhovlinova E, Niccolai L, Barbour R, White E, etal. Hepatitis C virus infection among drug injectors in St Petersburg, Russia: social and molecular epidemiology of an endemic infection. Addiction. 2009; 104: 1881–90.
- Aitken CK, McCaw RF, Bowden DS, Tracy SL, Kelsall JG, Higgs PG, etal. Molecular epidemiology of hepatitis C virus in a social network of injection drug users. J Infect Dis. 2004; 190: 1586–95.
- Magiorkinis G, Sypsa V, Magiorkinis E, Paraskevis D, Katsoulidou A, Belshaw R, etal. Integrating phylodynamics and epidemiology to estimate transmission diversity in viral epidemics. PLoS Comput Biol. 2013; 9: e1002876.
- Samimi-Rad K, Nasiri Toosi M, Masoudi-Nejad A, Najafi A, Rahimnia R, Asgari F, etal. Molecular epidemiology of hepatitis C virus among injection drug users in Iran: a slight change in prevalence of HCV genotypes over time. Arch Virol. 2012; 157: 1959–65.
- Li C, Lu L, Murphy DG, Negro F, Okamoto H. Origin of hepatitis C virus genotype 3 in Africa as estimated through an evolutionary analysis of the full-length genomes of nine subtypes, including the newly sequenced 3d and 3e. J Gen Virol. 2014; 95: 1677–88.
- Akkarathamrongsin S, Hacharoen P, Tangkijvanich P, Theamboonlers A, Tanaka Y, Mizokami M, etal. Molecular epidemiology and genetic history of hepatitis C virus subtype 3a infection in Thailand. Intervirology. 2013; 56: 284–94.
- Hillgren K, Sarkar K, Elofsson S, Britton S. [Widespread risk behavior among injecting drug users. Over 80 percent HCV-infected – 7 percent have HIV, as demonstrated by the first baseline study]. Lakartidningen. 2012; 109: 1221–5. [PubMed Abstract].