
Abstract
Background
Whole genome sequencing (WGS) is becoming a routine tool for infectious disease outbreak investigations. The Swedish situation provides an excellent opportunity to test the usefulness of WGS for investigation of outbreaks with Salmonella Dublin (S. Dublin) as epidemiological investigations are always performed when Salmonella is detected in livestock production, and index isolates from all detected herds are stored and therefore available for analysis. This study was performed to evaluate WGS as a tool in forward and backward tracings from herds infected with S. Dublin.
Material and methods
In this study, 28 isolates from 26 cattle herds were analysed and the WGS results were compared with results from the epidemiological investigations, for example, information on contacts between herds. The isolates originated from herds in three different outbreaks separated geographically and to some extent also in time, and from the only region in Sweden where S. Dublin is endemic (Öland).
Results
The WGS results of isolates from the three non-endemic regions were reliably separated from each other and from the endemic isolates. Within the outbreaks, herds with known epidemiological contacts generally showed smaller differences between isolates as compared to when there were no known epidemiological contacts.
Conclusion
The results indicate that WGS can provide valuable supplemental information in S. Dublin outbreak investigations. The resolution of the WGS was sufficient to distinguish isolates from the different outbreaks and provided additional information to the investigations within an outbreak.
To access the supplementary material for this article, please see Supplementary files under ‘Article Tools’
Salmonella Dublin (S. Dublin) is one of the most commonly occurring serotypes in cattle. It is adapted to cattle, and infections usually cause clinical disease (Citation1, Citation2) and decreased milk production (Citation3), resulting in substantial economic losses for the farmer (Citation4). S. Dublin also has zoonotic potential. Infection in humans is uncommon, but when occurring, a large proportion of patients will suffer from invasive infection (Citation5).
In Sweden, legislated Salmonella control was initiated in the 1960s. The programme covers all serotypes and all animal species along the entire chain from feed to food with one exception, the sheep-adapted S. diarizonae serovar 61: (k):1, 5Citation7, (Citation6). The programme has resulted in a very low prevalence of Salmonella in Swedish cattle Citation7–(Citation9). All herds detected with Salmonella are put under restrictions and forward and backward tracings are performed. Despite this, the source of infection is often not determined. Genotyping can provide valuable supporting information in such investigations
Previous studies using pulsed-field gel electrophoresis (PFGE) have provided unsatisfactory resolution among Swedish S. Dublin isolates (unpublished results) whereas a Danish study showed some ability to separate isolates based on PFGE and other typing methods (Citation10). Whole genome sequencing (WGS) has been successfully used for other Salmonella serotypes (Citation11) and could provide a better ability to distinguish between closely related isolates. However, to fully realise the potential of WGS as a typing tool, an increased knowledge about the single-nucleotide polymorphism (SNP) variation that occurs within and between outbreaks and endemic regions is needed in order to properly interpret the data in an epidemiological context. Reference databases with WGS results of historical isolates are also needed to understand the SNP variation that occurs over time.
The Swedish situation provides an excellent opportunity to test the usefulness of WGS in investigations of outbreaks with S. Dublin. This is because the prevalence of S. Dublin is very low with the exception of one region in the southeast with endemic occurrence (Citation8, Citation9), epidemiological investigations are always performed when Salmonella is detected, and index isolates from all detected herds are stored and therefore available for analyses.
The aspects of both laboratory procedures and data analysis of WGS are becoming routine measures. The next big challenge, in order to recognise the potential of these techniques in supporting outbreak investigations, is the interpretation of the data. The aim of this study was to investigate if WGS can be used as a tool in forward and backward tracings from herds infected with S. Dublin.
Material and methods
From January 1999 to December 2013, S. Dublin was isolated from 59 Swedish cattle herds. The first isolate from an infected herd is always frozen and stored at the Swedish National Veterinary Institute (SVA). Thereafter, if the herd remains under restrictions, at least two isolates from each positive sampling occasion are stored. Due to limited economic resources only a limited number of isolates could be analysed in this study. Isolates for WGS were chosen aiming at evaluating the genomic differences between isolates from herds within the same outbreak and between different outbreaks. In addition, isolates were chosen from herds in the island of Öland, the only region in Sweden with endemic infection. In total, isolates from 26 different herds were selected, originating from herds within three geographically well separated regions (Västergötland, Östergötland, and Skåne) and from the endemic region (Öland) (, ). The infected herds in the non-endemic regions were detected within a limited time period. The isolates from the endemic region were selected in order to gain information on the genomic variation occurring in this region; therefore, herds without known epidemiological contacts were selected. From two of the selected herds in Skåne, sequencing analysis had already been performed on previous isolates collected 1 year earlier, and these results were also included in this study.
Fig. 1 Map over southern Sweden showing the geographic location of the herds from which isolates were analysed. Green=Västergötland, Yellow=Östergötland, Red=Skåne, Blue=Endemic region (Öland).
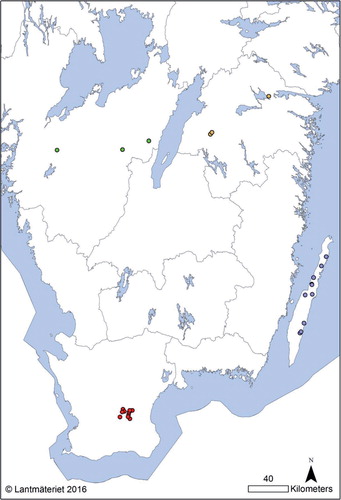
Table 1 Salmonella Dublin isolates selected for WGS
Data on herd identities, geographical location, and animal trade were retrieved from the national cattle database and registers of animal holdings kept at the Swedish Board of Agriculture (SBA). Registration of cattle is mandatory in Europe, with unique identification numbers for all cattle. Farmers are obliged to continuously report animal events such as births, deaths, animals sent to slaughter, sales, and purchases.
Directly following detection of a Salmonella-positive herd, an epidemiological investigation including forward and backward tracings is always performed by the SBA and SVA in cooperation. Information on contacts is collected via repeated conversations with the farmer, employees at the farm, farm veterinarians, and if relevant, other sources such as feed suppliers, companies spreading manure, and carcass collectors. This information is stored in notes and protocols which were used in this study.
Deoxyribonucleic acid (DNA) was extracted using the DNA Tissue Kit automated with an EZ1 biorobot (Qiagen). Sequencing libraries were prepared with the Nextera XT kit (Illumina) according to the manufacturer's instructions. Sequencing was performed with 300 bp V3 paired-end reads on an Illumina MiSeq instrument to a coverage of 41–57× per isolate. The sequence data set is available from the European Nucleotide Archive under study accession number PRJEB12354. The reads were assembled using MIRA v. 3.9.15 (Citation12). Variant SNP calling was performed using MUMmer 3.23 (Citation13), with the show-snps flags – lIrx 25, and the output filtered by BUFF >50 and SNP position assembly quality >80. The S. Dublin CT_02021853 genome (Genbank CP001144) (Citation14) was used as reference. Each assembly was queried with each SNP context from the MUMmer output using BLAST+ (Citation15), retaining only SNPs for which exactly one occurrence of either of the two alleles was found in all assemblies. A maximum likelihood phylogenetic tree was created in MEGA 5 (Citation16) based on the SNP states. The Kimura 2-parameter substitution model (Citation16) was used, selected in MEGA as minimising the Bayesian information criterium for the sequence data. One thousand bootstrap replicates were performed to test the reliability of branches. The analysis was repeated with the inclusion of several more reference isolates to further explore the relationship between Swedish S. Dublin and S. Dublin from other regions. Read data from four isolates from the United Kingdom were downloaded from the NCBI Short Read Archive: two isolated from animal sources in 2014 (Acc. no. SRR1965049 and SRR1965478) and two from food sources in 2015 (SRR3322264 and SRR3322365). These were assembled as described above. Furthermore, assemblies of sequence data from seven isolates from ground beef in the United States, all from different states (CA, MD, NM, OR, CO, PA, NY) isolated in 2015, were downloaded from Genbank (Acc. GCA_001480305.1, GCA_001480245.1, GCA_001247195.1, GCA_001245835.1, GCA_001246165.1, GCA_001480725.1, GCA_001481195.1). Finally, the assembly of the S. Dublin SC50 strain was included in the analysis (Acc. GCA_000953175.1). SNP position assembly quality filtering was not performed at this re-analysis stage as quality metrics was absent in some of the included sequences.
Results
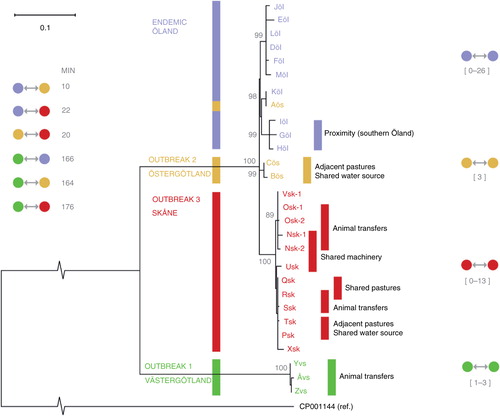
After filtering, a total of 537 variable genomic positions were available in all isolates, including the reference genome CT_02021853, for comparison in the SNP analysis. Based on SNP data, the isolates from the endemic region (Öland) formed a cluster together with the isolates from Östergötland and Skåne (). The Skåne isolates formed a distinct subgroup within this cluster, as did two of the three isolates from Östergötland. The third isolate from Östergötland clustered with the endemic region (Öland) isolates (Aös in ). Among the Öland isolates, the three isolates from the southern part of the island formed a cluster ( and ). The isolates from Västergötland were highly similar to each other but distinct from all other isolates in the study. With the exception of the isolate from Östergötland mentioned above (Aös in ), the isolates from the three non-endemic regions were separated from each other and from the endemic isolates in the tree, with 99–100% bootstrap support indicating a reliable separation. The isolates from Västergötland were a minimum of 164 SNPs from the other Swedish isolates, while the minimum between-cluster distance was 10–22 SNPs for the other three clusters (). The reference genome CT_02021853 was highly dissimilar to all the Swedish isolates (). When isolates from the United States and the United Kingdom were included in the analysis, these formed clusters in accordance with the country of origin, and were not similar to any Swedish isolates (Supplementary File).
Fig. 2 Maximum likelihood tree based on SNP variation among the 28 S. Dublin isolates included in the study. Ten herds were located in the one endemic region in Sweden, Öland (purple), three herds in Östergötland (yellow), ten herds in Skåne (red), and three herds in Västergötland (green). From two of the herds in Skåne, there were two isolates from samplings performed 1 year apart, Osk-1, Osk-2 and Nsk-1, Nsk-2. Distance to the reference sequence is not shown to scale as indicated by broken lines. Percent bootstrap support for branches is shown as grey numbers, only shown for >85%. Minimum pair-wise SNP distances between the clusters are shown to the left of the tree, and the range of pair-wise within cluster distances for each cluster is shown on the right side (Aös was excluded from these calculations). Known contacts from epidemiological investigations are described in the right part of the figure. Isolates from herds with known epidemiological contacts were generally closely related.
Epidemiological contacts between the herds are illustrated in . In Västergötland , there had been animal movements between the herds; a dairy herd (Åvs) had delivered calves to two specialised fattening units (Yvs, Zvs). In Östergötland, the two herds (Bös, Cös) in the subgroup with similar isolates were geographically very close (2 km) with adjacent pastures. There was also a river to which animals from one of these herds had access and water from the river was used for cleaning of stables and irrigation of pastures in the other herd. The third herd in Östergötland (Aös) was geographically separated, almost 50 km away from the two other herds (). Further, this herd had no known epidemiological contacts with the other two herds in Östergötland or with any herds in the endemic region (Öland).
Fig. 3 Known contacts between herds from which isolates were analysed. Distances in this figure are proportional to the real distance between the herds. The contacts were revealed during the epidemiological investigations in the three outbreak regions. From one of the included herds (sk), there was no isolate available; however, animals from Xsk had been grazing at a pasture on this farm that was located adjacent to Qsk. There were no known contacts between the herds in the endemic region (Öland).
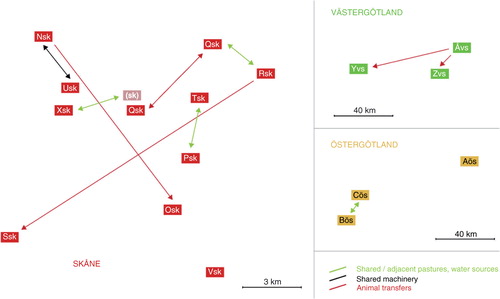
In Skåne, all herds were located geographically close to each other, within a range of 10 km. Two of the herds (Osk, Ssk) had purchased animals from two other herds (Nsk, Rsk) within the outbreak. Two herds had kept animals at adjacent pastures and the grazing animals were drinking from a common water course (Psk, Tsk). Small calves were able to move between the two pastures. Two herds had animals at the same pasture but during two consecutive grazing seasons (Qsk, Rsk). Two other herds had been sharing vehicles (Nsk, Usk); these vehicles were used for cleaning stables from manure and the same vehicles were also used for handling feed. One herd had no known contacts with the other herds, apart from geographical vicinity (Vsk).
The herds in the endemic region (Öland) had no known epidemiological contacts between them. Geographically the three herds in the southern part of the region were found to be colonised by a different WGS subgroup as compared with the herds located further north in the region ( and ). There were no known contacts between herds in the endemic region (Öland) and test-positive herds in any of the three non-endemic regions.
Discussion
In the present study, we compared S. Dublin isolates from four different regions, one region where the infection is endemic and three separate outbreaks, by SNP-typing based on WGS.
We found the variation between isolates from herds in the endemic region (Öland) to be comparatively small (0–26 SNPs), although substantially more variation was observed in this region compared with the variation observed within the studied outbreaks (). This suggests limited diversification of S. Dublin over time as the serotype has been present in this region for several decades (Citation7, Citation8). This is consistent with the very limited variation observed with PFGE in a previous Swedish study (unpublished observation). It also suggests that new subtypes have not been introduced into the region. The herds located in the southern part of the island formed a distinct SNP subgroup, supporting the notion of local spread of infection within this region. It is also in line with previous studies indicating that a local diffuse spread between herds exists (Citation9, Citation17–Citation19). The routes for this local spread have not been studied in detail and warrants further investigation as it is of importance in order to improve cost efficiency of control of S. Dublin (Citation20).
The isolates from Östergötland cluster with isolates from the endemic region (Öland). This suggests contacts between herds in these two regions that were not revealed during the epidemiological investigations; register data show that animal movements occur between these two regions. As S. Dublin has been shown to be able to persist in herds over long time periods Citation21–(Citation23), it cannot be excluded that epidemiological contact had occurred several years ago and was therefore not identified in the epidemiological investigations. Alternatively, the clustering of isolates between these regions could be due to a common original source of infection. However, the presumption of a common source for all isolates from Östergötland was not supported by WGS, as two distinct subgroups within the cluster were found.
The genetic distance between the isolates from herds within Västergötland and the isolates from herds in the endemic region is large (164 SNPs or more) and indicates that the cluster in Västergötland was not epidemiologically connected to the endemic region (Öland). The region Västergötland used to have more frequent occurrences of S. Dublin in cattle herds in the past, and it is possible that these subtypes of S. Dublin may have persisted at low levels in this region. As it is not known how fast SNP diversity develops, the infection in Västergötland could have originated from the endemic region (Öland) a long time ago. Alternatively, the herds in Västergötland might have been infected with a new subtype from a different, unknown source. Comparison with unrelated sets of isolates from the United Kingdom and the United States showed that the Västergötland isolates were no more similar to the other Swedish isolates than to isolates from other countries (Supplementary File), while all other Swedish isolates still formed a distinct cluster. This suggests the presence of distinct clones in the different countries, making identification of the source region of infections a possibility in the future when more reference genomic data are available.
The distance between isolates in Skåne and isolates from the endemic region (Öland) is smaller than the distance between isolates from Västergötland and the endemic region but still much larger than between the isolates within the endemic region. This suggests, either that the source of infection was not a subtype from the endemic region or that the introduction was a subtype from the endemic region that occurred many years ago. However, it cannot be excluded that subtypes similar to the isolates from Skåne also occur within the endemic region. Only a small number of isolates were analysed in this study, possibly failing to include the full genetic diversity in the endemic region. While the isolates from Skåne were similar to the endemic clone, WGS analysis could easily classify all isolates as Skåne/Öland with 100% bootstrap support and a minimum distance of 22 SNPs.
Within the outbreaks, there was good agreement between results from the epidemiological investigations and the WGS results. Herds with known epidemiological contacts generally showed smaller differences between isolates as compared with when there were no known epidemiological contacts ().
The results from Östergötland, with one isolate that was more closely related to isolates from the endemic region (Öland) than to the two other herds in the same region, show that WGS analyses also can be helpful in distinguishing herds that are not part of the same outbreak.
The quality of epidemiological information can vary considerably. One issue is the difficulties in assessing the time frame of infection with Salmonella in cattle herds. This means that detected contacts might falsely be considered to be the source of infection, and other contacts might be missed if they fall outside the time window examined. Also, the quality of collected information will depend heavily on the investigators understanding of potential routes of infection and the memory of involved persons. In these cases, all outbreak investigations were performed directly after detection of a positive herd by experienced veterinarians at the SBA and SVA.
In this study, only a limited number of isolates were analysed and therefore the results need to be interpreted with caution. Our understanding of the rate of SNP change in natural S. Dublin populations is limited, although the rate has been estimated at roughly one SNP per year genome wide in the closely related Enteritidis serotype (Citation24), and to 1.4 in S. Typhimurium DT104 (Citation25). However, the serotypes differ in their ecology (e.g. host range), and global evolution rates may not correspond well to those of smaller isolated populations. In addition, S. Dublin can survive extended periods in the environment, especially under cool conditions (Citation26), to later infect animals from a different farm or re-infect animals from the farm of origin, which will further complicate the relationship between time and SNP distance in smaller populations. Despite this, WGS analyses provided useful supplemental information to the epidemiological investigations that had been performed previously. However, to increase its usefulness, more data are needed on the expected change in diversity that is expected over time as well as the diversity in different geographical regions, also including other countries.
Conclusions
The results of this study show that WGS can provide valuable supplemental information to S. Dublin outbreak investigations. The resolution of WGS typing data was sufficient to distinguish isolates from the different outbreaks and provide additional information within outbreaks. Using WGS thus provides additional information that can improve epidemiological investigations concerning herds infected with S. Dublin. Future studies are needed to further improve the usefulness of WGS analysis of S. Dublin, in particular regarding genetic changes over time and diversity within regions.
Conflict of interest and funding
The Swedish Board of Agriculture financed the WGS analyses in this study. The authors declare no conflicts of interests.
Supplementary Material
Download PNG Image (29.3 KB){kind=link}
Acknowledgements
The authors would thank Lena Falkenäs, Sevinc Ferrari, and Heiður Loftsdóttir for excellent technical assistance with WGS analyses.
Notes
To access the supplementary material for this article, please see Supplementary files under ‘Article Tools’
References
- Carrique-Mas JJ, Willmington JA, Papadopoulou C, Watson EN, Davies RH. Salmonella infection in cattle in Great Britain, 2003 to 2008. Vet Rec. 2010; 167: 560–5. [PubMed Abstract].
- Richardson A, Watson WA. A contribution to the epidemiology of Salmonella Dublin infection in cattle. Br Vet J. 1971; 127: 173–83. [PubMed Abstract].
- Nielsen TD, Green LE, Kudahl AB, Ostergaard S, Nielsen LR. Evaluation of milk yield losses associated with Salmonella antibodies in bulk tank milk in bovine dairy herds. J Dairy Sci. 2012; 95: 4873–85. [PubMed Abstract].
- Nielsen TD, Kudahl AB, Ostergaard S, Nielsen LR. Gross margin losses due to Salmonella Dublin infection in Danish dairy cattle herds estimated by simulation modelling. Prev Vet Med. 2013; 111: 51–62. [PubMed Abstract].
- Jones TF, Ingram LA, Cieslak PR, Vugia DJ, Tobin-D'Angelo M, Hurd S, etal. Salmonellosis outcomes differ substantially by serotype. J Infect Dis. 2008; 198: 109–14. [PubMed Abstract].
- Sören K, Lindblad M, Jernberg C, Eriksson E, Melin L, Wahlström H, etal. Changes in the risk management of Salmonella enterica subspecies diarizonae serovar 61:(k):1, 5, (7) in Swedish sheep herds and sheep meat due to the results of a prevalence study 2012. Acta Vet Scand. 2015; 57: 6.
- National Veterinary Institute. Surveillance of infectious diseases in animals and humans in Sweden. 2014. SVA:s Report Series, No 26 ISSN 1654-7098. Uppsala 2015. Available from: http://www.sva.se/smittlage/zoonosrapporter [cited 10 June 2016].
- Lewerin SS, Skog L, Frössling J, Wahlström H. Geographical distribution of Salmonella infected pig, cattle and sheep herds in Sweden 1993–2010. Acta Vet Scand. 2011; 53: 51. [PubMed Abstract] [PubMed CentralFull Text].
- Ågren ECC, Sternberg Lewerin S, Wahlström H, Emanuelson U, Frössling J. Low prevalence of Salmonella in Swedish dairy herds highlight differences between serotypes. Prev Vet Med. 2016; 125: 38–45.
- Olsen JE, Skov M. Genomic lineage of Salmonella enterica serovar Dublin. Vet Microbiol. 1994; 40: 271–82. [PubMed Abstract].
- den Bakker HC, Allard MW, Bopp D, Brown EW, Fontana J, Iqbal Z, etal. Rapid whole-genome sequencing for surveillance of Salmonella enterica serovar enteritidis. Emerg Infect Dis. 2014; 20: 1306–14. [PubMed Abstract] [PubMed CentralFull Text].
- Chevreux B , Wetter T , Suhai S . Genome sequence assembly using trace signals and additional sequence information. 1999. Computer science and biology: Proceedings of the German conference on 430 bioinformatics (GCB) 99.
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, etal. Versatile and open software for comparing large genomes. Genome Biol. 2004; 5: R12. [PubMed Abstract] [PubMed CentralFull Text].
- Fricke WF, Mammel MK, McDermott PF, Tartera C, White DG, LeClerc JE, etal. Comparative genomics of 28 Salmonella enterica isolates: evidence for CRISPR-mediated adaptive sublineage evolution. J Bacteriol. 2011; 193: 3556–68. [PubMed Abstract] [PubMed CentralFull Text].
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, etal. BLAST plus: architecture and applications. BMC Bioinformatics. 2009; 10: 421. [PubMed Abstract] [PubMed CentralFull Text].
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011; 28: 2731–9. [PubMed Abstract] [PubMed CentralFull Text].
- Wedderkopp A, Stroger U, Lind P. Salmonella Dublin in Danish dairy herds: frequency of change to positive serological status in bulk tank milk ELISA in relation to serostatus of neighbouring farms. Acta Vet Scand. 2001; 42: 295–301. [PubMed Abstract] [PubMed CentralFull Text].
- Ersbøll AK, Nielsen LR. The range of influence between cattle herds is of importance for the local spread of Salmonella Dublin in Denmark. Prev Vet Med. 2008; 84: 277–90.
- Nielsen LR, Dohoo I. Survival analysis of factors affecting incidence risk of Salmonella Dublin in Danish dairy herds during a 7-year surveillance period. Prev Vet Med. 2012; 107: 160–9. [PubMed Abstract].
- Nielsen LR . Salmonella Dublin in cattle [Dr. med. vet. thesis]. 2013; University of Copenhagen.
- Boqvist S, Vågsholm I. Risk factors for hazard of release from Salmonella-control restriction on Swedish cattle farms from 1993 to 2002. Prev Vet Med. 71; 2005: 35–44.
- Nielsen LR, Kudahl AB, Ostergaard S. Age-structured dynamic, stochastic and mechanistic simulation model of Salmonella Dublin infection within dairy herds. Prev Vet Med. 2012; 105: 59–74. [PubMed Abstract].
- Nielsen LR, Nielsen SS. A structured approach to control of Salmonella Dublin in 10 Danish dairy herds based on risk scoring and test-and-manage procedures. Food Res Int. 2012; 45: 1158–65.
- Deng X, Desai PT, den Bakker HC, Mikoleit M, Tolar B, Trees E, etal. Genomic epidemiology of Salmonella enterica serotype enteritidis based on population structure of prevalent lineages. Emerg Infect Dis. 2014; 20: 1481–9. [PubMed Abstract] [PubMed CentralFull Text].
- Leekitcharoenphon P, Hendriksen RS, Le Hello S, Weill F-X, Baggesen DL, Jun S-R, etal. Global genomic Epidemiology of Salmonella enterica Serovar Typhimurium DT104. Appl Environ Microbiol. 2016; 82: 2516–26. [PubMed Abstract].
- Kirchner MJ, Liebana E, McLaren I, Clifton-Hadley FA, Wales AD, Davies RH. Comparison of the environmental survival characteristics of Salmonella Dublin and Salmonella Typhimurium. Vet Microbiol. 2012; 159: 509–14. [PubMed Abstract].