
Abstract
Extracellular vesicles (EV) are small membrane-bound vesicles enriched in a selective repertoire of mRNA, miRNA, proteins and cell surface receptors from parental cells and are actively involved in the transmission of inter and intracellular signals. Cancer cells produce EV that contain cargo including DNA, mRNA, miRNA and proteins that allow EV to create epigenetic changes in target cells both locally and systemically. Cancer-derived EV play critical roles in tumorigenesis, cancer cell migration, metastasis, evasion of host immune defense, chemoresistance, and they promote a premetastatic niche favourable to micrometastatic seeding. Their unique molecular profiles acquired from originator cells and their presence in numerous body fluids, including blood and urine, make them promising candidates as biomarkers for prostate, renal and bladder cancers. EV may ultimately serve as targets for therapy and as platforms for personalized medicine in urology. As urologic malignancy comprises 28% of new solid tumour diagnoses and 15% of cancer-related deaths, EV-related research is rapidly emerging and providing unique insights into disease progression. In this report, we review the current literature on EV in the setting of genitourinary fertility and malignancy.
Extracellular vesicles (EV) are small membrane-bound vesicles ranging in size from 40 to 1,000 nm and are released by most cell types. Enriched in a selective repertoire of mRNA, miRNA, proteins and cell surface receptors from parental cells, they are actively involved in conferring inter and intracellular signals. Cell-released vesicles are heterogeneous in size and composition. According to their biogenesis, non-apoptotic vesicles are classified as exosomes, originating from the membrane of the endosomal compartment, and microvesicles derived from direct cell surface “budding” (Citation1). Given the overlapping characteristics of exosomes and microvesicles, and their concomitant release from several cell types, the term EV has been suggested to include the different types of vesicles (Citation2) (). During EV formation, the membrane vesicle incorporates bioactive lipids and receptors as well as cytosolic proteins and nucleic acids characteristic of the originator cells (Citation4). EV may stay in the local microenvironment or enter biological fluids such as cerebrospinal fluid (CSF), plasma, milk and urine. In the past decade, there has been an explosion of the role of EV in inflammation, coagulation, infectious disease, immunology, stem cell renewal/expansion and cancer. Tumour-derived EV also possesses immunosuppressive properties and can facilitate tumour growth, metastasis and the development of drug resistance. Their unique molecular profiles acquired from parental cells and their presence in numerous body fluids, including blood and urine, make them promising candidates as successful biomarkers, targets for therapy, and may serve as platforms for personalized medicine.
Fig. 1. Extracellular vesicle (EV) origin: EV may originate from the endosomal compartment by exocytosis of vesicles formed within the multivesicular bodies or (a) from the cell surface by budding of plasma membrane. These shedding vesicles, sorted from the cell surface by budding of cell plasma membrane, are also named microvesicles. (b) Exocytic multivesicular bodies fuse with membrane after cell stimulation and release by exocytosis vesicles named exosomes. (b) These multivesicular bodies are created within the Golgi apparatus as a result of endosome compartmentalization. The insets are representative transmission electron microscopy of exosome generation from a multivesicular body and of vesicle generation by budding of plasma membrane (modified, in part, from Refs. (Citation3) and 7).
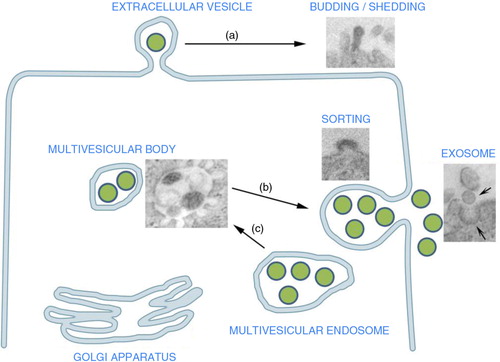
EV are key mediators of intercellular communication in both physiologic and pathophysiologic conditions (Citation1, Citation5) (Citation6). Originally thought to be “inert cellular debris,” EV facilitate cell-to-cell communication through direct stimulation of target cells via ligand binding, membrane receptor transfer, protein delivery and epigenetic reprogramming of target cells through mRNA, microRNA (miRNA), long non-coding RNA, or transcription factors (Citation7). Cancer cells also produce EV containing DNA, mRNA, miRNA and proteins that allow them to create epigenetic changes in target cells both locally and systemically. Cancer-derived EV play critical roles in tumorigenesis, cancer cell migration, metastasis, evasion of host immune defense and promote a premetastatic niche favourable to micrometastatic seeding.
Over the past decade, the study of EV in genitourinary (GU) malignancy has emerged and altered our understanding of complex microenvironments. The GU system consists of the urinary tract and the genital area covering the bladder, kidneys and prostate. Causes for GU disease can range from congenital anomalies and infections to trauma and cancer, which is the second leading cause of death in the United States. GU malignancy comprises 28% of new solid tumour diagnoses and 15% of cancer-related deaths (Citation8, Citation9). EV are particularly relevant in their role in the transfer of genetic material, as potential biomarkers for prostate, renal and bladder cancer, and may ultimately serve as therapeutic targets and as therapeutic vehicles. In this report, we review the current literature on EV in the setting of GU fertility and malignancy.
Prostasomes and fertility
In 1977, Ronquist first discovered extracellular membrane-bound microvesicles in prostatic fluid and seminal plasma (Citation10–Citation12). These EV were termed prostasomes and are similar to exosomes secreted by other cell types. The mean size of prostasomes is approximately 150 nm consistent with size characteristics of other EV reported in the literature and are present in multivesicular bodies of late endosomal origin. The protein composition of prostasomes secreted by prostatic acinar cells includes transport and structural proteins, GTP-binding proteins, signal transduction proteins, chaperone proteins and enzymes (Citation13). EV secreted into the seminal plasma by immobile prostatic acinar cells facilitate male fertility by promoting spermatozoa motility, protecting spermatozoa from female factor immune attack, supplying antioxidants, promoting capacitation and promoting the acrosome reaction (Citation14–Citation16) (Table ).
Table I. Extracellular vesicle mechanisms of action with respect to fertility
Three mechanisms of prostasome–spermatozoa communication have been postulated: direct contact between the EV and spermatozoa plasma membrane, fusion of membranes and internalization of the EV by the spermatozoa (Citation14, Citation17). Prostasomes likely exert a regulatory function on spermatozoa by creating portable microenvironments to facilitate sperm motility and hyperactivation resulting in vigorous beating of the sperm tail. Upon fusion, membrane receptors of prostasomes specifically localize to the sperm neck delivering progesterone receptors, cyclic adenosine diphosphoribose (cADPR) synthesizing enzymes, ryanodine receptors (RyR) and other calcium signalling tools. Progesterone-induced spermatozoa motility is reliant upon prostasomal transfer of cADPR-mediated calcium (Ca2 + ) mobilization via RyR (Citation15). EV also contain membrane attack complex (MAC) inhibitory protein CD59 and, therefore, allow evasion of sperm from female reproductive tract complement attack mediated cell lysis (Citation18). Furthermore, studies have also suggested that prostasomechromogranin B is bactericidal and that prostasome fusion with spermatozoa inhibits immune cell phagocytosis (Citation16, Citation17). Additionally, EV impart an antioxidant capacity to sperm, which prevents sperm damage from reactive oxygen species (ROS). This is of particular importance given sperm's intrinsic lack of repair mechanisms, and that ROS is proven to be a major cause of idiopathic male factor infertility (Table ).
Additional studies have shown that fusion of prostasomes with spermatozoa transiently decapitates after cholesterol transfer. This transient decapitation is thought to prevent untimely activation in the lower female reproductive tract (Citation17). The essential acrosome reaction, enabling a single sperm to penetrate and fertilize the ovum, also appears to be enhanced by prostasomal fusion with spermatozoa (Table ). Both progesterone and prostasome fusion independently stimulate the spermatozoa to undergo the acrosome reaction. However, prostasome-fused spermatozoa were activated at significantly lower levels of progesterone than non-fused spermatozoa (Citation14). It has also been shown that oviductal exosomes, along with uterosomes and vaginal exosomes, play an important role in post-testicular sperm acquisition of plasma membrane Ca2 + -ATPase 4a (PMCA4a), which is essential for hyperactivated motility and fertility (Citation19).
Prostate cancer
Prostate cancer is the most common solid malignancy affecting males in the United States with 186,000 new diagnoses and 28,000 deaths each year (Citation8). It affects the prostate gland that is responsible for the secretion of seminal fluid that nourishes and protects sperm. As previously discussed, prostasomes are EV created by the acinar cells of the male prostate gland. EV contribute to the pathogenesis of prostate cancer via immune evasion, enhancement of local tumour invasion and promotion of bone metastasis.
Prostasomes promote immune evasion by delivering complement inhibitory protein CD59 to normal and autologous cancer cells in addition to phosphorylation and inactivation of C3. EV created by prostate cancer cells have elevated quantities of protein kinase A, C and casein kinase. Protein kinase A phosphorylates complement C3 rendering it incapable of physiologic activation and subsequent formation of the MAC, which induces cell lysis. Babiker et al. demonstrated that human prostate cancer-derived EV and their unregulated protein kinase A inactivate the complement cascade, thus protecting prostate cancer cells from complement-mediated cell lysis (Citation20). Similarly, EV secreted by prostate cancer cells overexpress CD59 compared to benign prostate cells further protecting cells in the prostate cancer microenvironment from destruction (Citation21).
Beyond the protection provided from immune attack, EV from prostate cancer cells also appear to enhance local tumour invasion. Matrix degradation and fibroblast activation are central processes in local tumour invasion. Urokinase plasminogen activator (uPA) is associated with invasive potential in a number of tumour types and is present within EV derived from prostate cancer cells. In vitro studies of EV from PC-3 prostate cancer cell lines demonstrated elevated levels of uPA resulting in increased EV adherence to and degradation of collagen IV and basal membrane (). When EV from PC-3 cell lines were then added to the LnCaP (a poorly invasive prostate cell line), LnCaP cells acquired an enhanced ability to adhere and invade (Citation22).
Fig. 2. Tumour-derived EV and local invasion and metastasis: EV derived from primary tumour act to enhance matrix remodelling via a) matrix metalloproteinase (MMP) and urokinase-type plasminogen activator (uPA), enhance b) endothelial angiogenesis via vascular endothelial growth factor (VEGF) and c) EV released from PC3 cells activate fibroblasts sending antiapoptotic signals and growth signals (Citation23, Citation25). Ultimately, EV tumour release leads to downstream enhanced tumour cell migration, adhesion and invasion.
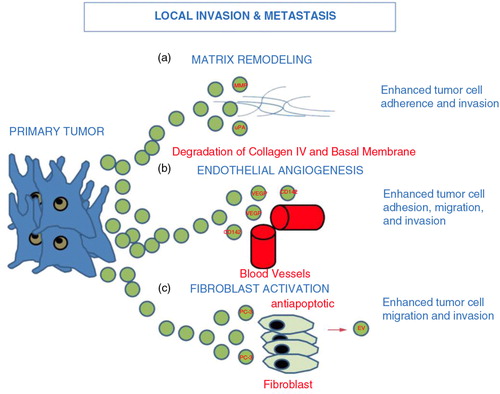
In 2009, Castellana et al. demonstrated a complex interaction between prostate cancer cells and fibroblasts creating a favourable prostatic tumour niche (Citation23). The authors were able to isolate matrix metalloproteinases (MMP) -9 and -14 from EV derived from the highly metastatic PC-3 prostate cancer cell line. These MMPs are matrix-degrading proteases critically involved in angiogenesis by allowing endothelial cells to migrate through basement membranes and form organized tubular structures that become new blood vessels.
Prostate tumour-derived EV can also regulate the immune response. EV derived from tumours have been shown to downregulate NKG2D-mediated cytotoxic response in PCa patients, thus promoting immune suppression and tumour escape (Citation24). Furthermore, EV derived from prostate cancer cells contain significantly higher levels of tissue factor CD142, which enhances cancer cell growth and proliferation via promotion of angiogenesis through vascular endothelial growth factor (VEGF) (Citation25). CD142 also alters cell adhesion, migration and tumour cell invasion properties (). Additionally, PC-3 EV were shown to promote MMP-9 expression and extracellular signal-regulated kinase (ERK1/2) phosphorylation in stimulated fibroblasts, which increased chemotherapy resistance. The ERK pathway is known to regulate a number of fundamental processes within cells including cellular differentiation, proliferation and survival. In turn, activated fibroblasts released EV that were able to promote migration and invasion of prostate cancer cells (Citation23) ().
Bone metastasis is common in advanced prostate cancer and generates tremendous morbidity in affected patients. Osteoblastic metastatic lesions are the result of a complex interplay between prostate cancer cells, osteoblasts and osteoclasts. Prostate cancer cells deliver osteoblastic factors including platelet-derived growth factor, endothelin-1 (ET1) and parathyroid hormone related protein, as well as osteolytic factors such as MMPs and VEGF. Bone marrow supports prostate cancer cells with growth factors such as transforming growth factor beta and insulin-like growth factors (Citation26, Citation27). Interestingly, PC-3 microvesicles stimulate osteoblastic differentiation, while those from LnCaP cell lines do not. Proteomic analysis of the EV from both cell lines demonstrated that PC-3 cell line EV contain erythroblast transformation specific transcription factor (Ets 1), also known as ETS-related gene (ERG) (Citation28). Renzulli et al. demonstrated that human prostate cancer-derived EV induced prostate cancer specific gene expression in human bone marrow cells (Citation29). Numerous genes including prostate-specific antigen (PSA) and transmembrane protease serine 2 (TMPRSS2) are expressed in human bone marrow cells following co-culture with prostate cancer cell EV.
Prostasomes as biomarkers
Much controversy currently exists regarding PSA screening, as it is not a cancer-specific biomarker. PSA is not a dichotomous biomarker; it reflects a continuum of risk for prostate cancer with no absolute value reflecting a negligible risk of malignancy. Its impact on mortality in screened populations remains a matter of intense debate. Two large prospective randomized trials investigated the impact of PSA screening on mortality in screened and unscreened populations. The Prostate, Lung, Colorectal, Ovarian (PLCO) trial in the United States found no reduction in mortality, while the European Randomized Study of Screening for Prostate Cancer found a 20% reduction in prostate cancer mortality in the screened group (Citation30–Citation32) Given the limitations of PSA, there is widespread interest in the search for screening tests with improved sensitivity and specificity for prostate cancer. A number of new biomarkers for prostate cancer are undergoing validation including prostate cancer antigen 3 (PCA3), proenzyme PSA (proPSA), TMPRSS2–ERG complex, alpha-methylacyl coenzyme A racemase, early PCA, human kallikrein 2, hespin, prostate stem cell antigen and glutathione S-transferase (GSTP1) (Citation33, Citation34).
During the transition from benign to neoplastic, the polarized columnar prostate cells become more cuboidal in architecture and lose their cellular polarity. The altered architecture of neoplastic prostate glands is hypothesized to account for the ability to measure serum levels of EV in men with prostate cancer. Tavoosidana et al. demonstrated that direct measurements of EV in blood plasma of men with prostate cancer were elevated compared to non-cancer controls (Citation35). Additionally, they demonstrated that prostatitis, benign prostatic hyperplasia and indolent or low-risk prostate cancer did not elevate blood plasma levels of EV. Total prostasome levels were statistically significantly higher in patients with intermediate (Gleason Grade 7 (GG7)) and high-risk disease (GG8 and 9) compared to controls but not in low-risk (GG6) disease compared to controls. In contrast, PSA levels were statistically significantly higher in high-risk disease (GG8 and 9) compared to low- and intermediate-risk disease. This is especially important given the ability of benign prostatic processes to elevate serum PSA values limiting its specificity as a marker for malignancy. Additionally, given the controversy surrounding the clinical relevance of low-risk prostate cancer, EV may provide a novel ability to differentially screen specifically for clinically significant prostate cancer. This “proof of concept study” looked only at total serum prostasome concentration as detected utilizing a novel multiple recognition assay. Modifications to the multiple recognition assay may allow detection of markers on prostasomes that denote prognostic significance in the future.
In 2012, Sandvig identified a number of new candidate prostate cancer biomarkers using PC-3 cell microvesicles (Citation36). The membrane glycoprotein CUB domain-containing protein 1 (CDCP1) was identified within the EV using proteomics. CDCP1 appears to be an anti-apoptotic factor that facilitates tumour cell survival during metastasis (Citation37). Interestingly, Siva et al. demonstrated that a monoclonal antibody against CDCP1 inhibited metastasis of prostate cancer (Citation38). When compared to benign prostate (RWPE-1) and non-metastatic prostate cancer (LnCaP) EV, the metastatic PC-3 cell line EV contained significantly higher levels of CDCP1 making it an attractive candidate for assessment of prostate cancer metastasis (Citation36). Furthermore, EV from patient's plasma have been shown to contain prostate cancer specific proteins such as survivin and phosphatase and tensin homolog (PTEN). Enhanced EV levels of survivin have been detected in prostate cancer patients compared with healthy subjects or patients with benign prostate hypertrophy, while EV containing PTEN have been detected only in patients with prostate cancer (Citation39, Citation40). Another candidate biomarker identified in PC-3 EV is tetraspanin CD151. This protein is upregulated in a number of cancers and is hypothesized to induce tumorigenesis via associations with MMP and integrins. While CD151 is expressed in EV of normal prostate cell lines, it is significantly overexpressed in the PC-3 EV. In fact, CD151 protein expression was found to be a better predictor of clinical outcomes in low-grade prostate cancer than histologic grade (Citation41). As a result, CD151 may serve as a prognostic factor for prostate cancer progression in the future.
There has also been considerable research performed on the use of miRNAs as potential biomarkers for PCa. Plasma EV miRNAs may be utilized for prognosis in castration-resistant prostate cancer and decreased miR-34a levels showed substantial clinical relevance with prostate cancer progression and poor prognosis and response to docletaxel (Citation42, Citation43). Bryant et al. have shown a differential expression of 12 EV-associated miRNAs in plasma and serum of patients with prostate cancer. In particular, miR-375 and miR-141 were significantly increased in EV isolated from metastatic patients in comparison with patients without metastasis (Citation44). Furthermore, miR141 has been suggested to discriminate prostate cancer patients from healthy subjects and to correlate to Gleason score and tumour progression (Citation45, Citation46).
Renal disease
EV in renal physiology and disease
The renal system consists of the kidneys, bladder and urethra and is responsible for the elimination of waste, regulation of blood volume and pressure, and regulation of pH. EV may serve as biomarkers of acute kidney injury (AKI), ischemia reperfusion injury, membranous glomerulonephritis and transplant rejection. Fetuin-A, present in urine exosomes, is a potential biomarker for renal injury models including cisplatin-induced nephrotoxicity and AKI in ICU patients. Fetuin-A levels were elevated up to 50-fold in these clinical scenarios compared to controls. In addition, urine EV with elevated levels of Fetuin-A were detected prior to changes in serum creatinine in patients with eventual AKI (Citation47). Therefore, the presence of specific EV in the urine may serve as an early marker for impending AKI. Furthermore, activating transcription factor 3 (ATF3) was elevated in urine EV after AKI in contrast to patients with CKD or in normal control patients (Citation48). Similarly, aquaporin-1 levels are decreased in urine EV following ischaemia reperfusion injury. In another study, 8 of 9 patients with focal glomerular sclerosis had elevated levels of Wilms tumour 1 (WT1) in their urine EV compared to 0 of 9 controls suggesting a role for EV in diagnosing podocyte effacement. Finally, mi-R-210 is reduced in EV in acute T-cell-mediated rejection in renal allograft patients (Citation49).
EVs derived from human mesenchymal stem cells (MSCs) have also been shown to accelerate recovery following AKI in vivo. Furthermore, in mice with glycerol-induced AKI, labelled MSC EV accumulated specifically in the kidneys at the damaged site of the mice with AKI compared with the healthy controls. This provides the basis for the examination and detection of AKI using EV derived from MSCs (Citation50). With further elucidation of these findings, it may be possible to reduce the need for renal biopsy to detect changes at the glomerular level (Citation7).
Renal cell cancer
The kidneys are a part of the urinary system that is responsible for the production of urine and the regulation of electrolytes, blood pressure and pH. Kidney cancer is a common urologic malignancy accounting for 3% of adult cancers and 90,000 deaths worldwide each year (Citation51). Clear cell renal cell carcinoma (ccRCC) is the most common subtype accounting for 70–80% of all renal cell carcinoma (RCC). The pathophysiology of ccRCC has been extensively studied and is linked to an altered regulation of hypoxia inducible factor. In variants of chromophobe RCC, and eosinophilic variants of ccRCC, all of the tumours display abundant mitochondria with EV present in the outpouchings (Citation52). These results suggest a close relationship between the EV and mitochondria, and indicate that defective mitochondriogenesis may be the source of EV in chromophobe RCCs that lead to disease progression.
Many studies have shown the angiogenic potential of EV released from tumour cells. For example, studies of the cells of origin in RCC have shown that EV released from human RCC stem cell populations both stimulated locoregional angiogenesis and created a favourable microenvironment for lung metastasis. EV released by a CD105+ cancer stem cell population stimulated angiogenesis and contained a different array of mRNAs than their CD105− counterparts. This renal cancer stem cell population released EV-containing mRNAs of genes coding for growth factors such as VEGF, fibroblast growth factor 2 (FGF2), angiopoietin1 and ephrin A3. Additionally, these EV contained mRNAs of genes coding for MMP including MMP2 and MMP9 (Citation53).
In vitro studies using human umbilical vein endothelial cells (HUVEC) demonstrated capillary-like structure production in cells treated with EV from CD105+ renal cell cancer stem cells. Furthermore, the EV from CD105+ renal cell cancer stem cells imparted greater resistance to apoptosis and increased endothelial cell invasion and tumour cell adhesion in pretreated HUVEC cells. In vivo studies using SCID mice injected with EV-stimulated HUVECs showed formation of dense clusters that organized into capillaries communicating with the murine vascular supply. A separate experiment injected SCID mice with EV from CD105+ RCC stem cells. These mice were then injected with renal tumour cells and organs were examined after 5 weeks. Metastasis was only found in lung tissues of the CD105+ mice, confirming that CD105+ EV were significantly more efficient in inducing metastasis than EV from unsorted cells. Lung VEGFR1, MMP9, VEGF and MMP2 expression were also enhanced by CD105+ EV (Citation54).
Urinary EV may also serve as a diagnostic tool for RCC. Del Boccio et al. have performed a comparative analysis on a hyphenated microLC-Q-TOF-MS platform of urinary EV showing differential composition of lipids in RCC-derived EV (Citation54). Moreover, a differential protein profile has been described in urinary EV from RCC patients suggesting the expression of a specific protein pattern that may be exploited for diagnosis (Citation55).
Bladder cancer
The bladder is a part of the urinary system that is responsible for the storage of urine produced by the kidneys. In the United States, transitional cell carcinoma (TCC) of the bladder is the 4th and 11th most common malignancy diagnosed in males and females, respectively (Citation8). Diagnosis is typically based on urinalysis, urine cytology, cystoscopy and upper urinary tract imaging consisting of retrograde pyelograms or computerized tomography. Urine cytology is expensive, lacks sensitivity in detecting low-grade tumours (4–31% with the median of 12%) and requires trained cytopathologists (Citation56). Cystoscopy, the gold standard for diagnosis, is invasive, expensive and may be inconclusive or falsely negative secondary to operator error, inexperience, or grossly abnormal bladder mucosa as a result of infection or chronically indwelling catheters. Currently, there are 3 types of urine markers: protein, cellular and genetic. The protein markers include bladder tumour antigen (BTA) stat and nuclear mitotic apparatus protein 22 (NMP22). BTA stat detects human complement factor H related protein in urine produced from bladder cancer cells. It is hypothesized that human complement factor H production by tumour cells helps in evasion of immune system mediated cell lysis. NMP22 is a NMP that is upregulated in bladder cancer cells. It is released into the urine via apoptosis. ImmunoCyt is the only cellular marker available and combines cytology and immunocytochemistry to detect bladder cancer in exfoliated urothelial cells via 3 fluorescent monoclonal antibodies to carcinoembryonic antigen and bladder tumour mucins. Finally, UroVysion is a genetic test that uses multitargeted fluorescence in situ hybridization to detect polysomy 3, 7 and 17 and loss of the 9p21 locus. New investigational biomarkers include the urinary UBC test to detect cytokeratin 8 and 18, bladder urothelial carcinoma (BLCA1/BLCA4), hyaluronic acid, hyaluronidase, Lewis X antigen, soluble Fas, survivin, human telomerase reverse transcriptase polymerase and aurora kinase A (Citation54). There is tremendous work going into identification of biomarkers for early cancer detection, screening for tumour recurrence and prognostic markers.
Smalley et al. identified 8 proteins as potential biomarkers for bladder cancer in a comparison of EV in urine from patients with bladder cancer and control patients (Citation57). Most of the proteins identified are either directly or indirectly associated with the plasma membrane and serve as key signal transduction molecules in regulating cell growth, differentiation, survival and apoptosis. Five of these proteins are associated with the epidermal growth factor receptor (EGFR) pathway, which is deregulated in bladder cancer carcinogenesis. These proteins include NRas, whose mutations have been identified in bladder cancer cells, EGFR kinase substrate 8 like protein 1 and 2 (EPS8L1 and EPS8L2), Mucin 4 and EH domain-containing protein 4 (EDH4). EPS8L1 and 2 are structurally similar to EPS8, which acts as a substrate for the EGFR. It is localized to the lamellipodia regulating generation of filopodia and EV formation. Mucin 4, a transmembrane glycoprotein, serves as a ligand to ErbB2, human epidermal growth factor receptor (HER2) and Neu, which are all members of the EGFR family. Once bound, Mucin 4 auto-phosphorylation occurs, activating cellular proliferation processes and possibly contributing to EV formation. EDH4 participates in the regulation of plasma membrane receptor and endocytic recycling compartment receptor. The 3 other proteins identified include the alpha subunit of GsGTP binding protein, retinoic acid inducible protein 3 [both associated with G-protein-coupled receptor (GPCR)] and resistin (Citation57). Welton et al. (on the basis of gene ontology analysis) extended the proteomic analysis and showed a strong association between EV proteome and bladder carcinoma (Citation58).
Most recently, Chen et al. compared the EV proteome of bladder cancer patients to age-matched hernia patients (Citation59). They found that 107 differentially expressed proteins were initially identified as potential biomarkers, 29 of which were precisely quantified from urine samples of bladder cancer, hernia control and patients with urinary tract infections (UTIs) or hematuria. Apolipoprotein A1 (APOA1), CD5L, fraction of genome altered, fibrinogen β chain precursor, fibroblast growth factor receptor 3, N-(4-hydroxyphenyl) retinamide and Helicobacter pylori protein (HP) exhibited statistically significant differences in concentration between patients with high-grade and low-grade bladder cancer, thus serving as potential grade discriminators. However, it should be noted that all of these proteins are plasma-associated microparticle proteins; therefore, blood-derived particles may account for their presence. The authors postulate that the increased levels of these 7 proteins in the high-grade patients were the result of increased numbers of blood-derived microparticles in the urine. Tumour-associated calcium signal transducer 2 (TACSTD2) was expressed at a 6.5-fold higher level in bladder cancer patients compared to hernia, UTI, or hematuria patients. While TACSTD2 is a cell-surface glycoprotein with little to no expression in normal tissues, it is overexpressed in a number of carcinomas including gastric, oral, pancreatic, colorectal and ovarian cancers. A recent study demonstrated that the differential methylation status of TACSTD2 was able to discriminate prostate cancer (methylated) from prostatic intraepithelial neoplasia (Citation60, Citation61). Given the challenge of large-scale ultracentrifugation to purify EV, Chen's group sought another method as a proof of this concept. Using a commercially available TACSTD2 ELISA kit and unprocessed urine, TACSTD2 was able to discriminate between low- and high-grade bladder cancer. They found, using different cut-off values, that they could differentiate all bladder cancer groups from hernia controls with a sensitivity, specificity, positive predictive value and negative predictive value of 73.6, 76.5, 84.4 and 62.6%, respectively. While more validation is clearly needed, TACSTD2 is certainly a potential biomarker for bladder cancer (Citation59).
Recently, Perez et al. generated a list of genes differentially expressed in bladder cancer versus control urinary EV by microarray technology, followed by polymerase chain reaction validation. They found expression of genes involved in cancer progression and metastasis such as LASS2 and GALT1 in cancer patient urinary EV and the unique presence of ARHGEF39 and FOXO3 genes in healthy controls. Since several other miRNAs detectable in the urinary pellets (miR-1224-3p, miR-135b, miR-15b and miR-126/miR-152 ratio) correlate with bladder cancer diagnosis and/or prognosis, their detection in urinary EV may provide diagnostic information (Citation62).
EV derived from the urine of patients with bladder cancer also contain bioactive molecules such as EGF-like repeats and discoidin I-like domains 3 (EDIL-3). EDIL-3 activates EGFR signalling and promotes angiogenesis and migration of bladder cancer cells and endothelial cells (Citation63). Bladder cancer cell-derived EV can inhibit tumour cell apoptosis, which is associated with the activation of protein kinase B (Akt) and extracellular signal-regulated kinases pathway genes, suggesting that tumour-derived EV are involved in bladder cancer progression. Therefore, inhibition of EV formation and release may be a novel strategy in future treatment of bladder cancer (Citation64). Conversely, EV-secreted miRNAs have been shown to inhibit bladder cancer progression, miR23b-inhibited invasion, anoikis, angiogenesis and pulmonary metastasis (Citation65).
EV purification methods
Due to the growing influence of EV, it is necessary to analyse EV isolation methods. The classic method for isolating vesicles excludes larger microvesicles from the extracted vesicle population. According to the classic method, samples are initially centrifuged at 300g for 10 minutes to remove cells, repeated at 10,000g for 30 minutes to remove larger vesicles (microvesicles) and then followed by centrifugation at 100,000g for 1 hour to isolate exosomes (Quesenberry Laboratory, unpublished results). On the other hand, it was necessary for Zhou's group to centrifuge at 17,000g for 15 minutes in order to remove urinary sediment, and then at 200,000g for 1 hour in order to isolate exosomes (Citation47). Meanwhile, in Grange et al., EV purified by differential ultracentrifugation were characterized by electron microscopy, FACS and microarray analysis (Citation53).
The discrepancies in isolation techniques in spin number, direct or indirect exosome isolation, and lack of standardization can lead to various outcomes in vesicle experiments. Welton et al. published a study on purification methods for isolating EV from the bladder cancer cell line HT1376 that originated from a high-grade T2 transitional cell cancer of the bladder identifying 353 proteins from EV (Citation58). The majority of proteins were involved in exosome biosynthesis. Among these were the endosomal sorting complex required for transport (ESCRT) proteins, numerous proteins involved in membrane trafficking and fusion processes, markers of endosomes and lysosomes and several proteins with chaperone functions. However, when compared to the data from Smalley et al., there was only 7.5% protein overlap. Welton hypothesized that the difference is due to source material and sample preparation approaches used by the 2 groups (Citation58). Due to the relative novelty of the field, there is a need of standardization in vesicle isolation methods.
EV as targeted pharmacotherapy and personalized drug delivery vehicles
In 2010, Renzulli et al. provided 2 main strategies for modifying effects of EV on effector cells: chemical blockade preventing EV from leaving cancer cells and antibody blockade of EV already released by cancer cells preventing interaction with effector cells (Citation29). As previously discussed, prostate cancer tumour EV create pro-invasive microenvironments in the local tumour site facilitating angiogenesis, matrix degradation via MMPs and uPA, cell adhesion and fibroblast activation (). Prostate cancer EV are also likely involved in the vicious cycle of bone metastasis via direct interaction with osteoblasts and osteoclasts. Renal cancer EV, on the other hand, promote endothelial cell invasion, confer resistance to apoptosis, enhance tumour cell adhesion and promote angiogenesis in vitro and in vivo at a local tumour level. Additionally, Grange's experiment using CD105+ renal cancer stem cell EV demonstrated ability to enhance angiogenesis, increase invasion of matrix, increase cellular adhesion, decrease apoptosis and create a premetastatic niche for tumour development in lung tissue (Citation53). Blocking the release of these EV could have a tremendous impact on local and systemic tumour biology and alter the natural history of disease ().
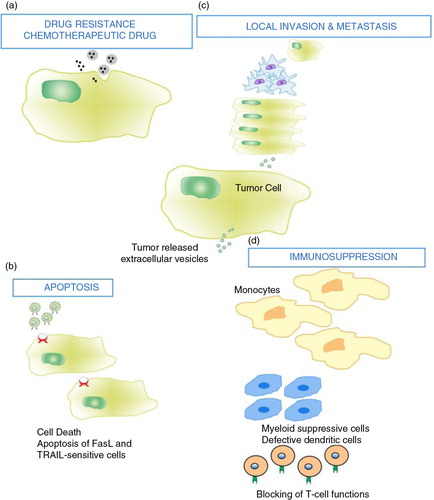
Fig. 3. Tumour cells release extracellular vesicles that can influence the malignant phenotype. Various examples include and are not limited to: (a) drug resistance; EV can influence the efflux of chemotherapeutic drugs via various mechanisms including ATPase and drug transporters. (b) apoptosis; through FasL and TRAIL, EV can induce apoptosis in activated antitumor T cells, abrogating T-cell-mediated apoptosis of tumour cells. (c) Local invasion and metastasis; EV can promote local invasion by activating fibroblasts and reducing fibroblast apoptosis, enhancing extracellular matrix degradation (mRNAs for MMP2 and MMP9), promoting angiogenesis (mRNA VEGF, FGF2, angiopoietin1) and increasing tumour cell adhesion. EV may enhance metastasis by promoting a pre-metastatic niche in lung tissue via upregulation of VEGFR1 expression, MMP2 in lung blood vessels and MMP9 in alveolar epithelial cells and blood vessels. (d) Immunosuppression; EV can alter monocyte differentiation into myeloid suppressive cells. This inhibits T-cell proliferation. Inhibiting T-cell responses upstream would abrogate antitumor immune potential. This figure is modified from (Citation66).
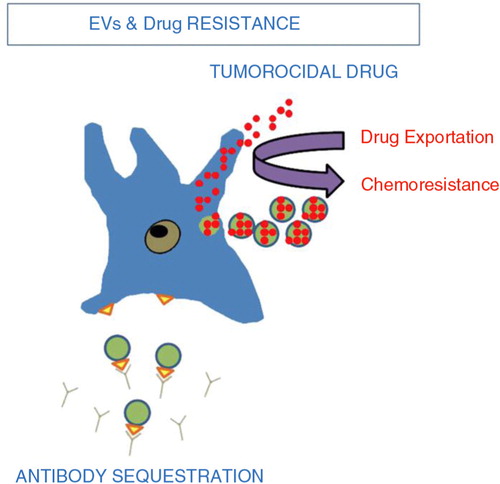
Blocking EV release from tumour cells can also battle drug resistance. To decrease the therapeutic action of chemotherapy agents, tumour cells shuttle drugs into shedding vesicles (Citation67) (). For example, PC-3 cells selectively package and secrete doxorubicin resulting in drug resistance (Citation68). Preventing efflux of these drugs from tumour cells would make these cells more susceptible to chemotherapy. Decreasing systemic doses needed to kill a given tumour cell reduces side effects that typically limit chemotherapy dosing.
Fig. 4. Tumour extracellular vesicles and chemoresistance (Citation67). Tumour EV act in 2 ways to reduce the efficacy of chemotherapy. (Citation1) intracytoplasmic chemotherapy exportation via shedding vesicles. (Citation2) Antibody sequestration.
Panagopoulos et al. demonstrated that EV isolated from camptothecin-resistant prostate cancer cells cause DU145 prostate cancer cells to become camptothecin resistant. Interestingly, non-malignant prostate epithelial cell EV can alter DU145 cell phenotype making them less “adherent” and vice versa. Significantly, this study has demonstrated that GG8 patient biopsied tumour EV can shift non-malignant prostate epithelial cells to a malignant phenotype. Proteins responsible for this shift have also been identified such as 14-3-3 zeta, prohibitin and Raf kinase inhibitor protein (Citation69).
Antibodies directed against cancer-specific cell receptors are a commonly used strategy in oncology. For example, RCCs are treated with drugs such as sunitinib and sorafenib that utilize antibodies directed against oncogenic receptors (Citation70, Citation71). These drugs block signal transduction at these receptor pathways. On the other hand, EV are known to contain a subset of the parental cancer cell membrane receptors and thus may act to neutralize antibody-based cancer therapies by binding the drug and acting as a “sink.” One classic example is breast cancer patients overexpressing HER2 treated with transtuzamab. These breast cancer tumour cells release EV with intact HER2 receptor and have been shown to reduce the efficacy of transtuzamab in vitro and in vivo. Thus, by inhibiting the release of EV with intact receptors that can act to neutralize antibody-based therapy, these treatments may become more efficacious.
Two viable strategies to combat this issue block EV in circulation from fusion, endocytosis and cell surface receptor signalling with target cells. The first method involves an antibody directed against EV-specific cell surface receptors that interact with effector cells to prevent membrane fusion, endocytosis, or receptor activation. The second method involves the removal of immunosuppressive, pro-infiltrative and pro-metastatic EV from circulation via a hollow-fibre cartridge (Hemopurifier), which is currently used to reduce HIV particle circulation in HIV patients. While transient in nature, this may be a viable option for malignancies responsible for high EV concentrations in the blood (Table ).
Table II. Proposed therapeutic strategies to mitigate extracellular vesicle effects in carcinogenesis
Although there have been studies in prostate cancer and other tumour models describing the characterization of proteins contained in EV derived from human cancer cell lines, there has not been an analysis performed of EV derived from patient tissue (Citation37). It is imperative to evaluate the content and capacity of tissue-derived EV to alter the genetic phenotype of recipient cells. This will provide a more relevant strategy for therapeutic intervention.
Overall, EV possess 5 characteristics making them ideal drug delivery vehicles for treating prostate, renal and bladder cancer. They are cell type specific; they exhibit predictable endocytosis or fusion with effector cells; they are lipophilic and cross membranes, including the blood–brain barrier; they are not filtered by the glomerulus and thus remain in circulation longer than most drug delivery molecules; they carry diverse array of molecules including cell surface receptors, cell surface proteins, cytosolic proteins, mRNA, miRNA and long non-coding RNA (Table ).
Table III. Extracellular vesicle characteristics that are favourable as a vehicle of therapy
Conclusions
EV are emerging as key players in both normal physiology and pathophysiology. They play essential roles in enhancing local tumour invasion and promoting metastatic disease. Given that EV share crucial membrane, protein and nucleic acid properties with their parental cells, are released in significant quantities by tumour cells, and are present in numerous body fluids including blood, plasma, CSF and urine, they may serve as ideal biomarkers for a wide range of urological diseases. Considering the growing role of EV, it is necessary to create a standardization process concerning EV isolation. Some mechanisms for prostate cancer development and management such as targeting androgen receptor signalling with drugs, that is abiraterone acetate and enzalutamide (MDV3100), have been discovered (Citation72, Citation73). Unfortunately in most patients, progression of this disease still occurs, as tumour cells become resistant to currently available therapies. Therefore, the on-going challenge is to identify rational targets to improve the therapeutic efficacy of treatment regimens. For example, a recent study demonstrated that EV released from mesenchymal stromal cells can be primed with paclitaxel and delivered in vivo to inhibit tumour growth (Citation74). New insights into the function and regulation of EV indicate cellular-based mechanisms of disease progression with the potential to translate this knowledge into innovative approaches for cancer diagnostics and personalized therapy (Citation75). There is also a need to begin an evaluation of the significance and mechanism of EV-mediated genetic transfer therapeutic options for blocking or manipulating this transfer in order to influence phenotype switching and disease progression. Although there have been studies in prostate cancer and other tumour models describing the characterization of proteins contained in EV derived from human cancer cell lines, there has not been an analysis of EV derived from patient tissue (Citation37). It is imperative to evaluate the content and capacity of tissue-derived EV to alter the genetic phenotype of recipient cells to provide a more relevant strategy for therapeutic intervention. As our understanding of their roles in tumour biology in vivo evolves, EV will become clear targets of drug therapy and serve as drug delivery vehicles.
Conflict of interest and funding
The authors declare no conflict of interest. This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number P20GM103421. The previous segment of this project was supported by the National Center for Research Resources (NCRR) under P20RR017695 (DC) and P20GM103468 (PQ).
Acknowledgements
The authors thank Kate Brilliant and Virginia Hovanessian for figure and manuscript preparation.
References
- Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009; 19: 43–51. [PubMed Abstract].
- Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. 2014; 3: 26913. doi: http://dx.doi.org/10.3402/jev.v3.26913.
- Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009; 9: 581–93. [PubMed Abstract].
- El Andaloussi S, Mäger I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013; 12: 347–57. [PubMed Abstract].
- Quesenberry PJ, Aliotta JM. The paradoxical dynamism of marrow stem cells: considerations of stem cells, niches, and microvesicles. Stem Cell Rev. 2008; 4: 137–47. [PubMed Abstract].
- Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia. 2006; 20: 1487–95. [PubMed Abstract].
- Camussi G, Deregibus MC, Bruno S, Cantaluppi V, Biancone L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010; 78: 838–48. [PubMed Abstract].
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T et al. Cancer statistics, 2008. CA Cancer J Clin. 2008; 58: 71–96. [PubMed Abstract].
- Horwich A, Huddart R. Retroperitoneal lymph-node dissection after chemotherapy for germ cell cancer in patients with elevated tumor markers. Nat Clin Pract Urol. 2006; 3: 250–1. [PubMed Abstract].
- Ronquist G, Hedstrom M. Restoration of detergent-inactivated adenosine triphosphatase activity of human prostatic fluid with concanavalin A. Biochim Biophys Acta. 1977; 483: 483–6. [PubMed Abstract].
- Ronquist G, Brody I, Gottfries A, Stegmayr B. An Mg2+ and Ca2 + -stimulated adenosine triphosphatase in human prostatic fluid: part I. Andrologia. 1978; 10: 261–72. [PubMed Abstract].
- Ronquist G, Brody I, Gottfries A, Stegmayr B. An Mg2+ and Ca2 + -stimulated adenosine triphosphatase in human prostatic fluid: part II. Andrologia. 1978; 10: 427–33. [PubMed Abstract].
- Ronquist G, Brody I. The prostasome: its secretion and function in man. Biochem Biophys Acta. 1985; 822: 203–18. [PubMed Abstract].
- Arienti G, Carlini E, Saccardi C, Palmerini CA. Role of human prostasomes in the activation of spermatozoa. J Cell Mol Med. 2004; 8: 77–84. [PubMed Abstract].
- Park KH, Kim BJ, Kang J, Nam TS, Lim JM, Kim HT et al. Ca2+ signaling tools acquired from prostasomes are required for progesterone-induced sperm motility. Sci Signal. 2011; 4: ra31. [PubMed Abstract].
- Skibinski G, Kelly RW, Harkiss D, James K. Immunosuppression by human seminal plasma – extracellular organelles (prostasomes) modulate activity of phagocytic cells. Am J Reprod Immunol. 1992; 28: 97–103. [PubMed Abstract].
- Ronquist G. Prostasomes are mediators of intercellular communication: from basic research to clinical implications. J Intern Med. 2012; 271: 400–13. [PubMed Abstract].
- Rooney IA, Atkinson JP, Krul ES, Schonfeld G, Polakoski K, Saffitz JE et al. Physiologic relevance of the membrane attack complex inhibitory protein CD59 in human seminal plasma: CD59 is present on extracellular organelles (prostasomes), binds cell membranes, and inhibits complement-mediated lysis. J Exp Med. 1993; 177: 1409–20. [PubMed Abstract].
- Al-Dossary AA, Strehler EE, Martin-Deleon PA. Expression and secretion of plasma membrane Ca2 + -ATPase 4a (PMCA4a) during murine estrus: association with oviductalexosomes and uptake in sperm. PLoS One. 2013; 8: e80181. [PubMed Abstract] [PubMed CentralFull Text].
- Babiker AA, Ronquist G, Nilsson B, Ekdahl KN. Overexpression of ecto-protein kinases in prostasomes of metastatic cell origin. Prostate. 2006; 66: 675–86. [PubMed Abstract].
- Babiker AA, Nilsson B, Ronquist G, Carlsson L, Ekdahl KN. Transfer of functional prostasomal CD59 of metastatic prostatic cancer cell origin protects cells against complement attack. Prostate. 2005; 62: 105–14. [PubMed Abstract].
- Angelucci A, D'Ascenzo S, Festuccia C, Gravina GL, Bologna M, Dolo V et al. Vesicle-associated urokinase plasminogen activator promotes invasion in prostate cancer cell lines. Clin Exp Metastasis. 2000; 18: 163–70. [PubMed Abstract].
- Castellana D, Zobairi F, Martinez MC, Panaro MA, Mitolo V, Freyssinet JM et al. Membrane microvesicles as actors in the establishment of a favorable prostatic tumoral niche: a role for activated fibroblasts and CX3CL1-CX3CR1 axis. Cancer Res. 2009; 69: 785–93. [PubMed Abstract].
- Lundholm M, Schröder M, Nagaeva O, Baranov V, Widmark A, Mincheva-Nilsson L et al. Prostate tumor-derived exosomes down-regulate NKG2D expression on natural killer cells and CD8+ T cells: mechanism of immune evasion. PLoS One. 2014; 9: e108925. [PubMed Abstract] [PubMed CentralFull Text].
- Abe K, Shoji M, Chen J, Bierhaus A, Danave I, Micko C et al. Regulation of vascular endothelial growth factor production and angiogenesis by the cytoplasmic tail of tissue factor. Proc Natl Acad Sci USA. 1999; 96: 8663–8. [PubMed Abstract] [PubMed CentralFull Text].
- Casimiro S, Guise TA, Chirgwin J. The critical role of the bone microenvironment in cancer metastases. Mol Cell Endocrinol. 2009; 310: 71–81. [PubMed Abstract].
- Clarke NW, Hart CA, Brown MD. Molecular mechanisms of metastasis in prostate cancer. Asian J Androl. 2009; 11: 57–67. [PubMed Abstract] [PubMed CentralFull Text].
- Itoh T, Ito Y, Ohtsuki Y, Ando M, Tsukamasa Y, Yamada N et al. Microvesicles released from hormone-refractory prostate cancer cells facilitate mouse pre-osteoblast differentiation. J Mol Histol. 2012; 43: 509–15. [PubMed Abstract] [PubMed CentralFull Text].
- Renzulli JF 2nd, Del Tatto M, Dooner G, Aliotta J, Goldstein L, Dooner M et al. Microvesicle induction of prostate specific gene expression in normal human bone marrow cells. J Urol. 2010; 184: 2165–71. [PubMed Abstract].
- Andriole GL, Crawford ED, Grubb RL 3rd, Buys SS, Chia D, Church TR et al. Prostate cancer screening in the randomized prostate, lung, colorectal, and ovarian cancer screening trial: mortality results after 13 years of follow-up. J Natl Cancer Inst. 2012; 104: 125–32. [PubMed Abstract] [PubMed CentralFull Text].
- Carlsson SV, Holmberg E, Moss SM, Roobol MJ, Schroder FH, Tammela TL et al. No excess mortality after prostate biopsy: results from the European Randomized Study of Screening for Prostate Cancer. BJU Int. 2011; 107: 1912–17. [PubMed Abstract].
- Ilic D, Neuberger MM, Djulbegovic M, Dahm P. Screening for prostate cancer. Cochrane Database Syst Rev. 2013; 1: CD004720. [PubMed Abstract].
- Ercole B, Parekh DJ. Methods to predict and lower the risk of prostate cancer. Sci World J. 2011; 11: 742–8.
- Reed AB, Parekh DJ. Biomarkers for prostate cancer detection. Expert Rev Anticancer Ther. 2010; 10: 103–14. [PubMed Abstract].
- Tavoosidana G, Ronquist G, Darmanis S, Yan J, Carlsson L, Wu D et al. Multiple recognition assay reveals prostasomes as promising plasma biomarkers for prostate cancer. Proc Natl Acad Sci USA. 2011; 108: 8809–14. [PubMed Abstract] [PubMed CentralFull Text].
- Sandvig K, Llorente A. Proteomic analysis of microvesicles released by the human prostate cancer cell line PC-3. Mol Cell Proteomics. 2012; 11: M111.012914. [PubMed Abstract] [PubMed CentralFull Text].
- Deryugina EI, Conn EM, Wortmann A, Partridge JJ, Kupriyanova TA, Ardi VC et al. Functional role of cell surface CUB domain-containing protein 1 in tumor cell dissemination. Mol Cancer Res. 2009; 7: 1197–211. [PubMed Abstract] [PubMed CentralFull Text].
- Siva AC, Wild MA, Kirkland RE, Nolan MJ, Lin B, Maruyama T. Targeting CUB Domain Containing Protein 1 with a Monoclonal Antibody Inhibits Metastasis in a Prostate Cancer Model. Cancer Res. 2008; 68: 3759. 66.
- Khan S, Jutzy JM, Valenzuela MM, Turay D, Aspe JR, Ashok A et al. Plasma-derived exosomal survivin, a plausible biomarker for early detection of prostate cancer. PLoS One. 2012; 7: e46737. [PubMed Abstract] [PubMed CentralFull Text].
- Gabriel K, Ingram A, Austin R, Kapoor A, Tang D, Majeed F et al. Regulation of the tumor suppressor PTEN through exosomes: a diagnostic potential for prostate cancer. PLoS One. 2013; 8(7): 70047.
- Ang J, Lijovic M, Ashman LK, Kan K, Frauman AG. CD151 protein expression predicts the clinical outcome of low-grade primary prostate cancer better than histologic grading: a new prognostic indicator?. Cancer Epidemiol Biomarkers Prev. 2004; 13: 1717–21. [PubMed Abstract].
- Huang X, Yuan T, Liang M, Du M, Xia S, Dittmar R. Exosomal miR-1290 and miR-375 as prognostic markers in castration-resistant prostate cancer. Eur Urol. 2015; 67: 33–41. [PubMed Abstract].
- Corcoran C, Rani S, O'Driscoll L. miR-34a is an intracellular and exosomal predictive biomarker for response to docetaxel with clinical relevance to prostate cancer progression. Prostate. 2014; 74: 1320–34. [PubMed Abstract].
- Bryant RJ, Pawlowski T, Catto JWF, Marsden G, Vessella RL, Rhees B et al. Changes in circulating microRNA levels associated with prostate cancer. British Journal of Cancer. 2012; 106(4): 768–774. [PubMed Abstract] [PubMed CentralFull Text].
- Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA. 2008; 105: 10513–18. [PubMed Abstract] [PubMed CentralFull Text].
- Brase JC, Johannes M, Schlomm T, Fälth M, Haese A, Steuber T et al. Circulating miRNAs are correlated with tumortumour progression in prostate cancer. Int. J. Cancer. 2011; 128: 608–616. [PubMed Abstract].
- Zhou H, Pisitkun T, Aponte A, Yuen PS, Hoffert JD, Yasuda H et al. Exosomal fetuin-A identified by proteomics: a novel urinary biomarker for detecting acute kidney injury. Kidney Int. 2006; 70: 1847–57. [PubMed Abstract] [PubMed CentralFull Text].
- Sonoda H, Takase H, Dohi Y, Kimura G. Uric acid levels predict future development of chronic kidney disease. Am J Nephrol. 2011; 33: 352–7. [PubMed Abstract].
- Fleissner F, Goerzig Y, Haverich A, Thum T. Microvesicles as novel biomarkers and therapeutic targets in transplantation medicine. Am J Transplant. 2012; 12: 289–97. [PubMed Abstract].
- Grange C, Tapparo M, Bruno S, Chatterjee D, Quesenberry PJ, Tetta C et al. Biodistribution of mesenchymal stem cell-derived extracellular vesicles in a model of acute kidney injury monitored by optical imaging. Int J Mol Med. 2014; 33: 1055–63. [PubMed Abstract] [PubMed CentralFull Text].
- Chow TF, Youssef YM, Lianidou E, Romaschin AD, Honey RJ, Stewart R et al. Differential expression profiling of microRNAs and their potential involvement in renal cell carcinoma pathogenesis. Clin Biochem. 2010; 43: 150–8. [PubMed Abstract].
- Tickoo SK, Lee MW, Eble JN, Amin M, Christopherson T, Zarbo RJ et al. Ultrastructural observations on mitochondria and microvesicles in renal oncocytoma, chromophobe renal cell carcinoma, and eosinophilic variant of conventional (clear cell) renal cell carcinoma. Am J Surg Pathol. 2000; 24: 1247–56. [PubMed Abstract].
- Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC et al. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 2011; 71: 5346–56. [PubMed Abstract].
- Del Boccio P, Raimondo F, Pieragostino D, Morosi L, Cozzi G., Sacchetta P et al. A hyphenated microLC-Q-TOF-MS platform for exosomallipidomics investigations: application to RCC urinary exosomes. Electrophoresis. 2012; 33: 689–696. [PubMed Abstract].
- Raimondo F, Morosi L, Corbetta S, Chinello C, Brambilla P, Della Mina P et al. Differential protein profiling of renal cell carcinoma urinary exosomes. Mol Biosyst. 2013; 9: 1220–33. [PubMed Abstract].
- Shariat SF, Chromecki TF, Cha EK, Karakiewicz PI, Sun M, Fradet Y et al. Risk stratification of organ confined bladder cancer after radical cystectomy using cell cycle related biomarkers. J Urol. 2012; 187: 457–62. [PubMed Abstract].
- Smalley DM, Sheman NE, Nelson K, Theodorescu D. Isolation and identification of potential urinary microparticle biomarkers of bladder cancer. J Proteome Res. 2008; 7: 2088–96. [PubMed Abstract].
- Welton JL, Khanna S, Giles PJ, Brennan P, Brewis IA, Staffurth J et al. Proteomics Aanalysis of Bbladder Ccancer Eexosomes. Molecular & Cellular Proteomics: MCP. 2010; 9(6): 1324–1338. [PubMed Abstract] [PubMed CentralFull Text].
- Chen CL, Lai YF, Tang P, Chien KY, Yu JS, Tsai CH et al. Comparative and targeted proteomic analyses of urinary microparticles from bladder cancer and hernia patients. J Proteome Res. 2012; 11: 5611–29. [PubMed Abstract].
- Ibragimova I, Ibanez de Caceres I, Hoffman AM, Potapova A, Dulaimi E, Al-Saleem T et al. Global reactivation of epigenetically silenced genes in prostate cancer. Cancer Prev Res (Phila). 2010; 3: 1084–92. [PubMed Abstract].
- Jeronimo C, Esteller M. DNA methylation markers for prostate cancer with a stem cell twist. Cancer Prev Res (Phila). 2010; 3: 1053–5. [PubMed Abstract].
- Perez A, Loizaga A, Arceo R, Lacasa I, Rabade A, Zorroza K et al. A pilot study on the potential of RNA-associated to urinary vesicles as a suitable non-invasive source for diagnostic purposes in bladder cancer. Cancers. 2014; 6(1): 179–192. [PubMed Abstract] [PubMed CentralFull Text].
- Beckham CJ, Olsen J, Yin PN, Wu CH, Ting HJ, Hagen FK et al. Bladder cancer exosomes contain EDIL-3/Del1 and facilitate cancer progression. J Urol. 2014; 192: 583–92. [PubMed Abstract].
- Yang L, Wu XH, Wang D, Luo CL, Chen LX. Bladder cancer cell-derived exosomes inhibit tumour cell apoptosis and induce cell proliferation in vitro. Mol Med Rep. 2013; 8: 1272–8. [PubMed Abstract].
- Ostenfeld MS, Jeppesen DK, Laurberg JR, Boysen AT. Cellular disposal of miR23b by RAB27-dependent exosome release is linked to acquisition of metastatic properties. Cancer Res. 2014; 74: 5758–71. [PubMed Abstract].
- Yang C, Robbins PD. The roles of tumor-derived exosomes in cancer pathogenesis. Clin Dev Immunol. 2011; 2011: 1–11.
- Safaei R, Larson BJ, Cheng TC, Gibson MA, Otani S, Naerdemann W et al. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol Cancer Ther. 2005; 4: 1595–604. [PubMed Abstract].
- Shedden K, Xie XT, Chandaroy P, Chang YT, Rosania GR. Expulsion of small molecules in vesicles shed by cancer cells: association with gene expression and chemosensitivity profiles. Cancer Res. 2003; 63: 4331–7. [PubMed Abstract].
- Panagopoulos K, Cross-Knorr S, Dillard C, Pantazatos D, Del Tatto M, Mills D et al. Reversal of chemosensitivity and induction of cell malignancy of a non-malignant prostate cancer cell line upon extracellular vesicle exposure. Mol Cancer. 2013; 12: 118. [PubMed Abstract] [PubMed CentralFull Text].
- Rini BI, Atkins MB. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009; 10: 992–1000. [PubMed Abstract].
- Rini BI. Metastatic renal cell carcinoma: many treatment options, one patient. J Clin Oncol. 2009; 27: 3225–34. [PubMed Abstract].
- Kohli M, Qin R, Jimenez R, Dehm SM. Biomarker-based targeting of the androgen-androgen receptor axis in advanced prostate cancer. Adv Urol. 2012; 2012: 781459. [PubMed Abstract] [PubMed CentralFull Text].
- Richter E, Masuda K, Cook C, Ehrich M, Tadese AY, Li H et al. A role for DNA methylation in regulating the growth suppressor PMEPA1 gene in prostate cancer. Epigenetics. 2007; 2: 100–9. [PubMed Abstract].
- Pascucci L, Coccè V, Bonomi A. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: a new approach for drug delivery. J Control Release. 2014; 192: 262–70. [PubMed Abstract].
- D'Souza-Schorey C, Clancy JW. Tumor-derived microvesicles: shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012; 26: 1287–99. [PubMed Abstract] [PubMed CentralFull Text].