
Abstract
Background
Our group has previously demonstrated that murine whole bone marrow cells (WBM) that internalize lung-derived extracellular vesicles (LDEVs) in culture express pulmonary epithelial cell–specific genes for up to 12 weeks. In addition, the lungs of lethally irradiated mice transplanted with lung vesicle–modulated marrow have 5 times more WBM-derived type II pneumocytes compared to mice transplanted with unmanipulated WBM. These findings indicate that extracellular vesicle modification may be an important consideration in the development of marrow cell–based cellular therapies. Current studies were performed to determine the specific marrow cell types that LDEV stably modify.
Methods
Murine WBM-derived stem/progenitor cells (Lin-/Sca-1+) and differentiated erythroid cells (Ter119+), granulocytes (Gr-1+) and B cells (CD19+) were cultured with carboxyfluorescein N-succinimidyl ester (CFSE)-labelled LDEV. LDEV+ cells (CFSE+) and LDEV− cells (CFSE−) were separated by flow cytometry and visualized by fluorescence microscopy, analyzed by RT-PCR or placed into long-term secondary culture. In addition, murine Lin-/Sca-1+ cells were cultured with CFSE-labelled LDEV isolated from rats, and RT-PCR analysis was performed on LDEV+ and – cells using species-specific primers for surfactant (rat/mouse hybrid co-cultures).
Results
Stem/progenitor cells and all of the differentiated cell types studied internalized LDEV in culture, but heterogeneously. Expression of a panel of pulmonary epithelial cell genes was higher in LDEV+cells compared to LDEV− cells and elevated expression of these genes persisted in long-term culture. Rat/mouse hybrid co-cultures revealed only mouse-specific surfactant B and C expression in LDEV+ Lin-/Sca-1+cells after 4 weeks of culture, indicating stable de novo gene expression.
Conclusions
LDEV can be internalized by differentiated and more primitive cells residing in the bone marrow in culture and can induce stable de novo pulmonary epithelial cell gene expression in these cells for several weeks after internalization. The gene expression represents a transcriptional activation of the target marrow cells. These studies serve as the basis for determining marrow cell types that can be used for cell-based therapies for processes that injure the pulmonary epithelial surfaces.
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
It has been well-described in multicellular organisms that intercellular communication is mediated by processes that include direct cell-to-cell contact and transfer of secreted molecules. However, an additional mechanism for intercellular communication, involving the transfer of extracellular vesicles (EVs), has recently emerged in the literature. The simplest and most inclusive definition of EVs is that they are spherical, cell-derived structures limited by a lipid bilayer of similar structure to that of the cell membrane of origin. They are shed spontaneously, but also in response to exogenous stressors including hypoxia, shear stress, irradiation, chemotherapeutic agents and cytokines (Citation1). EVs originating from platelets and red blood cells have been known about for decades and were initially felt to represent cellular cast-offs. Not only has their cellular source expanded to virtually every known cell type, their biological relevance is also gaining greater recognition.
EVs were first identified nearly 60 years ago and were described as microparticles with procoagulant activity (Citation2). Here, investigators demonstrated that the high-speed centrifugate of human cell and platelet-free plasma was capable of normalizing the clotting of blood from a patient suffering from haemophilia. Pro-thrombotic particles derived from platelets were later visualized by electron microscopy by Wolf in 1967 (Citation3). This “platelet dust” was shown to be capable of facilitating thrombin formation similarly to platelets. Their role, in vivo, was later defined when activated platelets were shown to release microparticles after attaching to the blood vessel wall (Citation4). These observations led to the belief, that in the setting of vascular injury, pro-thrombotic platelet and leukocyte-derived microparticles appear to play an integral role in thrombus formation (Citation5–Citation10). However, it was only recently that microparticles were believed to not only participate in normal homeostatic processes but also in the pathogenesis of a variety of human diseases. Platelet, monocyte and lymphocyte-derived microparticles with high tissue factor (TF) activity can be isolated from human atherosclerotic plaques, suggesting that they may participate in the pathogenesis of coronary artery disease (Citation11). In parallel with these observations, studies over the past several decades have yielded the discovery of several other sub-populations of EVs derived from a variety of cell types contributing to the notion that any given biological fluid is composed of a vastly heterogeneous collection of biologically active EVs.
Several distinct sub-populations of EVs have been described in the literature including exosomes (Citation12), microparticles (Citation13), ectosomes (Citation14), microvesicles (Citation15), membrane particles (Citation16) and apoptotic vesicles (Citation17). Common to all sub-populations is that their components are a reflection of the cell from which they were derived. They can form either at the plasma membrane or from internal compartments of the cell and contain cytoplasm as well as cell membrane components. As their membrane orientation is the same as that of the donor cell, they can be considered to be miniature versions of a cell (Citation18). Different sub-populations can be distinguished from one another based on size, density in sucrose, centrifugation force required for isolation, appearance by electron microscopy, mechanisms by which they are formed and surface protein and lipid composition (Citation18).
Our group has demonstrated that bone marrow cells are capable of contributing to the epithelial lung cellular component after transplantation into lethally irradiated mice (Citation19). A potential mechanism of this observation is that injured pulmonary epithelial cells release EVs which are internalized by cells of the bone marrow which, in turn, help to reconstitute the injured epithelium. Our previous work has shown that EVs from murine lung cells are internalized by cells of the bone marrow resulting in persistent pulmonary epithelial cell gene and protein expression in vitro and, after transplantation into lethally irradiated mice, an increase in the number of bone marrow-derived pulmonary epithelial cells in the lungs of transplant recipients (Citation20–Citation22). It is not known which type of cell residing in the bone marrow internalizes lung-derived extracellular vesicles (LDEVs) and if this results in a stable change of cellular phenotype. To address this, we performed experiments to determine if a bone marrow cell population enriched with hematopoietic stem/progenitor cells (lineage depleted, stem cell antigen-1+ or Lin-/Sca-1+) or if various differentiated hematopoietic cells of the bone marrow (cells of a granulocytic, erythrocytic and B cell lineage) are capable of internalizing LDEV in culture and stably expressing pulmonary epithelial cells genes.
Methods
Experimental animals
All mouse studies were approved by the Institutional Animal Care and Use Committee at Rhode Island Hospital (CMTT numbers 0131-11, 0080-13). Six- to eight-week-old male C57BL/6 mice (Jackson Laboratories) were used for all studies. Animals had ad libitum access to food and water and were given 1 week to acclimate prior to experimental treatment or euthanasia. Euthanasia was performed using CO2 inhalation or isoflurane inhalation followed by cervical dislocation.
Tissue harvest
For lung harvest, blood was flushed from the pulmonary vasculature through the right ventricle using Dulbecco's phosphate-buffered saline (PBS, Invitrogen). Lungs were placed in ice-cold PBS supplemented with 5% heat-inactivated foetal calf serum (HICFS, Hyclone) and 1% penicillin-streptomycin (PS, Invitrogen). For whole bone marrow cell (WBM) harvest, tibiae, femurs, iliac crests and spines were collected, and all surrounding muscles were removed with sterile gauze. Bones were placed in ice-cold PBS/5%HIFCS/1%PS and crushed using mortar and pestle. Cells were strained through 40 µm cell strainer (Allegiance) then centrifuged at 300 g for 10 minutes at 4°C.
Lineage depletion
Mononuclear cells were isolated from WBM by discontinuous density centrifugation at 1,000 g for 30 minutes (room temperature) using OptiPrep (Accurate Chemical). Mononuclear cells were lineage depleted (Lin-) by adding the following rat–anti-mouse antibodies: anti-Ter119, B220, Mac-1, Gr-1, CD4 and CD8 (BD Biosciences). Cells were incubated on ice for 15 minutes. Dynabead M450 anti-rat IgG (Dynal) was added, and lineage positive cells were removed by a magnetic column. Remaining Lin- cells were counted, and percent viability was determined using trypan blue stain (Gibco).
Isolation of Lin-/Sca-1+cells
Allophycocyanin (APC)-conjugated anti-mouse LY-6AE (Sca-1) (BD Biosciences) was added to a final concentration of 1 µg/106 Lin- cells, then incubated for 30 minutes on ice. Cells were washed with PBS, centrifuged at 300 g for 10 minutes and then passed through a 40-µm filter. Propidium iodide (0.05 mg/mL) was added (1:1,000 dilution), and Lin-/Sca-1+ cells were then separated using a 5 laser Becton Dickinson/Cytopeia Influx High Speed Cell Sorter.
Isolation of differentiated bone marrow cells
WBM cells (isolated as described above) at 1×107 cells/ml PBS were labelled with either of the following antibodies (0.25 µg/1×106 cells, Invitrogen) on ice for 15 minutes: Anti-Ter119-APC (erythroid cells), CD19-pacific blue (B cells) or Gr-1-APC (granulocytes). Cells were washed with PBS, centrifuged at 300 g for 10 minutes and then passed through a 40-µm filter. Propidium iodide (0.05 mg/mL) was added (1:1,000 dilution), and erythroid cells, B cells and granulocytes were then separated using a 5 laser Becton Dickinson/Cytopeia Influx High Speed Cell Sorter.
LDEV isolation and characterization
Lungs were filled with dispase (Sigma) and incubated for 45 minutes on ice. Lungs were then mechanically dissociated with forceps into a single cell suspension, filtered through a 40 µm cell strainer and washed with PBS by centrifugation (300 g for 10 minutes, 4°C). Lung cells were cultured (1×106 cells/ml) in Bronchial Epithelial Growth Media (BEGM, Lonza), supplemented with 0.5 µg/ml epinephrine, 10 µg/ml transferrin, 5 µg/ml insulin, 0.1 ng/ml retinoic acid, 52 µg/ml bovine pituitary extract, 0.5 µg/ml hydrocortisone, 0.5 pg/ml human recombinant epidermal growth factor and 6.5 ng/ml triiodothyronine, at 37°C/5% CO2 for 7 days. Cultured lung cells were then removed from the conditioned media by centrifugation at 300 g for 10 minutes at 4°C (performed twice). Cell-free media was ultracentrifuged at 100,000 g for 1 hour at 4°C in a Thermo Scientific Sorval WX Ultra series ultracentrifuge. The supernatant was discarded, and the pellet was resuspended in PBS supplemented with 5 mM Hepes [4-(2-hydroxyethyl) piperazine-1-ethanesulfonic acid, N-(2-hydroxyethyl) piperazine-N′-(2-ethanesulfonic acid)] (Sigma). The pelleted material, LDEVs, was ultracentrifuged again at 100,000 g for 1 hour at 4°C, resuspended in PBS. LDEVs isolated by these methods have been fully characterized, and these qualities have been described in a recent publication by our group (Citation23). Proteomic analysis of our LDEVs demonstrated the presence of proteins characteristic of both exosomes (including various tetraspanins) and microvesicles (including CD40 ligand). LDEVs were quantified by BCA assay and by NanoSight NS500 analysis. NanoSight and transmission electron microscopy (Supplementary Fig. 1) analyses revealed EV in the size range of exosomes and microvesicles.
Isolation of CFSE positive cells after exposure to CFSE-labelled LDEV in culture
LDEVs were labelled with the cell cytoplasm dye CFSE (carboxyfluorescein N-succinimidyl ester, Molecular Probes), final concentration of 0.02 µM, for 15 minutes at 37°C. An equal volume of 10% foetal bovine serum solution in PBS was then added, and the samples were ultracentrifuged at 100,000 g for 1 hour at 4°C. 5×105 erythroid cells, B cells, granulocytes or Lin-/Sca-1+ cells were plated in 6-well plastic culture plates (USA Scientific) in 5 ml of DMEM-glutamax at 5.3×104 cells/cm2. CFSE-labelled LDEV isolated from 1 mouse lung (25 µg EV protein by BCA protein assay) were added to culture wells so that LDEVs isolated from 1 mouse lung was co-cultured with 5×105 cells. As a control, no LDEVs were added to other culture wells. Co-cultures were maintained at 37°C for 48 hours. Cells were then harvested, washed with PBS and centrifuged at 300 g for 10 minutes at 4°C and then passed through a 40-µm filter. Cells were then separated using a 5 laser Becton Dickinson/Cytopeia Influx High Speed Cell Sorter based on their presence or absence of CFSE. CFSE was excited at 488 nm and detected through a 528/38 bandpass filter. CFSE+ events were defined as cells that internalized CFSE-labelled LDEV whereas CFSE negative events were defined as cells exposed to LDEV in culture that did not internalize them. Cells were then analyzed by RT-PCR or placed into long-term culture.
Imaging of CFSE positive cells after isolation by flow cytometry
Cells were counterstained with the nuclear label 4′-6-diamidino-2-phenylindole (DAPI), visualized using conventional and deconvolution fluorescence microscopy (Zeiss Axioplan 2 microscope; Carl Zeiss) at room temperature and photographed at 63×magnification using the AxioVision software package (Carl Zeiss). Three dimensional images were created from a 25-layer (0.4 micron/layer) z stack to demonstrate co-localization of fluorescent signal. No photosubtraction or processing of artefact was performed.
Long-term cell cultures
Cells that internalized LDEV, cells that had been exposed to LDEV in culture that did not internalize them and cells that were not exposed to LDEV (control) were plated in 6-well plastic culture plates and cultured in DMEM-glutamax supplemented with 15% foetal bovine serum, 1% PS and recombinant murine stem cell factor (SCF, final concentration 50 ng/ml) at 37°C (5.3×104 cells/cm2). Cells were split into separate wells when they reached 80–100% confluence and were removed at 2 weeks intervals (up to 8 weeks) for RT-PCR analysis.
RT-PCR analysis
Total RNA extracted from cells was measured for quantity and quality (260/280 ratio) using a Nanodrop ND/1000 spectrophotometer (Thermo Scientific). For each sample, 10 ng of RNA was used to amplify cDNA using the High Capacity cDNA transcription kit (Applied Biosystems). cDNA amplification reactions were performed on a 9800 Fast Thermal Cycler (Applied Biosystems) and consisted of 1 cycle (10 minutes, 25°C), 1 cycle (120 minutes, 37°C) and 1 cycle (5 minutes, 85°C). All Real Time RT-PCR reactions were performed 7900HT Fast RT PCR System using the following murine primers: β2 microglobulin (Mm00437762_m1), TTF-1 (Mm00447558_m1), c/EBP alpha (Mm00514283_s1), HNF-3 beta (FOXa2) (Mm01976556_s1), Ppp3r1 (Mm01187904_m1), sftpa1 (Mm00499170_m1), Sftpb (Mm00455681_m1), Sftpc (Mm00488144_m1), Sftpd (Mm00486060_m1), Scgb1a1 (Mm00442046_m1), Aqp5 (Mm00437578_m1), CD34 (Mm00519283_m1), Ptprc (Mm00448463_m1), CD150 (Mm00443316_m1), ckit (Mm00445212_m1), sca-1, (Mm00726565_s1), mac-1 (Mm00434455_m1), gr-1 (Mm00439151_m1), Fcgr2b (CD32) (Mm00438875_m1), Fcgr4 (CD16) (Mm00519988_m1), EGF (Mm00500151_m1), ABCa3 (Mm00550501_m1) and pdpn (Mm00494716_m1). Duplicate reactions of the target and housekeeping genes were performed simultaneously for each cDNA template. The PCR reaction consisted of an initial enzyme activation step at (10 minutes, 95°C) followed by 40 cycles (15 seconds, 95°C; 1 minute, 60°C). A cycle threshold value (CT) value was obtained for each sample, and duplicate sample values were averaged. The 2−ΔΔCT method was used to calculate relative expression of each target gene (Citation24). To calculate 2−ΔΔCT for target genes with no expression in the control group, a CT value of 40 was assigned to the control group so that a relative quantity of the target gene could be reported.
Cell culture experiments used for RT-PCR analysis were performed a total of 3 times, and fold expression values were averaged from all experiments. A fold difference of greater than or less than 2 compared with the comparison group is considered biologically significant. Gene expression changes are displayed as heat maps using a base 2 logarithmic scale such that a change in colour shade represents a 2-fold difference in gene expression. Genes with increased expression relative to the control group (≥2-fold increase) are displayed as shades of red whereas genes with decreased expression relative to the control group (≥2-fold decrease) are displayed as shades of green. Genes with no significant change in gene expression relative to the control group (<2-fold increase or <2-fold decrease) are displayed using a red/green colour. Genes with no expression are displayed in black. Gene expression in cells that achieved statistical significance (Student's t-test) compared with its control group is displayed with an asterisk in all heat maps.
Rat/mouse hybrid co-culture
LDEVs were isolated from 1 Fischer-344 rat and labelled with CFSE, as described above. Lin-/Sca-1+ bone marrow cells isolated from male C57BL/6 mice were plated in 6-well plastic culture plates (USA Scientific) in 5 ml of DMEM-glutamax at 5.3×104 cells/cm2. Twenty-five micrograms of CFSE-labelled rat LDEV were added to this flask. As a control, Lin-/Sca-1+ bone marrow cells were cultured in the absence of LDEV. Co-cultures were maintained at 37°C for 48 hours separated by flow cytometry into cells that internalized LDEV and cells that had been exposed to LDEV in culture that did not internalize them. All cell populations, including cells not exposed to LDEV (control) were plated in 6-well plastic culture plates and cultured in DMEM-glutamax supplemented with 15% foetal bovine serum, 1% PS and recombinant murine (SCF, final concentration 50 ng/ml) at 37°C (5.3×104 cells/cm2) for 1 or 4 weeks. For 4 week cultures, cells were split into new culture wells when 80–100% confluence was achieved. This occurred at culture day 7 (passage 1), day 9 (passage 2), day 12 (passage 3), day 16 (passage 4) and day 22 (passage 5). All passages were cultured for a total of 4 weeks. After culture, cells were analyzed by RT-PCR using mouse or rat-specific primers for the pulmonary epithelial cell–specific genes surfactant B (Sp-B) and C (Sp-C). Murine assays used were as follows: β2 microglobulin (Mm00437762_m1), surfactant B (Mm00455681_m1) and surfactant C (Mm00488144_m1). Rat assays included β2 microglobulin (Rn00560865_m1), surfactant B (Rn00684785_m1), surfactant C (Rn01466216_g1) (all from Applied Biosystems). Each passage from the 4 week cell cultures was analyzed separately.
Statistical analysis
Data were analyzed using the Student's t-test when there were fewer than 6 measurements within 2 parent groups. Alternatively, Wilcoxon rank sum test was performed when there were 6 or more measurements within 2 parent groups; p≤0.05 represented statistical significance. Data were presented as mean±standard error.
Results
LDEVs are internalized by bone marrow–derived stem/progenitor cells and differentiated cells in culture
To determine if both bone marrow-derived stem/progenitor cells and differentiated cells are capable of internalizing LDEV in culture, bone marrow–derived Lin-/Sca-1+ cells and differentiated erythroid cells (Ter119+), granulocytes (Gr-1+) and B cells (CD19+) were cultured with CFSE-labelled LDEV for 48 hours. Cultured cells were then separated by flow cytometry into cells that internalized LDEV (LDEV+ cells) and cells exposed to LDEV that did not internalize them (LDEV− cells, ). By fluorescence microscopy, CFSE+ LDEV could be visualized within all 4 populations flow cytometry–separated cells and were noted to be adjacent to the nuclei of all cell types (). LDEV+ cells, expressed as a percentage of all cells cultured with CFSE-labelled LDEV, were 25.2% for Ter119+, 29.5% for CD19+ and 29.2% for Gr-1+ cells, but these were not significantly different from one another. However, there were significantly fewer LDEV+ Lin-/Sca-1+ cells (15.3%) compared with other cell populations (p<0.05, Table ). These data indicate that stem/progenitor cells and differentiated cells derived isolated from the bone marrow are capable of internalizing LDEV in culture.
Fig. 1. Separation of LDEV+ and LDEV− bone marrow cells cultured with CFSE-labelled LDEV. (a) Whole bone marrow (WBM) cells (5.3×104 cells/cm2) cultured without CFSE-labelled LDEV for 48 hours. (b) Lin-/Sca-1+, (c) CD19+, (d) Ter119+ and (e) Gr-1+ cells (all 5.3×104 cells/cm2) cultured with CFSE-labelled LDEV (one mouse lung equivalent) for 48 hours. LDEV+ cells (R3) and LDEV− (**) could be detected and separated by flow cytometry. Y-axis, CFSE (FITC); X-axis, forward scatter (FSC).
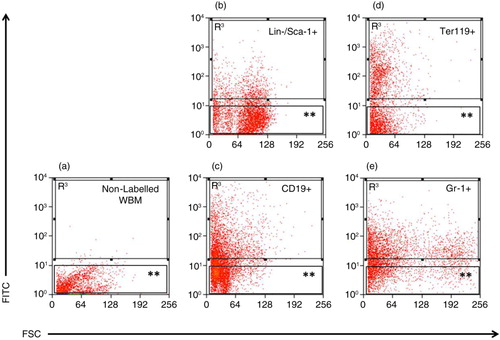
Fig. 2. Flow cytometry–separated LDEV+ cells. (a) Lin-/Sca-1+, (b) CD19+, (c) Ter119+ and (d) Gr-1+ cells that have internalized CFSE-labelled LDEV. CFSE-labelled LDEV (FITC, green) can be found adjacent to the nucleus (DAPI, blue). Red bar=10 µm.
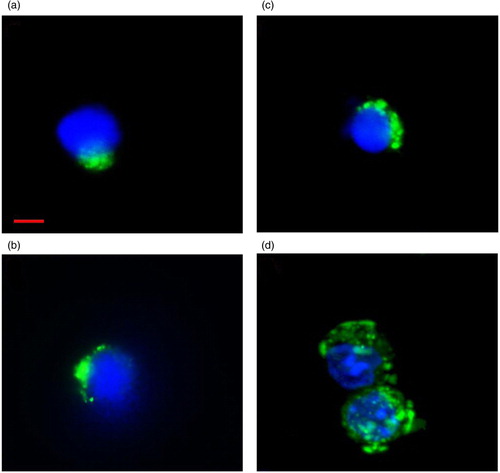
Table I. LDEV+ bone marrow cell populations after culture with CFSE-labelled LDEV
Cells that internalize LDEV contain pulmonary epithelial cell gene mRNA transcripts
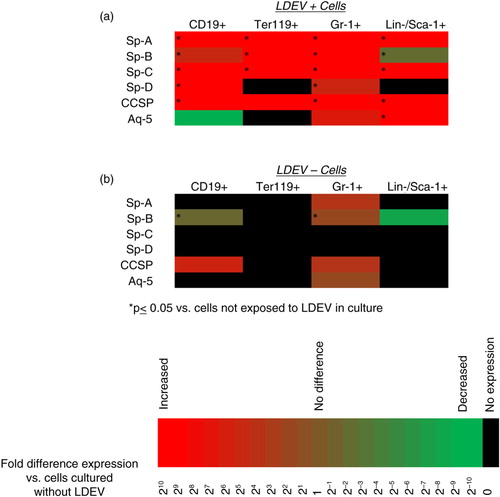
To determine if cells that internalize LDEVs contain mRNA species that are typically expressed by pulmonary epithelial cells, RT-PCR analysis was performed on flow cytometry–separated LDEV+ and LDEV− cells, focusing on genes expressed by type I pneumocytes (aquaporin-5; Aq-5), type II pneumocytes (surfactants A-D; Sp-A, Sp-B, Sp-C, Sp-D) and clara cells (clara cell-specific protein; CCSP). We found that there was high and statistically significant expression of most of these genes in LDEV+ cell populations whereas expression in LDEV− cells was relatively low or absent (). These data indicate that pulmonary epithelial cell mRNA species can be detected in abundance in cells that internalize LDEVs.
Fig. 3. Pulmonary epithelial cell mRNA transcripts detected in cells cultured with CFSE-labelled LDEV. (a) Cells cultured with CFSE-labelled LDEV (5.3×104 cells/cm2 with one mouse lung equivalent of LDEV for 48 hours) that are LDEV+ and (b) LDEV−. Gene expression is relative to cells not exposed to LDEV in culture (control) and is displayed as heat maps using a base 2 logarithmic scale. Cell culture experiments performed 3 times, and fold expression values are an average of all 3 experiments. *p>0.05 (Student's t-test), gene expression versus control cells.
Persistent pulmonary epithelial cell gene expression in cells that internalize LDEV
To determine if cells that have internalized LDEV in culture have persistent expression of pulmonary epithelial cell genes, LDEV+ and LDEV− Lin-/Sca-1+ cells and Gr-1+ cells were separated by flow cytometry and placed into secondary culture. Compared to LDEV− Lin-/Sca-1+ cells and Gr-1+ cells, those that were LDEV+ generally had significantly increased expression of a variety of pulmonary epithelial cells genes. For some of these genes, expression remained elevated for up to 8 weeks in culture ().
Fig. 4. Gene expression profile of LDEV+ Gr-1+ and Lin-/Sca-1+ cells in long-term culture. Expression of pulmonary epithelial cell genes, stem/progenitor cell genes and granulocyte genes in (a) Gr-1+ cells and (b) Lin-/Sca-1+ cells that are LDEV+ relative to cells that are LDEV−. Cells were cultured for 2, 4, 6 or 8 weeks (5.3×104 cells/cm2). Gene expression changes relative to LDEV− cells are displayed as heat maps using a base 2 logarithmic scale. Cell culture experiments performed 3 times, and fold expression values are an average of all 3 experiments. *p > 0.05 (Student's t-test), gene expression versus control cells.
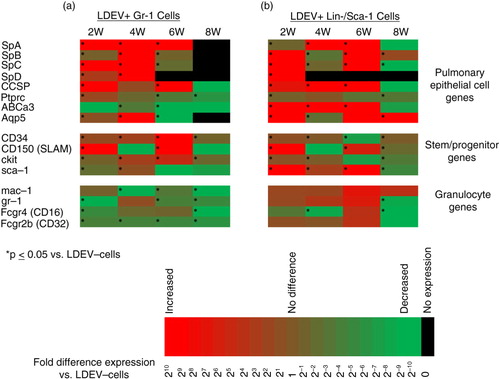
In addition, LDEV+ Lin-/Sca-1+ cells had significantly increased expression of a panel of stem/progenitor cell genes compared to LDEV− Lin-/Sca-1+ cells, and this expression persisted in culture. Although granulocyte gene expression was also detected in these cells, expression was generally less than or not different than expression in LDEV− Lin-/Sca-1+ cells. These data suggest that Lin-/Sca-1+cell internalization of LDEV induces persistent expression of stem/progenitor cell genes. Granulocyte gene expression in LDEV+ Gr-1+cells was generally less than expression in LDEV− Gr-1+cells. However, stem/progenitor cell gene expression in these cells was significantly increased compared to LDEV− Gr-1+ cells, and this expression persisted in culture. These data suggest that Gr-1+cell internalization of LDEV induces loss of granulocyte gene expression and induction of stem/progenitor cell gene expression ().
De novo pulmonary epithelial cell gene expression in cells that internalize LDEV
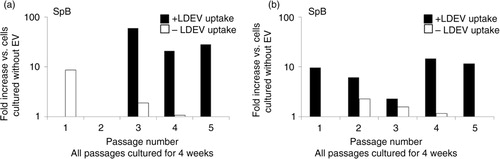
To determine if pulmonary epithelial cell gene mRNA species detected in cells that internalized LDEVs is directly transferred by LDEV or transcribed de novo, we cultured murine Lin-/Sca-1+ cells with CFSE-labelled EVs isolated from rat lungs. LDEV+ and LDEV− cells isolated by flow cytometry then cultured for up to 4 weeks, and cells were analyzed by RT-PCR using mouse and rat-specific primers for Sp-B and Sp-C. Immediately after separation into LDEV+ and LDEV− cells (day 0), we found only rat-specific Sp-B and Sp-C in LDEV+ cells, indicating that these mRNA species were transferred from rat LDEV. In addition, rat-specific Sp-B was also isolated from LDEV− cells. After 7 days of culture (day 7), only mouse-specific Sp-B and Sp-C was isolated from LDEV+ cells, indicating that these mRNA species were produced de novo by murine Lin-/Sca-1+ cells. No surfactant expression was detected in LDEV− cells after 7 days of culture (). Due to cell growth in culture, remaining cells were split into new culture wells at culture day 7 (passage 1), day 9 (passage 2), day 12 (passage 3), day 16 (passage 4) and day 22 (passage 5). All culture wells were maintained for a total of 4 weeks, and then cells were analyzed by RT-PCR. We found that after 4 weeks of culture, LDEV+cells isolated from each passage expressed Sp-B and/or Sp-C, and the surfactant expressed was only mouse-specific. LDEV− cells expressed Sp-B and Sp-C as well, although levels were lower compared with LDEV+ cells (). These data indicate that de novo pulmonary epithelial gene expression occurs in Lin-/Sca-1+ cells that have internalized LDEV, and that this expression persists for weeks in culture.
Fig. 5. Sp-B and Sp-C expression in the mouse Lin-/Sca-1 cell/rat LDEV hybrid co-culture, short term. Expression (+ + +) or no expression (–) of mouse- or rat-specific Sp-B, Sp-C in mouse Lin-/Sca-1+ cells that were cultured for 48 hours with CFSE-labelled rat LDEV (5.3×104 cells/cm2 with 25 µg of rat LDEV). Shown is the presence or absence of expression in cells immediately after flow cytometry separation into LDEV+ cells and LDEV− cells (Day 0) and after 7 days of culture (Day 7).
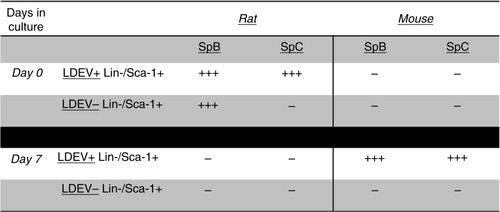
Fig. 6. Sp-B and Sp-C expression in the mouse Lin-/Sca-1 cell/rat LDEV hybrid co-culture, long term. Mouse-specific (a) Sp-B and (b) Sp-C expression in LDEV+ (black bars) and LDEV− (white bars) cells from each passage (X-axis) after 4 weeks of culture. No rat-specific Sp-B or Sp-C expression was found in either cell population (not shown). Fold difference of gene expression is compared to cells cultured without LDEV (Y-axis).
Discussion
We have previously demonstrated that unfractionated murine WBM cells cultured with EVs isolated from murine lung tissue express a variety of pulmonary epithelial cell genes in vitro for up to 12 weeks (Citation22). In these studies, we also demonstrated that the lungs of lethally irradiated mice transplanted with WBM cells cultured with LDEV have up to 5 times more bone marrow-derived type II pneumocytes when compared to mice transplanted with unmanipulated WBM cells. These studies did not investigate the specific cell type residing in the bone marrow that is responsible for these stable changes in cellular phenotype. The bone marrow serves as an attractive source of cells for the development of cell-based therapies given their ability to engraft a recipient's bone marrow, and thus provide a stable reservoir of reparative cells. Exposure of cells to EVs prior to transplantation may augment their effects; however, many critical variables must be defined including the cellular source of the EVs and the marrow cells that EVs are specifically targeting. Importantly, these variables must be considered in the context of the disease process of interest. The goal of the present study is to define the bone marrow cell type which may be subject to stable EV-induced phenotypic changes.
We report that after exposure to CFSE-labelled LDEV in culture, bone marrow–derived hematopoietic stem/progenitor cells (Lin-/Sca-1+ cells) and differentiated hematopoietic cells, including granulocytes (Gr-1+), erythrocytes (Ter119+) and B cells (CD19+), can be identified by flow cytometry as CFSE+ cells. Internalization of LDEV was further confirmed by fluorescence microscopy as CFSE+ LDEV could be identified within the cytoplasm of all cell types. Distribution of LDEV within all cell types appeared similar as they clustered in a peri-nuclear pattern. However, the biological effects, that is, induction of different mRNAs, differed between cell types. This indicates that while uptake of LDEVs may be similar, the cellular responses are dependent upon specific target cell features.
Further biologic complexity is indicated by the fact that LDEVs used in this study represent a heterogeneous mixture of vesicle cell populations and were likely to have been derived from several of the dozens of cells present in the murine lung. We chose a panel of mRNAs expressed in pulmonary epithelial cells including type I (Aq-5) and type II (Sp-A-D) pneumocytes and clara cells (CCSP) to demonstrate LDEV/bone marrow cell interactions as we have previously demonstrated that the technique we use to isolate LDEV from cultured murine lung cells contain these transcripts (Citation20–Citation22). Detection of pulmonary epithelial cell mRNA in cells that ordinarily harbour few to none of these transcripts further demonstrates that LDEV are capable of transferring their mRNA cargo to bone marrow cells in culture. Importantly, cells that were separated by flow cytometry based on CFSE fluorescence had substantially more of these mRNA transcripts than cells exposed to LDEV in culture that were CFSE negative, supporting the notion that internalization of LDEV leads to transfer of their mRNAs.
Although LDEV-based pulmonary epithelial cell–derived mRNA transcripts could be detected in all bone marrow cell types soon after exposure to LDEV in culture, these transcripts appeared to persist for weeks after placing cells into secondary culture. We have previously demonstrated in a hybrid rat–mouse co-culture system (using LDEV isolated from rats and marrow cells isolated from mice) that pulmonary epithelial cell mRNA transcripts isolated from co-cultured marrow cells at early time points were of both mouse and rat origin, indicating that originator cell mRNAs and a transcriptionally active factor or factors were transferred However, at later co-culture time points (beyond 2 weeks), pulmonary epithelial cell mRNA transcripts were mouse-specific, indicating that they were transcribed de novo (Citation22). In present studies, Lin-/Sca-1+ and Gr-1+ cells that had internalized LDEV, as well as cells that did not internalize LDEV, were placed into secondary culture for up to 8 weeks. We found that there was persistent expression of various pulmonary epithelial cell genes in both LDEV+ cell populations for up to 8 weeks. In most instances, expression of these genes was significantly higher compared to LDEV− cells. Interestingly, LDEV+ Lin-/Sca-1+ cells had high persistent expression of stem/progenitor cell genes compared to LDEV− cells. Granulocyte gene expression in LDEV+ Gr-1+ cells was generally lower compared with LDEV− cells but stem/progenitor cell gene expression in these cells was significantly increased, suggesting that Gr-1+ cell internalization of LDEV induces loss of granulocyte gene expression and induction of stem/progenitor cell gene expression. This phenotypic change may explain how differentiated bone marrow cells can be maintained in culture and stably express pulmonary epithelial cell genes.
Hybrid rat/mouse co-culture experiments were performed to determine if the presence of pulmonary epithelial cell-derived mRNA in bone marrow cells was due to transfer of LDEV-based mRNA or due to the transfer of LDEV-based factors that induce de novo transcription of pulmonary epithelial cell mRNA. These experiments demonstrated that soon after LDEV internalization, Lin-/Sca-1+ cell-based surfactant mRNA species were only rat-specific, indicating transfer of mRNA species via LDEV. After 4 weeks in culture, surfactant mRNA species isolated were only mouse-specific, indicating de novo transcription. Although LDEV+ cells and LDEV− cells both expressed varying degrees of surfactant in long-term culture, LDEV− cells appeared to lose surfactant expression with successive culture passages. By passages 4 and 5, LDEV− cell surfactant expression was very low or absent while LDEV+ cell surfactant expression remained elevated. These data further emphasize the importance of LDEV internalization for long-term, stable, de novo expression of pulmonary epithelial cell genes.
In summary, differentiated and more primitive cells residing in the bone marrow are capable of internalizing LDEV in culture. Pulmonary epithelial cell mRNA is transferred to these cells and, presumably, LDEV-based transcription factors which govern persistent expression of these genes for several weeks in culture. Persistent LDEV-induced gene expression in different cell types that can be maintained and expanded in culture present the opportunity to broaden our understanding of bone marrow cell–based cellular therapies. The optimal cell type used to repair the pulmonary epithelium is likely dependent on many factors, including the disease model being tested, and the mechanism of repair may depend on cellular engraftment or non-engraftment related paracrine effects. These important variables require further investigation and are outside of the scope of these studies. These studies do serve as the basis for determining the optimal cell type that can be used for cell-based therapies for processes that injure the pulmonary epithelial surfaces.
Conflict of interest and funding
The authors have not received any funding or benefits from industry or elsewhere to conduct this study.
Authors' contributions
JMA, MP, EHS and MSD performed the research; JMA, MP, EHS, MSD and PJQ designed the research study; JMA, MP, EHS, MSD and PJQ analyzed the data; JMA, SW, LRG and PJQ wrote the paper.
Supplementary figure 1
Download PDF (92.2 KB)Acknowledgements
This project was supported by the National Heart Lung and Blood Institute (NHLBI) of the National Institutes of Health (NIH) through the grant numbers 5K08HL086868 and 1R01HL103726 and by the National Center for Research Resources (NCRR) and the National Institute of General Medical Sciences (NIGMS) of the NIH through Grant Numbers 8P20GM103468 and 8P20GM103421.
Notes
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
Related Research Data
References
- Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia. 2006; 20: 1487–95.
- Chargaff E, West R. The biological significance of the thromboplastic protein of blood. J Biol Chem. 1946; 166: 189–97.
- Wolf P. The nature and significance of platelet products in human plasma. Br J Haematol. 1967; 13: 269–88.
- Warren BA, Vales O. The release of vesicles from platelets following adhesion to vessel walls in vitro. Br J Exp Pathol. 1972; 53: 206–15.
- Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA. Tissue-factor bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood. 2005; 106: 1604–11.
- Mackman N. Tissue-specific hemostasis in mice. Arterioscler Thromb Vasc Biol. 2005; 25: 2273–81.
- Falati S, Liu Q, Gross P, Merrill-Skoloff G, Chou J, Vandendries E et al. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J Exp Med. 2003; 197: 1585–98.
- Hrachovinova I, Cambien B, Hafezi-Moghadam A, Kappelmayer J, Camphausen RT, Widom A et al. Interaction of P-selectin and PSGL-1 generates microparticles that correct hemostasis in a mouse model of hemophilia A. Nat Med. 2003; 9: 1020–5.
- Andre P, Hartwell D, Hrachovinova I, Saffaripour S, Wagner DD. Pro-coagulant state resulting from high levels of soluble P-selectin in blood. Proc Natl Acad Sci USA. 2000; 97: 13835–40.
- Cambien B, Wagner DD. A new role in hemostasis for the adhesion receptor P-selectin. Trends Mol Med. 2004; 10: 179–86.
- Mallat Z, Hugel B, Ohan J, Lesèche G, Freyssinet JM, Tedgui A. Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques: a role for apoptosis in plaque thrombogenicity. Circulation. 1999; 99: 348–53.
- Keller S, Sanderson MP, Stoeck A. Exosomes: from biogenesis and secretion to biological function. Immunol Lett. 2006; 107: 102–8.
- Morel O, Toti F, Hugel B, Freyssinet JM. Cellular microparticles: a disseminated storage pool of bioactive vascular effectors. Curr Opin Hematol. 2004; 11: 156–64.
- Wood CR, Huang K, Diener DR, Rosenbaum JL. The cilium secretes bioactive ectosomes. Curr Biol. 2013; 23: 906–11.
- Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood. 1999; 94: 3791–9.
- Lorizate M, Sachsenheimer T, Glass B, Habermann A, Gerl MJ, Kräusslich HG et al. Comparative lipidomics analysis of HIV-1 particles and their producer cell membrane in different cell lines. Cell Microbiol. 2013; 15: 292–304.
- Schiller M, Heyder P, Ziegler S, Niessen A, Claben L, Lauffer A et al. During apoptosis HMGβ1 is translocated into apoptotic cell-derived membraneous vesicles. Autoimmunity. 2013; 46: 342–6.
- Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009; 9: 581–93.
- Aliotta JM, Keaney P, Passero M, Dooner MS, Pimentel J, Greer D et al. Bone marrow production of lung cells: the impact of G-CSF, cardiotoxin, graded doses of irradiation, and subpopulation phenotype. Exp Hematol. 2006; 34: 230–41.
- Aliotta JM, Sanchez-Guijo FM, Dooner GJ, Johnson KW, Dooner MS, Greer KA et al. Alteration of marrow cell gene expression, protein production, and engraftment into lung by lung-derived microvesicles: a novel mechanism for phenotype modulation. Stem Cells. 2007; 25: 2245–56.
- Aliotta JM, Pereira M, Johnson KW, de Paz N, Dooner MS, Puente N et al. Microvesicle entry into marrow cells mediates tissue-specific changes in mRNA by direct delivery of mRNA and induction of transcription. Exp Hematol. 2010; 38: 233–45.
- Aliotta JM, Pereira M, Amaral A, Dooner MS, Sears EH, Brilliant K et al. Stable cell fate changes in marrow cells induced by lung-derived microvesicles. J Extracell Vesicles. 2012; 1: 18163. doi: http://dx.doi.org/10.3402/jev.v1i0.18163.
- Aliotta JM, Pereira M, Amaral A, Sorokina A, Igbinoba Z, Hasslinger A et al. Induction of pulmonary hypertensive changes by extracellular vesicles from monocrotaline-treated mice. Cardiovasc Res. 2013; 100: 354–62.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001; 25: 402–8.